

Reduction Chemistry of Natural Pyrethrins and Preliminary Insecticidal Activity of Reduced Pyrethrins
Todd E. Markham A B C , Andrew C. Kotze D , Peter J. Duggan A C E and Martin R. Johnston A B E
A C E and Martin R. Johnston A B E
A College of Science and Engineering, Flinders University, Adelaide, SA 5042, Australia.
B Flinders Institute for Nanoscale Science and Technology, Flinders University, Adelaide, SA 5042, Australia.
C CSIRO Manufacturing, Bag 10, Clayton South, Vic. 3169, Australia.
D CSIRO Agriculture and Food, St Lucia, Qld 4067, Australia.
E Corresponding authors. Email: peter.duggan@csiro.au; martin.johnston@flinders.edu.au
Australian Journal of Chemistry 74(4) 268-281 https://doi.org/10.1071/CH20302
Submitted: 9 October 2020 Accepted: 24 November 2020 Published: 11 December 2020
Journal Compilation © CSIRO 2021 Open Access CC BY
Abstract
The natural extract pyrethrum is an insecticidal oil derived from Tanacetum cinerariifolium that is commonly used in domestic and agricultural pesticides. The major constituents of the extract are the Pyrethrins, six esters that provide pyrethrum with its insecticidal properties. These Pyrethrins readily degrade through several environmental means and as such, there can be significant Pyrethrin losses during processing and long-term storage of pyrethrum-based insecticides. This work attempts to alleviate the effect of these degradative processes through the pursuit of stabilised Pyrethrins by chemically removing oxidatively sensitive functionality. Several reduced Pyrethrin analogues were produced and a method to convert the more sensitive Pyrethrins present in the pyrethrum concentrate into their respective more stable jasmolin counterparts, as a mixture with the over-reduced tetrahydropyrethrins, was developed. All other reduction processes abolished insecticidal activity against Lucilia cuprina larvae, whereas some isomerised analogues showed comparable potency with the individual natural pyrethrin esters. This work has revealed new insights into the structure–activity relationships in this unique class of insecticide.
Introduction
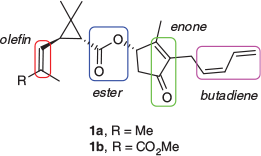
Pyrethrum, the natural extract of Tanacetum cinerariifolium (pyrethrum daisies), has long been utilised as an insect repellent and insecticidal agent.[1] The extract is used widely throughout the domestic and agricultural sectors for pest control and crop protection respectively, particularly where low mammalian toxicity and minimal environmental impact are paramount. The pyrethrum concentrate contains six esters, commonly known as the Pyrethrins,† which are the source of its repellent and insecticidal activity. These esters (Fig. 1) comprise a chrysanthemic acid (Pyrethrins I, 1a–3a) or pyrethric acid, also known as chrysanthemic diacid (Pyrethrins II, 1b–3b), linked with hydroxycyclopentenones, known as rethrolones.[2] The individual esters differ by the hydrocarbon chains on the rethrolone moiety where the pyrethrins (1a and 1b) contain a conjugated pentadienyl substituent, the jasmolins (2a and 2b) are substituted with a cis-pentenyl chain, and the cinerins (3a and 3b) have a shorter cis-butenyl alkyl group.[3]

|
The Pyrethrins are prone to degradation through several chemical routes, bestowing the concentrate with some attractive qualities, such as its short residence time in the environment and very limited propensity to induce insect resistance, which have allowed its continued widespread use. Despite these environmentally favourable qualities, the ready decomposition by thermal, photochemical, and oxidative means can detrimentally affect the long-term storage and applicability of pyrethrum.[2,4–6] Current measures in place to limit degradation include storage in dark, cool facilities and the presence of antioxidant additives like butylated hydroxytoluene (BHT). However, these preventative measures do not entirely alleviate the loss of Pyrethrins during the production and storage of the pyrethrum extract.
Previous synthetic chemical approaches aiming to reduce the rate of degradation of this class of insecticide led to the development of the pyrethroids. These fully synthetic analogues share structural similarities with the natural Pyrethrins and act on insects through similar modes.[7] However, the pyrethroids are accompanied by their own limitations such as increased environmental persistence and inducing insecticidal resistance.[8,9] The synthetic manipulation of the natural Pyrethrins still remains a fairly unexplored approach to increasing the stability of these environmentally sensitive natural products.
The pyrethrin scaffold is rich with chemical functionality (Fig. 2), which ultimately contributes to the instability and high reactivity of the pyrethrins under environmental conditions, but also provides avenues for synthetic manipulation.
|
Many of the functional groups within the pyrethrin structures contain unsaturated centres, which contribute to their photolytic and oxidative sensitivity.[2,5,10] Pyrethrins I (1a) and II (1b) are the predominant constituents of the concentrate, each contributing 30–40 % of the total Pyrethrin content. However, importantly, these two constituents are also more prone to the degradative processes than the minor constituents.[1,2] The jasmolins (2a and 2b) and cinerins (3a and 3b) exhibit greater stability to these chemical decomposition pathways but are present in much lower concentrations in the extract. As the jasmolins (2a and 2b) differ from pyrethrins (1a and 1b) through a single point of unsaturation, the selective transformation of the less stable pyrethrins (1a and 1b) to the more stable jasmolins (2a and 2b) through reductive chemistry may result in the desired stability increase while potentially maintaining many of the advantageous qualities of the original pyrethrum extract. Described herein is the exploration of several protocols for the chemo- and regioselective reduction of pyrethrins (1a and 1b) and a preliminary evaluation of the insecticidal activity of the various reduction products using larvae of the Australian sheep blowfly (Lucilia cuprina) as a test organism. As such, the current knowledge of structure–activity relationships within this unique class of natural product has been expanded.
Experimental
General Methods
Pyrethrum concentrate (80 % Pyrethrins) was supplied by Botanical Resources Australia, reagents were purchased from Sigma–Aldrich, and solvents were purchased from Chem-Supply. THF was dried by distillation over sodium benzophenone ketyl and DCM was dried by distillation over calcium hydride. Thin-layer chromatography (TLC) was performed using Chem-Supply silica gel 60 F254 TLC plates and visualised under UV light (254 nm). Column chromatography was undertaken using Sanpont silica gel of 230–400 mesh (0.040–0.063 mm) at atmospheric pressure unless otherwise stated.
The pyrethrum concentrate was separated by dry column vacuum chromatography (DCVC) using silica gel of 230–400 mesh (0.040–0.063 mm) and a gradient elution from 1 to 25 % ethyl acetate in hexane to give the Pyrethrins I and Pyrethrins II subsets. The individual ester pyrethrin I (1a, 91 % purity) was obtained by subjecting the Pyrethrins I subset to column chromatography on a three-tiered glass column eluting in 8 % ethyl acetate in hexane and pyrethrin II (1b, 91 % purity) was obtained by repeated DCVC of the Pyrethrins II subset. Individual jasmolins (2a and 2b) and cinerins (3a and 3b) were provided by CSIRO in >98 % purity. For synthetic protocols, BHT was removed from pyrethrum concentrate by application to a short silica gel plug with hexane, then eluting the Pyrethrins off the plug with ethyl acetate.
All 1H and 13C NMR spectra were recorded on a Bruker 600 MHz Avance III NMR spectrometer at 25°C using CDCl3 as the solvent and internal lock. All spectra were referenced to the residual solvent peak (CDCl3: 1H 7.26 ppm; 13C 77.0 ppm) and are recorded as follows: (1) chemical shift (ppm); (2) integration; (3) multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; quin, quintet; dd, doublet of doublets; dt, doublet of triplets; ddd, doublet of doublet of doublets; m, multiplet; brs, broad singlet; *, multiplicity assigned based on prior literature,[11] signals overlap owing to two similar-magnitude coupling constants); (4) coupling constant (Hz). Stereochemistry was assigned through NOESY correlation.
High-performance liquid chromatography (HPLC) analysis was performed on an Agilent Technologies 1200 series system with a photodiode array detector at 223, 229, and 235 nm with a Phenomenex Phenosphere-Next C18 column (150 × 4.6 mm ID, 5 µm). Solvent A was 1 % acetic acid in water and solvent B was acetonitrile. The column was kept at 40°C with a constant flow rate of 0.8 mL min−1 and a 10 µL injection. The solvent program was amended from Wang et al.[12] as follows: 50 % solvent A for 10 min followed by a linear gradient to 40 % solvent A over 5 min. Solvent A was held at 40 % for a further 10 min before a linear gradient to 35 % solvent A over 5 min; 35 % solvent A was held for 10 min after which a linear gradient to 20 % solvent A was undertaken over 5 min; 20 % solvent A was then held for an additional 5 min.
Liquid chromatography–mass spectrometry (LC-MS) analysis was conducted on a Shimadzu LCMS-2020 with a photodiode array detector at 254 nm and a single quadrupole mass spectrometer. Solvent A was 0.1 % formic acid in water and solvent B was 0.1 % formic acid in acetonitrile. The column was a Phenomenex Kinetex C18 column (100 × 2.1 mm ID, 2.1 µm) maintained at 30°C with a constant flow rate of 0.4 mL min−1 and a 1 µL injection. The solvent program was as follows: 30 % solvent A for 0.5 min before a linear gradient to 5 % solvent A over 5.5 min. Solvent A was held at 5 % for 2 min before a linear gradient to 30 % solvent A over 0.5 min. Solvent A was held at 35 % for an additional 3.5 min.
