

Multi-Bond Forming Processes in Efficient Synthesis*
Nicholas J. Green A and Michael S. Sherburn A BA Australian Research Council Centre of Excellence for Free Radical Chemistry and Biotechnology, Research School of Chemistry, Australian National University, Canberra, ACT 0200, Australia.
B Corresponding author. Email: sherburn@rsc.anu.edu.au

Nicholas Green is currently a Ph.D. student in the Sherburn research group at the Research School of Chemistry, the Australian National University (ANU). He studied a Ph.B. (Science) at the ANU and received an Honours degree with the University Medal in 2011, taking up doctoral studies as a Rodney Rickards scholar soon after. His research interests include the use of highly unsaturated molecules in synthesis and the catalytic, enantioselective Diels–Alder reaction. |

Professor Michael Sherburn studied chemistry at the University of Nottingham, UK, and received his Ph.D. in 1991 with John A. Murphy. He then moved to Australia and worked as a post-doctoral fellow in the Research School of Chemistry, the Australian National University (ANU) with Lewis N. Mander. He held academic positions at Massey University in New Zealand and the University of Sydney before being appointed at the Research School of Chemistry, ANU in 2002. His awards include the Le Fèvre Memorial Prize of the Australian Academy of Science (2006) and the A. J. Birch Medal of the RACI (2008). |
Australian Journal of Chemistry 66(3) 267-283 https://doi.org/10.1071/CH13003
Submitted: 2 January 2013 Accepted: 1 February 2013 Published: 18 March 2013
Abstract
An increasing number of synthetic organic chemists are embracing the philosophy of efficiency. Herein we highlight multi-bond forming processes, which form two or more new covalent bonds in a single synthetic operation. Such processes, which have the ability to rapidly increase structural complexity, are preeminent in contemporary synthetic organic chemistry. In this short review we classify, analyse, and contrast contemporary multi-bond forming processes, frame these cutting edge contributions within a historical context, and speculate on likely future developments in the area.
Introduction
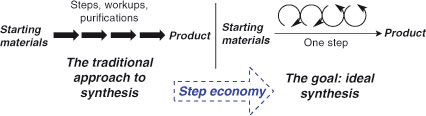
The techniques of synthetic chemistry should allow the construction of virtually any molecule, whether derived from nature or from our own imagination. Nevertheless, even a small molecule can represent a significant synthetic challenge, since the successful combination of just a few atoms requires a mastery of chemo-, regio-, and stereoselectivity. Synthetic techniques have naturally focussed, therefore, on solving the simplest problem of synthesis: the formation of one chemical bond at a time. In the past decade, two Nobel Prizes have been awarded for the development of new organic reactions and, in both cases, these reactions form one new covalent bond.[1] While our proficiency in solo bond formation has led to magnificent achievements in total synthesis,[2] even our best efforts appear somewhat feeble when compared with biological systems.[3] Frankly, synthetic chemists are presently working far from our true limits.[4] The argument for a move away from solo bond forming reactions is a simple one: since the organic structures that we target are significantly different from cheap and readily available starting materials, our reliance on a ‘one bond at a time’ strategy inevitably leads to long synthetic sequences (Fig. 1).
|
The pursuit of ideality[5] in synthesis is not merely academic, for the material fruits of chemistry can only be shared widely when there exist robust, scalable, economical, and logistically feasible methods of synthesis. A recurring (and growing) criticism of academic laboratory syntheses is their tendency to yield tiny quantities of products through a marathon of laboratory effort. Although always an impressive feat of intellect, skill, and perseverance, most published total syntheses could not be used directly to satisfy a large demand for a substance.[6] Even though many members of the pharmaceutical industry are trained by conducting total syntheses in academic laboratories, total synthesis – seen as lengthy and expensive – has been described as ‘out of fashion’ in this setting.[7] This is alarming given that this industry offers perhaps the greatest potential for total synthesis to contribute to the benefit of mankind.
It is clear that in order for synthesis to remain relevant, the problem of step economy must be addressed. Notwithstanding the enormous advances of the past two centuries, the science must continue to progress and evolve, and the invention of new, complexity generating reactions must be at the forefront of this evolution. Encouragingly, a growing movement of chemists is responding to the challenge of step economy by the development of enabling multi-bond forming processes,[8] the subject of this review.
Processes that form more than one chemical bond lead to a more rapid increase in target-relevant structural complexity.[5c] Such transformations are by no means new, with notable contributions dating back more than a century. Driven by the desire to deliver step-efficient syntheses and fuelled by increasingly sophisticated synthetic methods, the field has witnessed an accelerating expansion over the past couple of decades. The recent popularity of these processes can be seen as a consequence of our need to invent new science in synthesis, rather than operating merely as synthetic engineers who simply apply known methods.
An impressive selection of old and new multi-bond forming processes is presented in Scheme 1. We believe that an appreciation of the similarities and differences between distinct classes of process is critical to the development of the field. The aim of the present review is to classify the different types of multi-bond forming processes, to provide recent examples of their application, and to offer perspective on the state-of-the-art and future directions of more efficient syntheses.

|
Classification of Multi-Bond Forming Processes
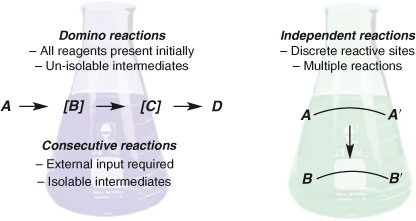
The common characteristic of every reaction depicted in Scheme 1 is the creation of two or more bonds without the traditional workup and/or purification interlude. Despite this similarity, all have significant differences. Tietze and Beifuss have defined ‘domino’ and ‘consecutive’ reactions[9] in order to draw a distinction between those processes that involve either a single or multiple synthetic operations.[10] Thus, a domino reaction has been defined as ‘