Ultraperformance convergence chromatography (UPCC) was used for chiral analysis on a Waters Acquity UPC2 system equipped with photodiode array detection at 230 nm. The column was maintained at 40°C with a constant flow rate of 1.2 mL min−1, a 1 µL injection, and convergence pressure of 13790 kPa. The solvent program was as follows: 97 % solvent A for 0.5 min followed by a linear gradient to 40 % solvent A over 2.5 min. Solvent A was held at 40 % for 3 min before a linear gradient to 97 % solvent A over 0.1 min. Solvent A was supercritical carbon dioxide (scCO2) and solvent B was either ethanol/isopropanol/acetonitrile (1 : 1 : 1) with 20 mM ammonium acetate, methanol/isopropanol (1 : 1) with 0.2 % v/v formic acid, ethanol/acetonitrile (1 : 1) with 0.2 % v/v formic acid, or ethanol/isopropanol (1 : 1) with 0.2 % v/v formic acid. The column was either a Waters Trefoil AMY1 (50 × 2.1 mm ID, 2.5 µm), Waters Trefoil CEL1 (50 × 2.1 mm ID, 2.5 µm), or Waters Trefoil CEL2 (50 × 2.1 mm ID, 2.5 µm).
Fourier-transform infrared spectroscopy (FTIR) was recorded using a PerkinElmer Spectrum100 spectrometer with an attenuated total reflectance (ATR) diamond crystal attachment. All spectra are reported in wavenumbers (ν, cm−1).
High-resolution mass spectrometry (HRMS) was performed on a PerkinElmer AxION Direct Sample Analysis (DSA) with an AxION®2 time of flight (ToF) mass spectrometer using atmospheric pressure chemical ionisation (APCI) in positive ion mode.
Optical rotations were recorded on a PolAAR 21 polarimeter referenced to the sodium D line (589 nm) at 20°C.
Chemistry
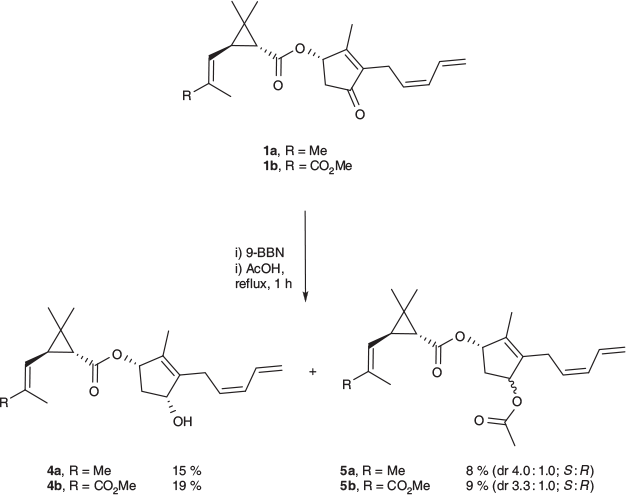
Hydroboration–Protonolysis of Pyrethrins (1a and 1b) with 9-Borabicyclo[3.3.1]none. Pyrethrin I 1a (117 mg, 0.357 mmol) was stirred in a solution of 9-borabicyclo[3.3.1]none (9-BBN) (0.65 mL, 0.5 M in THF, 0.3 mmol) under an atmosphere of nitrogen for 5 h. Acetic acid (200 µL, 3.49 mmol) was added and the resulting mixture refluxed for 1 h. Residual acid was quenched with 10 % sodium bicarbonate solution and the resulting mixture extracted with ethyl acetate. The solution was dried (Na2SO4) and solvent removed under vacuum, giving a pale yellow oil. The residue was purified by column chromatography (15 % ethyl acetate in hexane), yielding the allylic alcohol 4R-4a (18 mg, 15 %) and allylic ester 5a (11 mg, 8 %, dr 4.0 : 1.0 (S : R)).
Pyrethrin II 1b (79 mg, 0.212 mmol) was subjected to the above procedure using 9-BBN (0.6 mL, 0.5 M in THF, 0.3 mmol) and acetic acid (200 µL, 3.49 mmol). Column chromatography (15 % ethyl acetate in hexane) afforded the allylic alcohol 4R-4b (15 mg, 19 %) and allylic ester 5b (8 mg, 9 %, dr 3.3 : 1.0 (S : R)).
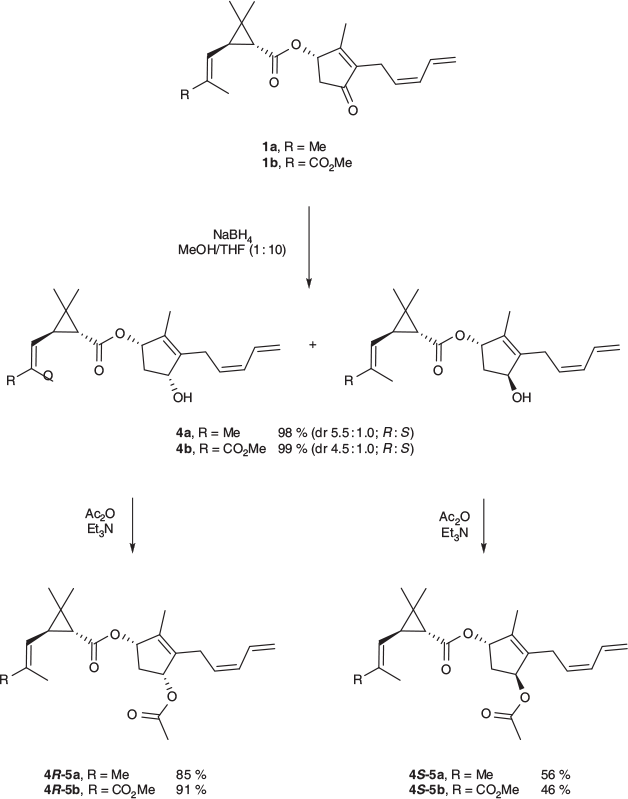
Reduction of Pyrethins (1a and 1b) with Sodium Borohydride. Sodium borohydride (90 mg, 2.4 mmol) was added to a solution of pyrethrin I 1a (310 mg, 0.95 mmol) in methanolic THF (10 % v/v, 5 mL) at 0°C. The resulting mixture was stirred for 3 h with continued cooling. Water was added and the aqueous mixture was extracted with ethyl acetate. The ethyl acetate was dried (Na2SO4) and the solvent removed under vacuum, giving a diastereomeric mixture of the allylic alcohol 4a as a colourless oil (305 mg, 98 %, dr 5.5 : 1.0 (R : S)). The individual diastereomers were isolated by silica gel column chromatography (15 % ethyl acetate in hexane) giving 4R-4a (202 mg) and 4S-4a (45 mg).
(1S, 4R)-4-Hydroxy-2-methyl-3-((2Z)-penta-2,4-dien-1-yl)cyclopent-2-en-1-yl (1R,3R)-2,2-Dimethyl-3-(2-methylprop-1-enyl)cyclopropane-1-carboxylate 4R-4a
δH (600 MHz, CDCl3) 6.75 (1H, ddd*, J 10.6, 16.9), 6.07 (1H, dd*, J 10.9), 5.42 (1H, dt, J 8.3, 9.7), 5.35 (1H, dd, J 3.4, 7.2), 5.23 (1H, d, J 16.7), 5.16 (1H, d, J 10.1), 4.90 (1H, d, J 7.7), 4.50 (1H, brs), 3.16 (1H, dd, J 8.5, 14.9), 3.05 (1H, dd, J 7.3, 14.9), 2.78 (1H, m), 2.04 (1H, m), 1.71 (3H, s), 1.70 (3H, s), 1.67 (1H, d, J 8.1), 1.57 (3H, s), 1.54 (1H, ddd*, J 3.7, 14.7), 1.39 (1H, d, J 5.3), 1.25 (3H, s), 1.12 (3H, s). δC (150 MHz, CDCl3) 172.61, 141.65, 135.70, 134.83, 131.83, 130.55, 128.34, 121.26, 118.30, 79.34, 75.84, 40.72, 35.04, 32.63, 28.73, 25.69, 24.83, 22.32, 20.56, 18.63, 11.76. [α]D20 73.7 (c 0.6, CHCl3).
(1S, 4S)-4-Hydroxy-2-methyl-3-((2Z)-penta-2,4-dien-1-yl)cyclopent-2-en-1-yl (1R,3R)-2,2-Dimethyl-3-(2-methylprop-1-enyl)cyclopropane-1-carboxylate 4S-4a
δH (600 MHz, CDCl3) 6.75 (1H, ddd*, J 10.3, 16.8), 6.09 (1H, dd*, J 10.9), 5.69 (1H, d, J 6.1), 5.45 (1H, dt, J 8.0, 9.9), 5.24 (1H, d, J 16.7), 5.16 (1H, d, J 10.2), 4.88 (1H, d, J 7.7), 4.81 (1H, brs), 3.12 (1H, dd, J 8.5, 14.9), 3.03 (1H, dd, J 7.3, 14.9), 2.15 (1H, ddd, J 3.2, 7.0, 14.9), 2.06 (1H, ddd, J 3.1, 7.0, 14.9), 2.03 (1H, m), 1.70 (3H, s), 1.69 (6H, s), 1.49 (1H, d, J 6.4), 1.36 (1H, d, J 5.3), 1.24 (3H, s), 1.11 (3H, s). δC (150 MHz, CDCl3) 172.80, 142.08, 135.67, 135.32, 131.79, 130.68, 128.24, 121.24, 118.33, 81.11, 76.85, 41.23, 35.02, 32.60, 28.67, 25.67, 24.89, 22.30, 20.53, 18.60, 11.84. [α]D20 286.7 (c 0.5, CHCl3). νmax/cm−1 3424, 2921, 1719, 1421, 1378, 1282, 1195, 1158, 1081, 1023, 904, 852. m/z (HRMS) 313.2162; calc. for C21H29O2 [M – OH]+ 313.2162.
Pyrethrin II 1b (330 mg, 0.89 mmol) was subjected to the same procedure using sodium borohydride (86 mg, 2.3 mmol). The diastereomeric mixture of the allylic alcohol 4b (330 mg, 99 %, dr 4.5 : 1.0 (R : S)) was resolved by silica gel column chromatography (30 % ethyl acetate in hexane) giving 4R-4b (163 mg) and 4S-4b (40 mg).
(1S,4R)-4-Hydroxy-2-methyl-3-((2Z)-penta-2,4-dien-1-yl)cyclopent-2-en-1-yl (1R,3R)-3-((E)-3-Methoxy-2-methyl-3-oxoprop-1-enyl)-2,2-dimethylcyclopropane-1-carboxylate 4R-4b
δH (600 MHz, CDCl3) 6.75 (1H, ddd*, J 10.7, 16.8), 6.46 (1H, d, J 9.7), 6.08 (1H, dd*, J 10.9), 5.42 (1H, dt, J 8.2, 9.7), 5.36 (1H, dd, J 3.1, 6.9), 5.54 (1H, d, J 16.8), 5.17 (1H, d, J 10.2), 4.51 (1H, brs), 3.73 (3H, s), 3.16 (1H, dd, J 8.6, 14.9), 3.05 (1H, dd, J 7.2, 14.9), 2.80 (1H, m), 2.20 (1H, dd, J 5.2, 9.6), 1.93 (3H, s), 1.72 (1H, d, J 5.2), 1.70 (3H, s), 1.69 (1H, d, J 7.4), 1.57 (3H, s), 1.54 (1H, ddd*, J 3.5, 14.7), 1.30 (3H, s), 1.22 (3H, s). δC (150 MHz, CDCl3) 171.47, 168.36, 141.96, 139.64, 134.54, 131.79, 130.64, 129.59, 128.20, 118.41, 79.87, 75.77, 51.93, 40.65, 36.35, 32.70, 24.84, 22.79, 22.55, 20.59, 13.00, 11.75. [α]D20 117.3 (c 0.5, CHCl3).
(1S,4S)-4-Hydroxy-2-methyl-3-((2Z)-penta-2,4-dien-1-yl)cyclopent-2-en-1-yl (1R,3R)-3-((E)-3-Methoxy-2-methyl-3-oxoprop-1-enyl)-2,2-dimethylcyclopropane-1-carboxylate 4S-4b
δH (600 MHz, CDCl3) 6.75 (1H, ddd*, J 10.8, 16.8), 6.45 (1H, d, J 9.7), 6.10 (1H, dd*, J 10.9), 5.70 (1H, d, J 6.3), 5.45 (1H, dt, J 8.2, 9.6), 5.26 (1H, d, J 16.7), 5.17 (1H, d, J 10.2), 4.82 (1H, brs), 3.72 (3H, s), 3.12 (1H, dd, J 8.7, 14.9), 3.04 (1H, dd, J 7.1, 14.9), 2.17 (2H, m), 2.06 (1H, ddd, J 3.1, 7.1, 14.8), 1.93 (3H, s), 1.70 (4H, m), 1.54 (1H, d, J 6.7), 1.29 (3H, s), 1.21 (3H, s). δC (150 MHz, CDCl3) 171.63, 168.36, 142.43, 139.65, 135.07, 131.74, 130.82, 129.58, 128.09, 118.48, 81.63, 76.86, 51.92, 41.20, 36.35, 32.69, 30.24, 24.91, 22.55, 20.57, 12.99, 11.85. [α]D20 –177.5 (c 0.4, CHCl3). νmax/cm−1 3489, 2924, 1713, 1642, 1434, 1261, 1222, 1176, 111, 1010, 940, 905, 831, 762. m/z (HRMS) 357.2077; calc. for C22H29O4 [M – OH]+: 357.2060.
General Procedure for the Acylation of Allylic Alcohols 4. Acetic anhydride (1 mL, 11 mmol) and triethylamine (1 mL, 7 mmol) were added to a solution of the allylic alcohol 4 in dry DCM (5 mL) under a nitrogen atmosphere. The mixture was heated under reflux for 4.5 h. The resulting solution was allowed to cool to room temperature and washed with water. The organic layer was collected and dried (Na2SO4). The solvent was removed under vacuum, yielding the allyl ester 5.
(1S,4R)-4-Acetoxy-2-methyl-3-((2Z)-penta-2,4-dien-1-yl)cyclopent-2-en-1-yl (1R,3R)-2,2-Dimethyl-3-(2-methylprop-1-enyl)cyclopropane-1-carboxylate 4R-5a
Yield: 110 mg, 85 %. δH (600 MHz, CDCl3) 6.64 (1H, ddd*, J 10.2, 16.8), 6.05 (1H, dd*, J 10.8), 5.48 (1H, m), 5.44 (1H, dd, J 3.5, 7.4), 5.35 (1H, dt, J 7.9, 10.6), 5.23 (1H, d, J 16.8), 5.14 (1H, d, J 10.2), 4.90 (1H, d, J 7.8), 3.04 (2H, m), 2.92 (1H, m), 2.05 (1H, m), 2.04 (3H, s), 1.74 (3H, s), 1.71 (3H, s), 1.70 (3H, s), 1.54 (1H, ddd*, J 3.7, 15.1), 1.41 (1H, d, J 5.3), 1.25 (3H, s), 1.13 (3H, s). δC (150 MHz, CDCl3) 172.47, 170.88, 137.78, 137.55, 135.73, 131.64, 130.56, 127.77, 121.23, 118.27, 79.05, 77.68, 38.20, 34.93, 32.72, 28.79, 25.70, 24.99, 22.32, 21.33, 20.58, 18.62, 11.92. [α]D20 –12.5 (c 0.4, CHCl3).
(1S,4S)-4-Acetoxy-2-methyl-3-((2Z)-penta-2,4-dien-1-yl)cyclopent-2-en-1-yl (1R,3R)-2,2-Dimethyl-3-(2-methylprop-1-enyl)cyclopropane-1-carboxylate 4S-5a
Yield: 68 mg, 56 %. δH (600 MHz, CDCl3) 6.65 (1H, ddd*, J 10.6, 16.9), 6.06 (1H, dd*, J 10.9), 5.72 (1H, d, J 6.9), 5.68 (1H, brs), 5.37 (1H, dt, J 8.2, 10.3), 5.22 (1H, d, J 16.8), 5.14 (1H, d, J 10.3), 4.88 (1H, d, J 7.8), 3.01 (2H, m), 2.20 (1H, ddd, J 3.7, 15.2), 2.12 (1H, ddd, J 3.4, 7, 15.2), 2.04 (1H, m), 2.01 (3H, s), 1.73 (3H, s), 1.71 (3H, s), 1.70 (3H, s), 1.38 (1H, d, J 5.3), 1.24 (3H, s), 1.12 (3H, s). δC (150 MHz, CDCl3) 172.68, 171.11, 138.39, 138.09, 135.72, 131.66, 130.63, 127.61, 121.22, 118.22, 80.63, 79.39, 38.59, 34.95, 32.66, 28.83, 25.69, 25.06, 22.29, 21.32, 20.52, 18.62, 11.89. [α]D20 183.3 (c 0.6, CHCl3). νmax/cm−1 2924, 1736, 1720, 1433, 1377, 1236, 1194, 1158, 1115, 1022, 997, 906. m/z (HRMS) 373.2382; calc. for C23H32O4 [M + H]+: 373.2373.
(1S,4R)-4-Acetoxy-2-methyl-3-((2Z)-penta-2,4-dien-1-yl)cyclopent-2-en-1-yl (1R,3R)-3-((E)-3-methoxy-2-methyl-3-oxoprop-1-enyl)-2,2-dimethylcyclopropane-1-carboxylate 4R-5b
Yield: 158 mg, 91 %. δH (600 MHz, CDCl3) 6.64 (1H, ddd*, J 10.6, 16.8), 6.45 (1H, d, J 9.7), 6.05 (1H, dd*, J 10.8), 5.49 (1H, m), 5.44 (1H, m), 5.33 (1H, dt, J 7.9, 10.7), 5.23 (1H, d, J 16.8), 5.14 (1H, d, J 10.1), 3.72 (3H, s), 3.04 (2H, m), 2.93 (1H, m), 2.19 (1H, dd, J 5.3, 9.7), 2.04 (3H, s), 1.93 (3H, s), 1.73 (4H, m), 1.53 (1H, ddd*, J 3.8, 15.1), 1.29 (3H, s), 1.21 (3H, s). δC (150 MHz, CDCl3) 171.30, 170.84, 168.32, 139.55, 138.13, 137.19, 131.58, 130.62, 129.62, 127.60, 118.34, 79.57, 77.58, 51.92, 38.17, 36.21, 32.73, 30.30, 24.98, 22.53, 21.30, 20.58, 12.98, 11.89. [α]D20 25.0 (c 0.4, CHCl3).
(1S,4S)-4-Acetoxy-2-methyl-3-((2Z)-penta-2,4-dien-1-yl)cyclopent-2-en-1-yl (1R,3R)-3-((E)-3-Methoxy-2-methyl-3-oxoprop-1-enyl)-2,2-dimethylcyclopropane-1-carboxylate 4S-5b
Yield: 44 mg, 46 %. δH (600 MHz, CDCl3) 6.65 (1H, ddd*, J 10.3, 16.8), 6.45 (1H, d, J 9.7), 6.07 (1H, dd*, J 10.9), 5.72 (1H, m), 5.67 (1H, m), 5.37 (1H, dt, J 7.9, 10.1), 5.23 (1H, d, J 16.8), 5.14 (1H, d, J 10.1), 3.72 (3H, s), 3.02 (2H, m), 2.19 (2H, m), 2.14 (1H, ddd, J 3.4, 7.1, 15.4), 2.02 (3H, s), 1.93 (3H, s), 1.72 (3H, s), 1.70 (1H, d, J 5.2), 1.29 (3H, s), 1.22 (3H, s). δC (150 MHz, CDCl3) 171.55, 171.11, 168.34, 139.57, 138.49, 137.99, 131.61, 130.72, 129.61, 127.47, 118.30, 81.71, 79.29, 51.92, 38.57, 36.25, 32.72, 30.36, 25.04, 22.52, 21.31, 20.54, 12.99, 11.89. [α]D20 –70.0 (c 0.4, CHCl3). νmax/cm−1 3668, 2970, 2924, 1715, 1643, 1434, 1381, 1259, 1222, 1174, 1150, 1111, 1050, 904, 804. m/z (HRMS) 357.2062; calc. for C22H28O4 [M – OAc]+: 357.2062.
Stereoselective Reduction of Pyrethrins (1a and 1b) with l-Selectride. l-Selectride (1.0 M in THF, 0.5 mL, 0.5 mmol) was added to a solution of pyrethrin I 1a (160 mg, 0.49 mmol) in dry THF 5 mL at 0°C under an atmosphere of nitrogen. The mixture was stirred with continued cooling for an additional 1 h. The reaction was quenched with water and extracted with ethyl acetate. The ethyl acetate was dried (Na2SO4) and the solvent removed under vacuum, giving a yellow oil. The oil was subjected to column chromatography (15 % ethyl acetate in hexane), giving allylic alcohol 4R-4a (62 mg, 38 %).
Pyrethrin II 1b (150 mg, 0.3 mmol) was subjected to the same procedure detailed above. The resulting oil was subjected to column chromatography (30 % ethyl acetate in hexane), giving allylic alcohol 4R-4b (30 mg, 20 %).
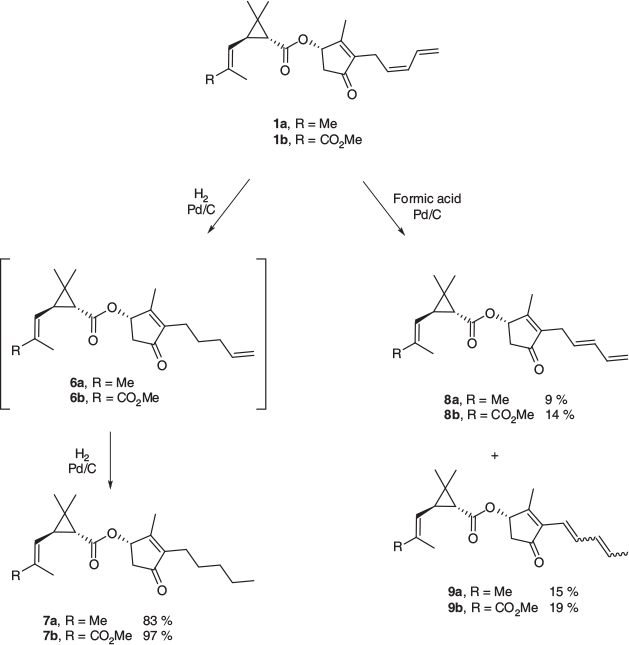
Catalytic Hydrogenation of the Pyrethrins (1a and 1b). Pyrethrin I 1a (100 mg, 0.305 mmol) in THF (2 mL) was stirred in the presence of palladium on carbon (10 wt-% loading, 10 mg) under an atmosphere of hydrogen at room temperature. After 4 h, the mixture was filtered over Celite and the solvent removed under vacuum, yielding 7a as a colourless oil (83 mg, 83 %).
(1S)-2-Methyl-4-oxo-3-pentylcyclopent-2-en-1-yl (1R,3R)-2,2-Dimethyl-3-(2-methylprop-1-enyl)cyclopropane-1-carboxylate 7a
δH (600 MHz, CDCl3) 5.65 (1H, d, J 6.2), 4.89 (1H, d, J 7.7), 2.83 (1H, dd, J 6.3, 18.7), 2.21 (1H, dd, J 1.7, 18.7), 2.18 (2H, m), 2.08 (1H, m), 2.01 (3H, s), 1.71 (3H, s), 1.69 (3H, s), 1.40–1.27 (7H, m), 1.26 (3H, s), 1.13 (3H, s), 0.88 (3H, t, J 7.3). δC (150 MHz, CDCl3) 204.73, 172.50, 164.67, 144.37, 136.02, 120.97, 73.09, 42.27, 34.72, 33.06, 31.91, 29.21, 27.89, 25.69, 23.17, 22.57, 22.25, 20.54, 18.63, 14.11 (two overlapping signals). νmax/cm−1 3675, 2971, 2922, 1713, 1655, 1420, 1380, 1282, 1235, 1192, 1151, 1114, 1065, 995, 964, 906, 849. m/z (HRMS) 333.2439; calc. for C21H33O3 [M + H]+: 333.2424. [α]D20 –27.4 (c 0.8, CHCl3).
Pyrethrin II 1b (136 mg, 0.366 mmol) was subjected to the procedure detailed above using palladium on carbon (1 wt-% loading, 12 mg). Tetrahydropyrethrin 7b was obtained as a colourless oil (133 mg, 97 %).
(1S)-2-Methyl-4-oxo-3-pentylcyclopent-2-en-1-yl (1R,3R)-3-((E)-3-Methoxy-2-methyl-3-oxoprop-1-en-1-yl)-2,2-dimethylcyclopropane-1-carboxylate 7b
δH (600 MHz, CDCl3) 6.45 (1H, d, J 9.6), 5.64 (1H, d, J 5.9), 3.72 (3H, s), 2.84 (1H, dd, J 6.3, 18.6), 2.22 (4H, m), 1.99 (3H, s), 1.84 (3H, s), 1.74 (1H, d, J 5.2), 1.38 (2H, m), 1.30 (3H, s), 1.27 (4H, m), 1.23 (3H, s), 0.87 (3H, t, J 6.9). δC (150 MHz, CDCl3) 204.45, 171.41, 168.27, 164.22, 144.63, 139.18, 129.89, 73.59, 51.96, 42.20, 35.96, 33.02, 31.90, 30.46, 27.88, 23.18, 22.57, 22.47, 20.55, 14.11, 13.01. νmax/cm−1 3675, 2954, 2928, 2872, 1710, 1649, 1435, 1385, 1260, 1221, 1173, 1148, 1055, 993, 831. m/z (HRMS) 377.2333; calc. for C22H31O5 [M + H]+: 377.2323. [α]D20 15.2 (c 1.3, CHCl3).
Isomerism of Pyrethrins (1a and 1b) Under Catalytic Transfer Hydrogenation Conditions. Pyrethrin I 1a (110 mg, 0.335 mmol) was heated under reflux in dry THF (2 mL) in the presence of palladium on carbon (10 wt-% loading, 15 mg) and formic acid (200 µL, 5.30 mmol) for 5 h under nitrogen. The mixture was allowed to cool, filtered over Celite, and the solvent removed under vacuum, yielding a pale yellow oil. The resulting mixture was purified by column chromatography (10 % ethyl acetate in hexane) giving trans-pyrethrin 8a (10 mg, 9 %) and iso-pyrethrin 9a as a mixture of diastereomers (17 mg, 15 %).
(1S)-2-Methyl-4-oxo-3-((E)-penta-2,4-dien-1-yl)cyclopent-2-en-1-yl (1R,3R)-2,2-Dimethyl-3-(2-methylprop-1-enyl)cyclopropane-1-carboxylate 8a
δH (600 MHz, CDCl3) 6.27 (1H, ddd*, J 10.3, 16.9), 6.05 (1H, dd, J 10.3, 15.2), 5.67 (1H, d, J 6.1), 5.64 (1H, dt, J 6.8, 15.2), 5.12 (1H, d, J 17.0), 5.00 (1H, d, J 10.1), 4.90 (1H, d, J 7.7), 3.00 (2H, d, J 6.8), 2.87 (1H, dd, J 6.3, 18.7), 2.24 (1H, dd, J 1.32, 18.7), 2.08 (1H, m), 2.03 (3H, s), 1.72 (3H, s), 1.71 (3H, s), 1.40 (1H, d, J 5.3), 1.26 (3H, s), 1.14 (3H, s). δC (150 MHz, CDCl3) 203.99, 172.46, 165.87, 141.67, 136.75, 136.08, 132.45, 129.64, 120.94, 116.22, 73.07, 42.19, 34.70, 33.14, 29.29, 26.18, 25.70, 22.25, 20.55, 18.64, 14.22. νmax/cm−1 3675, 2972, 2924, 1715, 1655, 1420, 1380, 1282, 1235, 1193, 1152, 1114, 1065, 1003, 963, 901, 849. m/z (HRMS) 329.2122; calc. for C21H29O3 [M + H]+: 329.2111. [α]D20 –44.7 (c 0.8, CHCl3).
(S)-2-Methyl-4-oxo-3-(penta-1,3-dien-1-yl)cyclopent-2-en-1-yl (1R,3R)-2,2-Dimethyl-3-(2-methylprop-1-enyl)cyclopropane-1-carboxylate 9a
δH (600 MHz, CDCl3) 7.64 (0.37H, dd, J 11.4, 15.5), 7.29 (0.63H, dd, J 10.7, 15.6), 6.09 (2H, m), 5.92 (0.63H, m), 5.67 (1.4H, m), 4.90 (1H, d, J 7.6), 2.90 (1H, m), 2.28 (1H, m), 2.09 (3H, s), 1.85 (2H, d, J 7.1), 1.80 (3H, d, J 6.7), 1.72 (3H, s), 1.71 (3H, s), 1.41 (2H, m), 1.26 (3H, s), 1.13 (3H, s) (some diastereomeric signals overlap). δC (150 MHz, CDCl3) 203.42, 172.50, 163.92, 163.38, 137.94, 136.21, 136.05, 133.31, 132.60, 130.97, 130.36, 130.17, 120.96, 119.68, 117.74, 72.71, 42.95, 34.75, 33.14, 30.47, 29.28, 25.70, 22.25, 20.58, 18.65, 14.36, 14.06 (some diastereomeric signals overlap). νmax/cm−1 2955, 2926, 1716, 1431, 1379, 1282, 1193, 1152, 1114, 994, 860. m/z (HRMS) 329.2122; calc. for C21H29O3 [M + H]+: 329.2111. [α]D20 –179.2 (c 0.5, CHCl3).
Pyrethrin II 1b (108 mg, 0.290 mmol) was treated following the above procedure using palladium on carbon (10 wt-% loading, 12 mg) and formic acid (200 µL, 5.30 mmol). The isolated yellow oil was purified by column chromatography (20 % ethyl acetate in hexane) giving 8b (15 mg, 14 %) and 9b as a mixture of diastereomers (20 mg, 19 %).
(1S)-2-Methyl-4-oxo-3-((E)-penta-2,4-dien-1-yl)cyclopent-2-en-1-yl (1R,3R)-3-((E)-3-Methoxy-2-methyl-3-oxoprop-1-en-1-yl)-2,2-dimethylcyclopropane-1-carboxylate 8b
δH (600 MHz, CDCl3) 6.46 (1H, d, J 9.6), 6.26 (1H, ddd*, J 10.3, 16.9), 6.05 (1H, dd, J 10.5, 15.1), 5.67 (1H, d, J 6.1), 5.64 (1H, dt, J 6.8, 15.2), 5.11 (1H, d, J 17.0), 4.99 (1H, d, J 10.3), 3.72 (3H, s), 3.01 (2H, d, J 6.8), 2.88 (1H, dd, J 6.2, 18.7), 2.23 (2H, m), 2.03 (3H, s), 1.94 (3H, s), 1.74 (1H, d, J 5.1), 1.30 (3H, s), 1.23 (3H, s). δC (150 MHz, CDCl3) 203.71, 171.35, 168.25, 165.40, 141.92, 139.10, 136.69, 132.52, 129.92, 129.48, 116.30, 73.54, 51.96, 42.10, 35.91, 33.05, 30.71, 26.17, 22.46, 20.55, 14.20, 13.02. νmax/cm−1 3662, 2952, 1709, 1649, 1435, 1383, 1340, 1260, 1221, 1173, 1147, 1111, 1055, 996, 904, 830. m/z (HRMS) 373.2022; calc. for C22H29O5 [M + H]+: 373.2010. [α]D20 17.8 (c 0.5, CHCl3).
(S)-2-Methyl-4-oxo-3-(penta-1,3-dien-1-yl)cyclopent-2-en-1-yl (1R,3R)-3-((E)-3-Methoxy-2-methyl-3-oxoprop-1-en-1-yl)-2,2-dimethylcyclopropane-1-carboxylate 9b
δH (600 MHz, CDCl3) 7.71 (0.33H, dd, J 11.5, 15.4), 7.35 (0.66H, dd, J 10.7, 15.6), 6.46 (1H, d, J 9.6), 6.09 (2H, m), 5.92 (0.66H, m), 5.66 (1.66H, m), 3.72 (3H, s), 2.90 (1H, m), 2.25 (2H, m), 2.08 (3H, s), 1.94 (3H, s), 1.84 (1H, d, J 7.2), 1.80 (2H, d, J 6.7), 1.74 (1H, m), 1.30 (3H, s), 1.24 (3H, s) (some diastereomeric signals overlap). δC (150 MHz, CDCl3) 203.12, 171.37, 168.24, 163.34, 162.80, 139.13, 138.08, 136.37, 133.45, 132.54, 131.11, 130.30, 129.88, 119.55, 117.61, 73.17, 51.94, 42.85, 35.95, 33.02, 30.66, 22.45, 20.56, 18.60, 14.32, 14.04, 13.00 (some diastereomeric signals overlap). νmax/cm−1 2952, 2928, 1710, 1643, 1435, 1385, 1260, 1221, 1173, 1147, 1111, 993, 830. m/z (HRMS) 373.2010; calc. for C22H29O5 [M + H]+: 373.2010. [α]D20 –56.9 (c 1.0, CHCl3).
Reduction of Pyrethrins (1a and 1b) by Diimide-Mediated Transfer Hydrogenation. Pyrethrin I 1a (110 mg, 0.335 mmol) in THF (5 mL) was stirred vigorously open to air in the presence of hydrazine monohydrate (800 mg, 15.98 mmol), copper(ii) sulfate pentahydrate (8.5 mg, 0.034 mmol, 10 mol-%), and acetic acid (20 mg, 0.33 mmol). The reaction was monitored by TLC (8 % ethyl acetate in hexane) until all of the pyrethrin starting material was consumed (typically 7–8 h). The reaction mixture was filtered and subsequently diluted with brine water. The resulting solution was extracted with ethyl acetate and dried (Na2SO4). Solvent was removed under vacuum, yielding a colourless oil (85 mg, 77 % mass recovery), which was determined by HPLC to be 68 % jasmolin I 2a and 25 % tetrahydropyrethrin I 7a. Characterisation was undertaken on the isolated mixture with signals assigned to jasmolin I by comparison with the natural ester.
Jasmolin I 2a
δH (600 MHz, CDCl3) 5.65 (1H, d, J 6.2), 5.42 (1H, dt, J 7.3, 17.8), 5.24 (1H, dt, J 7.3, 17.8), 4.90 (1H, d, J 7.7), 2.98 (2H, d, J 7.3), 2.85 (1H, dd, J 6.4, 18.7), 2.22 (1H, d, J 18.7), 2.15 (2H, m), 2.08 (1H, m), 2.03 (3H, s), 1.73 (3H, s), 1.71 (3H, s), 1.41 (1H, m) 1.26 (3H, s), 1.14 (3H, s), 0.99 (3H, t, J 7.5). δC (150 MHz, CDCl3) 204.12, 172.47, 164.95, 142.89, 136.04, 133.30, 124.05, 120.95, 73.13, 42.21, 34.71, 33.08, 29.23, 25.69, 22.23, 21.35, 20.73, 20.54, 18.63, 14.22, 14.17. νmax/cm−1 3675, 2965, 2928, 1714, 1655, 1447, 1420, 1380, 1282, 1235, 1193, 1152, 1114, 1046, 995, 850. m/z (HRMS) 331.2256; calc. for C21H31O3 [M + H]+: 331.2268. Isolated mixture [α]D20 –32.2 (c 0.9, CHCl3) (natural jasmolin I [α]D20 –56.0 (c 0.5, CHCl3)).
Pyrethrin II 1b (145 mg, 0.390 mmol) in THF (5 mL) was subjected to the same procedure as above using hydrazine monohydrate (270 mg, 5.4 mmol), copper(ii) sulfate pentahydrate (11 mg, 0.044 mmol, 10 mol-%), and acetic acid (10 mg, 0.17 mmol). The reaction was monitored by TLC (25 % ethyl acetate in hexane) until all of the pyrethrin starting material was consumed (typically 7–8 h). A colourless oil was obtained (120 mg, 83 % mass recovery), which was determined by HPLC to be 66 % jasmolin II 2a and 26 % tetrahydropyrethrin II 7b. Characterisation was undertaken on the isolated mixture with signals assigned to jasmolin II by comparison with the natural ester.
Jasmolin II 2b
δH (600 MHz, CDCl3) 6.45 (1H, d, J 9.6), 5.64 (1H, d, J 5.4), 5.42 (1H, dt, J 7.26, 10.6), 5.23 (1H, dt, J 7.3, 10.6), 3.73 (3H, s), 2.97 (2H, d, J 7.2), 2.86 (1H, dd, J 6.4, 18.7), 2.23 (2H, m), 2.16 (2H, quin, J 7.4), 2.03 (3H, s), 1.94 (3H, s), 1.74 (1H, d, J 5.2), 1.30 (3H, s), 1.23 (3H, s), 0.99 (3H, t, J 7.5). δC (150 MHz, CDCl3) 203.87, 171.39, 168.28, 164.51, 143.17, 139.19, 133.42, 129.91, 123.94, 73.63, 51.97, 42.20, 35.95, 33.03, 30.69, 22.47, 21.37, 20.75, 20.56, 14.23, 14.17, 13.02. νmax/cm−1 3675, 2956, 1712, 1648, 1435, 1383, 1324, 1261, 1221, 1174, 1148, 1111, 1056, 995, 830, 762. m/z (HRMS) 375.2162; calc. for C22H31O5 [M + H]+: 375.2166. Isolated mixture [α]D20 10.3 (c 1.1, CHCl3) (natural jasmolin II [α]D20 8.0 (c 0.5, CHCl3)).
Reduction of Pyrethrum Concentrate by Diimide-Mediated Transfer Hydrogenation. Pyrethrum concentrate (HPLC analysis: 43 % pyrethrin I 1a, 36 % pyrethrin II 1b, 4 % jasmolin I 2a, 4 % jasmolin II 2b) (500 mg, ~1.36 mmol) in THF (5 mL) was stirred vigorously open to air in the presence of hydrazine monohydrate (517 mg, 10.3 mmol), copper(ii) sulfate (19 mg, 0.12 mmol, 10 mol-%), and acetic acid (10 mg, 0.17 mmol). The reaction was stirred at room temperature for 8 h before being filtered and diluted with brine. The resulting solution was extracted with ethyl acetate and dried (Na2SO4). Solvent was removed under vacuum, yielding a pale yellow oil (HPLC analysis: 3 % pyrethrin I 1a, 2 % pyrethrin II 1b, 29 % jasmolin I 2a, 36 % jasmolin II 2b, 7 % tetrahydropyrethrin I 7a, 8 % tetrahydropyrethrin II 7b) (400 mg, 80 % mass recovery).
Biology
Australian sheep blowfly (Lucilia cuprina) larvae were prepared and used in insecticidal activity assays as detailed in published procedures.[13,14] Specifically, a plug of cotton wool (~0.2 g) on top of three layers of filter paper (within a 70-mL plastic pot) was loaded with 4 mL of a solution of the compound in ethanol, and the solvent allowed to evaporate. Controls were prepared in the same manner, by loading the cotton wool plug with ethanol or a solution of BHT. On Day 0 of the assay, a sheep serum-based medium was added to the cotton wool and 50 freshly hatched L. cuprina larvae were added. The plastic pots containing the larvae were incubated at 28°C over a period of 4 days. The larvae were fed with 1 mL of nutrient medium on Day 1, and 2 mL on Days 2 and 3. Late on Day 4, the larvae were transferred to larger pots with a layer of sand (a medium for pupation) and allowed to incubate further. On Day 9, the resulting pupae were collected by sieving the sand and the bioactivity calculated by pupation rate: the number of collected pupae in assays with experimental compounds was expressed as a percentage of the average number of pupae in the control assays. All tested materials had a purity >90 % and were prepared as solutions in hexane, stabilised with 5 wt-% BHT and subsequently diluted for testing in ethanol. BHT controls exhibited no inhibition of pupation on L. cuprina larvae at concentrations up to 5 µmol per assay, 10 times the amount present in the individual samples. Each assay was performed in duplicate.
The pupation rate dose–response data were analysed with GraphPad Prism® software. Non-linear regression was used with the ‘variable slope’ option to determine the concentration of the test compound that gives 50 % of the normal pupation rate (IC50), together with 95 % confidence intervals.
Results and Discussion
Borane and Borohydride Reducing Agents
Regioselective reduction of the terminal olefin of the pentadienyl unit in pyrethrins 1a and 1b was a challenge that was thought could potentially be overcome by application of sterically hindered reagents that should only access the least hindered alkene. Hydroboration with 9-BBN, and subsequent protonolysis with acetic acid, were chosen owing to the steric encumbrance imposed by the 9-BBN and its propensity to undergo monohydroboration with conjugated dienes.[15] Rather than obtaining the desired reactivity of the terminal olefin, however, the carbonyl of the enone was reduced (Scheme 1), with the isolation of both the allyl alcohols (4a and 4b) and allyl esters (5a and 5b) via column chromatography.
|
The allyl alcohols 4a and 4b were produced in 15–19 % yield as the (4R)-stereoisomers, as determined by NOE correlation, whereas the allyl esters 5a and 5b were isolated as diastereomeric mixtures in yields of 8–9 %. This hydroboration–protonolysis procedure was quite inefficient, with small amounts of the intermediate organoborane and the starting ketone also being isolated. The allylic esters 5a and 5b were likely formed through esterification of the allyl alcohols 4a and 4b with the excess acetic acid under the forcing conditions employed for protonolysis. This esterification reaction appears to proceed more readily for the 4S-isomers, with the 4S-acetates dominating over the 4R-acetates, and no 4S-alcohol being observed. The propensity to reduce the ketone functionality observed here is consistent with other reported outcomes with 9-BBN, which has previously been used for carbonyl reduction in ketones, aldehydes, and their α,β-unsaturated counterparts, the latter selectively leading to allylic alcohols in the presence of other reducible functionalities.[16,17]
Given the observed reactivity of 9-BBN on the enone unit, sodium borohydride was employed in an attempt to optimise the yield of the allyl alcohols 4a and 4b and in so doing, provide sufficient quantities for bioactivity testing and ultimately allow the further exploration of structure–activity relationships. It was found that subjecting pyrethrins 1a and 1b to reduction with sodium borohydride in a 1 : 10 methanol/THF solvent blend resulted in near-quantitative conversion to the allyl alcohols 4a and 4b as a mixture of diastereomers (Scheme 2).
|
The competing 1,4-enone reduction was not observed and its occurrence is likely limited by the hindrance enforced by both the methyl substituent on the cyclopentenone and the reactive sodium borohydride–methanol adduct employed.[18,19] Column chromatography of the diastereomeric mixture gave the individual diastereomers, which were in turn subjected to acetylation in acetic anhydride/triethylamine to give the individual diastereomers of the allyl esters 5a and 5b (Scheme 2), allowing investigation into the potential role of stereochemistry at the new stereocentre on insecticidal action. Single diastereomers of the allyl alcohols (4R-4a and 4R-4b) could be furnished from the pyrethrins 1a and 1b through reduction with the stereoselective borohydride l-selectride, albeit in low to moderate yields of 20 and 38 %, depending on the substituent on the chrysanthemic acid moiety of the starting pyrethrin.
Although the boron reagents did not selectively produce the jasmolins (2a and 2b) from pyrethrins (1a and 1b), the resultant allylic alcohols (4a and 4b) and esters (5a and 5b) were nonetheless subjected to preliminary insecticidal activity assays (see below).
Hydrogenation
Previous investigations into the reduction of pyrethrins (1a and 1b) employed catalytic hydrogenation, leading to the tetrahydropyrethrins (7a and 7b). It was reported that such analogues had significantly lower bioactivity than the natural Pyrethrins (1–3).[20–22] Attempts to regulate these hydrogenation procedures towards selective reduction of pyrethrins (1a and 1b) to their jasmolin counterparts (2a and 2b) has not previously been described.
In an effort to achieve selectivity in the reductive process, experiments with several catalyst loadings, ranging from 1 to 10 wt-% palladium on carbon, employing short reactions times and atmospheric pressure, were undertaken. Unfortunately, all of the conditions employed yielded the tetrahydropyrethrins (7a and 7b; Scheme 3) exclusively. Optimised reaction conditions with the highest catalyst loading (10 wt-% Pd/C) furnished the tetrahydropyrethrins (7a and 7b) in high yields of 83–97 %. The intermediate dihydropyrethrins (6a and 6b; Scheme 3) were detected by 1H NMR analysis of the reaction mixtures following 2-h hydrogen exposure with a 1 wt-% Pd/C catalyst loading, suggesting that these partially reduced compounds are intermediates in the full reduction of the pentadienyl side chains to give the tetrahydropyrethrins (7a and 7b). The dihydropyrethrins 6a and 6b could not be separated from the tetrahydropyrethrins 7a and 7b, which were also present in these reaction mixtures, and as a result the bioactivity of the partially reduced pyrethrins 6a and 6b could not be explored.
|
Transfer Hydrogenation
The increased control provided by catalytic transfer hydrogenation was of interest to alleviate the selectivity issues encountered with the standard hydrogenation procedures.[23] Initial experiments made use of cyclohexene or cyclohexadiene as hydrogen transfer reagents; however, neither resulted in reduction. This lack of reactivity is likely due to the temperature dependence of the competition between hydrogen transfer and disproportionation of these transfer reagents.[24] This issue is usually overcome by substitution of the reaction solvent with a higher-boiling liquid; however, in this case this was not feasible owing to the thermal instability of the Pyrethrins (1–3).[4]
Experiments were then performed with formic acid as the hydrogen transfer reagent using similar reaction conditions to those previously employed. Interestingly, instead of the desired olefin reduction, the combination of formic acid and palladium on carbon at elevated temperatures resulted in both cis to trans isomerism and olefin migration in the pentadienyl rethrolone side chain (Scheme 4) of the pyrethrins (1a and 1b). Such processes likely result from a palladium hydride, generated by the transfer process,[24–28] undergoing hydropalladation of the cis-olefin to give organopalladium species 10 (Scheme 4), and subsequent β-hydride elimination, giving the trans-pyrethrins 8 or double-migration isomers 9, presumably via the partially conjugated isomer 11. This isomerisation process was, however, ineffective at producing large quantities of these olefin isomers, with significant amounts of starting material recovered from reaction mixtures.
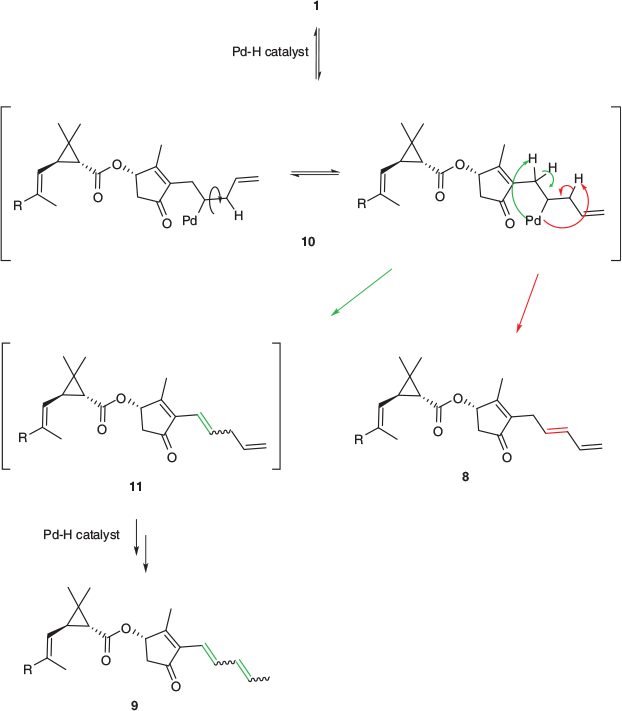
|
Diimide-Mediated Transfer Hydrogenation
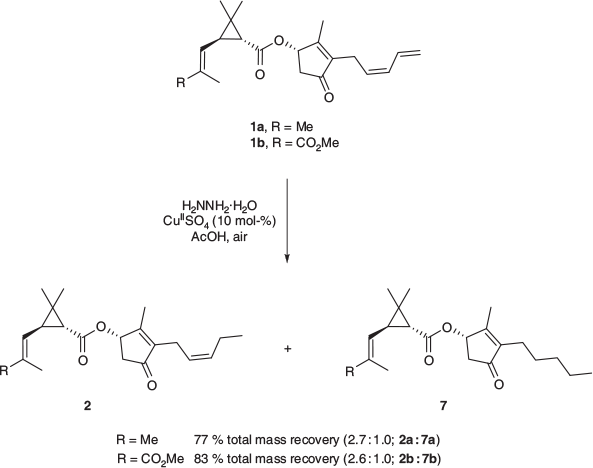
Diimide (HN=NH) is a reducing agent that is known to have a high chemoselectivity towards unpolarised olefins and a high sensitivity to steric hinderance, with reactivity significantly decreasing with increasing substitution around the double bond.[29] Reduction protocols that employ diimide use cheap, readily available materials and are operationally simple; the diimide is generated in situ from hydrazine in the presence of atmospheric oxygen, acetic acid, and catalytic quantities of copper(ii). When pyrethrin I (1a) and II (1b) were individually subjected to these conditions, the desired jasmolins (2a and 2b) were obtained as mixtures in 77 and 83 % mass recovery respectively (Scheme 5). The jasmolin components of these mixtures were found to be chemically identical to the natural jasmolins: in addition to being spectroscopically identical, analysis by UPCC found that the semisynthetic variants and the individual natural jasmolins (2a and 2b) had very similar retention times. Further, the stereocentres present in the pyrethrins (1a and 1b) appear to have been undisturbed under the conditions of the diimide reduction, as no evidence of the semisynthetic jasmolin signals resolving into more than a single peak could be found with a range of chiral columns and chromatographic conditions. Chromatographic analysis also revealed the presence of a single significant by-product in each of the diimide reduction mixtures, constituting ~27 % of the product. Further analysis by LC-MS identified these by-products as the corresponding tetrahydropyrethrins 7a and 7b, which resulted from reduction of the semisynthetic jasmolins 2a and 2b.
|
This methodology could then be applied to the pyrethrum concentrate, where all six of the Pyrethrin esters (1–3) are present. It was found that pyrethrins I (1a) and II (1b) could be reduced to their respective jasmolins (2a and 2b) in the presence of other minor pyrethrum constituents. HPLC analysis showed that very little pyrethrin I (1a) or II (1b) was detectable in the resulting mixture (Fig. 3b), with the jasmolins (2a and 2b) constituting ~50 % of the treated concentrate, as opposed to 7 % in the original concentrate. HPLC analysis also indicated the presence of the two minor reaction by-products, with the elution of tetrahydropyrethrin II 7b at 33.94 min and tetrahydropyrethrin I 7a at 49.80 min (Fig. 3b) constituting ~15 % of the resulting mixture.

|
This process was found to be amenable to hydrazine levels as low as 10 equiv. before the rate of reduction started to be adversely affected and reaction times needed to be increased. This necessity for an excess of hydrazine stems from the readiness of diimide to undergo disproportionation to hydrazine and molecular nitrogen.[30] The protocol also showed promise under catalyst-free conditions, potentially alleviating the issues associated with heavy metal disposal in an industrial application. The copper-free reaction was generally significantly slower than the catalysed variant, however, and required increased equivalents of hydrazine to combat the disproportionation process.[30] The copper-catalysed diimide-mediated reduction has shown promise up to gram scale with the pyrethrum concentrate in the conversion of the more susceptible pyrethrins (1a and 1b) and is potentially suitable for scale-up via large-scale continuous flow processes.[31]
Preliminary Insecticidal Activity and Structure–Activity Relationships
Pyrethrins, and pyrethroids, act on the voltage-gated sodium ion channels that regulate nerve stimulation in the insect nervous system.[32] The binding of Pyrethrins and pyrethroids to these channels causes them to remain open for longer periods of time, allowing a greater influx of sodium ions, ultimately resulting in overexcitation of the nerve cell.[7,32] The major consequence of this is the ‘knockdown’ of the insect, where it is heavily incapacitated by paralysis and overstimulation. Eventually the overexcitation results in the death of the organism.[32] Much of the data on the mode of action of the natural Pyrethrins is extrapolated from that of the pyrethroids, with little data existing for both the action and insecticidal activity of the individual Pyrethrin esters. The in vitro inhibition by these natural products on cockroach sodium ion channels expressed in Xenopus oocytes has, however, been recently reported, and the results suggest that Pyrethrins are also able to interact with the closed form of the channels.[30]
In the current study, a preliminary screen of the insecticidal activity of the individual natural Pyrethrins was performed by measuring their effects on the larval mortality of an economically important pest, Lucilia cuprina (Australian sheep blowfly), with the ability of larvae to develop to the pupal stage used as a measure of the larvicidal activity of the compounds. This is an operationally simple insecticidal assay, and assesses effects on larvae as this is the most relevant life stage for the control of this important pest in the Australian context. Commercial synthetic pyrethroids, α-cypermethrin and deltamethrin, were used as positive controls. The chemically reduced and isomerised pyrethrin analogues (4a, 4b, 5a, 5b, 7a, 7b, 8a, 8b, 9a, and 9b) were also tested, as well as products resulting from the diimide reduction reactions. Dose–response curves are shown in Figs 4 and 5 and IC50 data are shown in Table 1.

|
|

|
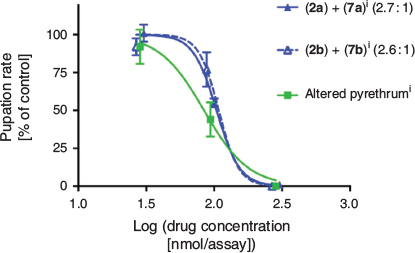
The individual Pyrethrins exhibited a range of toxicities towards L. cuprina, despite the minor differences between the acid moieties and/or the rethrolone side chain. Of the six isolated natural Pyrethrins, pyrethrin I 1a and cinerin I 3a were the most potent, with IC50 values for pupation inhibition of 11 and 13 nmol per assay respectively. Jasmolin I 2a was significantly less active than the other two esters in the series, requiring ~6.5 times the concentration of cinerin I 3a to inhibit pupation to 50 % (84 versus 13 nmol per assay). Pyrethrins II 1b–3b also showed this trend across the series. However, the Pyrethrins II 1b and 3b were less active than their Pyrethrin I counterparts 1a and 3a, whereas 2a and 2b showed similar activity. The insecticidal activities of the isolated pyrethrins 1a and 1b and cinerins 3a and 3b have previously been reported, using the house fly (Musca domestica)[33] and mustard beetle (Phaedon cochleariae)[34] as test organisms. Of these four natural Pyrethrins, cinerin II 3b was always found to be the least potent,[33,34] and the results with L. cuprina reported here are consistent with this finding. The other three more potent Pyrethrins (1a, 1b, and 3a) were reported to have varied relative activities, depending on the test organism and mode of administration. The isolated jasmolins I 2a and II 2b have only very recently been tested on live insects (common mosquito, Culex pipiens pallens).[35] The reported toxicity of the jasmolins against the mosquito are consistent with the trend in activity found with L. cuprina, being the weakest insecticides of the six natural Pyrethrin esters. This trend observed with the natural Pyrethrins (1–3) is also consistent with the in vitro electrophysiology activities with cockroach sodium channels recently reported by Dong and coworkers.[7] The individual semisynthetic jasmolin mixtures retained similar activities to that of their natural counterparts, likely due to the high jasmolin (2a and 2b) content.
The pyrethrum concentrate exhibited a similar IC50 to that of pyrethrin II (1b), presumably due to the majority of the mixture comprising the more active pyrethrins (1a and 1b), with only minor amounts of the less active esters being present. The altered pyrethrum concentrate obtained after diimide reduction retained insecticidal activity (IC50 84 nmol per assay); however, as expected, it had a reduced potency, very similar to that of the jasmolins 2a and 2b.
All the reduced pyrethrin analogues synthesised (4a, 4b, 5a, 5b, 7a, and 7b) via various reductive procedures were found to be inactive in the L. cuprina assay. Specifically, the ketone functionality appears to be vital for insecticidal activity. Consistent with this finding, acetylation of the allylic alcohols 4a and 4b to give 5a and 5b failed to restore biological activity. Unsaturation in the pentyl side chain of the Pyrethrins also greatly influenced insecticidal activity. The lack of observed activity of the tetrahydropyrethrins 7a and 7b is consistent with previous reports that indicated that these compounds had considerably less insecticidal activity than their natural, unsaturated analogues pyrethrin I and II (1a and 1b).[19–21] It is also consistent with the in silico model of a cockroach sodium ion channel developed by Dong and coworkers, in which unsaturation in the sidechain was predicted to be important for channel binding and inactivation by pyrethrin II (1b).[7] The importance of the unsaturated sidechain is further highlighted by the measured activities of the butadiene isomers 8a, 8b, 9a, and 9b. The trans-pyrethrin isomers 8a and 8b showed significant inhibition of pupation, albeit slightly decreased from that of pyrethrins 1a and 1b bearing the natural cis-geometry. The migratory isomers 9a and 9b, however, were found to be inactive. This is again consistent with Dong’s model, which requires the pyrethrin molecule to bend at the methylene that links the cyclopentenone ring and butadiene chain in order to make important contacts with particular protein helices of the channel. In the fully conjugated trienes 9a and 9b, the diene would be coplanar with the cyclopentenone ring and incapable of bending and making the required protein contacts.
Conclusion
In summary, several Pyrethrin analogues have been synthesised through the exploration of the reduction chemistry of the natural Pyrethrins. As a result, a procedure for the conversion of pyrethrins (1a and 1b) to jasmolins (2a and 2b) was developed, resulting in the removal of the more sensitive esters; however, some over-reduction was observed, resulting in the coproduction of the tetrahydropyrethrins 7a and 7b. The bioactivity of the synthesised analogues was assessed using the commercially important organism Lucilia cuprina and new structure–activity relationships relating to the ketone and unsaturated side chain functionalities have been revealed. From these assays, it was also found that the altered pyrethrum concentrate still exhibits significant insecticidal activity while being expected to have increased stability relative to the natural extract.[2]
Supplementary Material
NMR and IR spectra of all new compounds, NMR spectra of natural isolated jasmolins, NMR and IR spectra, and HPLC, LC-MS, and UPCC traces of mixtures produced from diimide reduction reactions and figures showing NOE correlations for epimers of 4a and 4b are available on the Journal’s website.
Conflicts of Interest
The CSIRO has previously undertaken commercial research for Botanical Resources Australia, a major commercial producer of pyrethrum.
Acknowledgements
This work was funded by co-investment by Botanical Resources Australia and CSIRO Manufacturing. T.E.M. acknowledges Flinders University for provision of a Flinders University Research Scholarship. The authors wish to thank Botanical Resources Australia for providing pyrethrum concentrate and CSIRO for providing purified individual Pyrethrins. The authors also thank Jason Smith, University of Tasmania, for knowledge and assistance with diimide-mediated reductions as well as Jason Young and Flinders Analytical for access to and insight into the DSA-ToF for HRMS analysis. The authors also thank Marc McEwan, CSIRO, for LC-MS and UPCC analysis.
References
[1] J. E. Casida, Environ. Health Perspect. 1980, 34, 189.| Crossref | GoogleScholarGoogle Scholar | 6993201PubMed |
[2] J. A. Freemont, S. W. Littler, O. E. Hutt, S. Mauger, A. G. Meyer, D. A. Winkler, M. G. Kerr, J. H. Ryan, H. F. Cole, P. J. Duggan, J. Agric. Food Chem. 2016, 64, 7134.
| Crossref | GoogleScholarGoogle Scholar | 27599033PubMed |
[3] L. Crombie, P. Hemesley, G. Pattenden, J. Chem. Soc. 1969, 1016.
[4] B. L. Atkinson, A. J. Blackman, H. Faber, J. Agric. Food Chem. 2004, 52, 280.
| Crossref | GoogleScholarGoogle Scholar | 14733509PubMed |
[5] M. J. Bullivant, G. Pattenden, Pestic. Sci. 1976, 7, 231.
| Crossref | GoogleScholarGoogle Scholar |
[6] E. V. Minello, F. Lai, M. T. Zonchello, M. Melis, M. Russo, P. Cabras, J. Agric. Food Chem. 2005, 53, 8302.
| Crossref | GoogleScholarGoogle Scholar | 16218679PubMed |
[7] M. Chen, Y. Du, G. Zhu, G. Takamatsu, M. Ihara, K. Matsuda, B. S. Zhorov, K. Dong, Pestic. Biochem. Physiol. 2018, 151, 82.
| Crossref | GoogleScholarGoogle Scholar | 30704718PubMed |
[8] D. G. Beddie, A. W. Farnham, B. P. S. Khambay, Pestic. Sci. 1996, 48, 175.
| Crossref | GoogleScholarGoogle Scholar |
[9] F. Zhu, J. Wigginton, A. Romero, A. Moore, K. Ferguson, R. Palli, M. F. Potter, K. F. Haynes, S. R. Palli, Arch. Insect Biochem. Physiol. 2010, 73, 245.
| 20301216PubMed |
[10] Y.-L. Chen, J. E. Casida, J. Agric. Food Chem. 1969, 17, 208.
| Crossref | GoogleScholarGoogle Scholar |
[11] A. F. Bramwell, L. Crombie, P. Hemesley, G. Pattenden, Tetrahedron 1969, 25, 1727.
| Crossref | GoogleScholarGoogle Scholar | 5796569PubMed |
[12] I.-H. Wang, V. Subramanian, R. Moorman, J. Burleson, J. Ko, J. Chromatogr. A 1997, 766, 277.
| Crossref | GoogleScholarGoogle Scholar |
[13] N. H. Bagnall, B. M. Hines, A. J. Lucke, P. K. Gupta, R. C. Reid, D. P. Fairlie, A. C. Kotze, Int. J. Parasitol. Drugs Drug Resist. 2017, 7, 51.
| Crossref | GoogleScholarGoogle Scholar | 28110187PubMed |
[14] A. C. Kotze, N. H. Bagnall, A. P. Ruffell, R. Pearson, Med. Vet. Entomol. 2014, 28, 297.
| Crossref | GoogleScholarGoogle Scholar | 24417268PubMed |
[15] H. C. Brown, R. Liotta, G. W. Kramer, J. Org. Chem. 1978, 43, 1058.
| Crossref | GoogleScholarGoogle Scholar |
[16] H. C. Brown, S. Krishnamurthy, N. M. Yoon, J. Org. Chem. 1976, 41, 1778.
| Crossref | GoogleScholarGoogle Scholar |
[17] S. Krishnamurthy, H. C. Brown, J. Org. Chem. 1977, 42, 1197.
| Crossref | GoogleScholarGoogle Scholar |
[18] R. S. Varma, G. W. Kabalka, Synth. Commun. 1985, 15, 985.
| Crossref | GoogleScholarGoogle Scholar |
[19] G. R. Meyer, J. Chem. Educ. 1981, 58, 628.
| Crossref | GoogleScholarGoogle Scholar |
[20] W. A. Gersdorff, J. Econ. Entomol. 1947, 40, 878.
| Crossref | GoogleScholarGoogle Scholar | 18858107PubMed |
[21] H. L. Haller, W. N. Sullivan, J. Econ. Entomol. 1938, 31, 276.
| Crossref | GoogleScholarGoogle Scholar |
[22] L. Crombie, M. Elliott, S. H. Harper, J. Chem. Soc. 1950, 971.
| Crossref | GoogleScholarGoogle Scholar |
[23] D. Wang, D. Astruc, Chem. Rev. 2015, 115, 6621.
| Crossref | GoogleScholarGoogle Scholar | 26061159PubMed |
[24] G. Brieger, T. J. Nestrick, Chem. Rev. 1974, 74, 567.
| Crossref | GoogleScholarGoogle Scholar |
[25] D. Gauthier, A. T. Lindhardt, E. P. K. Olsen, J. Overgaard, T. Skrydstrup, J. Am. Chem. Soc. 2010, 132, 7998.
| Crossref | GoogleScholarGoogle Scholar | 20481527PubMed |
[26] R. Shen, T. Chen, Y. Zhao, R. Qiu, Y. Zhou, S. Yin, X. Wang, M. Goto, L.-B. Han, J. Am. Chem. Soc. 2011, 133, 17037.
| Crossref | GoogleScholarGoogle Scholar | 21916436PubMed |
[27] E. H. P. Tan, G. C. Lloyd-Jones, J. N. Harvey, A. J. J. Lennox, B. M. Mills, Angew. Chem. Int. Ed. 2011, 50, 9602.
| Crossref | GoogleScholarGoogle Scholar |
[28] J. Yu, J. B. Spencer, J. Org. Chem. 1997, 62, 8618.
| Crossref | GoogleScholarGoogle Scholar |
[29] C. Smit, M. W. Fraaije, A. J. Minnaard, J. Org. Chem. 2008, 73, 9482.
| Crossref | GoogleScholarGoogle Scholar | 18975908PubMed |
[30] Y. Imada, H. Iida, T. Naota, J. Am. Chem. Soc. 2005, 127, 14544.
| Crossref | GoogleScholarGoogle Scholar | 16231886PubMed |
[31] B. Pieber, S. T. Martinez, D. Cantillo, C. O. Kapper, Angew. Chem. Int. Ed. 2013, 52, 10241.
| Crossref | GoogleScholarGoogle Scholar |
[32] T. G. E. Davies, L. M. Field, P. N. R. Usherwood, M. S. Williamson, IUBMB Life 2007, 59, 151.
| Crossref | GoogleScholarGoogle Scholar |
[33] R. M. Sawicki, M. Elliott, J. C. Gower, M. Snarey, E. M. Thain, J. Sci. Food Agric. 1962, 13, 172.
| Crossref | GoogleScholarGoogle Scholar |
[34] J. Ward, Chem. Ind. 1953, 586.
[35] M. Kawamoto, M. Moriyama, Y. Ashida, N. Matsudo, Y. Tanabe, J. Org. Chem. 2020, 85, 2984.
| Crossref | GoogleScholarGoogle Scholar | 31912734PubMed |
† The informal nomenclature used in the pyrethrin field is somewhat confusing. Collectively, the six esters shown in Fig. 1 are known as Pyrethrins, three of which are known as Pyrethrins I and the other three Pyrethrins II, but there are also individual esters that are known as pyrethrins – pyrethrin I (1a) and pyrethrin II (1b). The terms that refer to groups of compounds tend to be capitalised (e.g. Pyrethrins), whereas the terms that refer to the individual pyrethrin esters are not (e.g. pyrethin I).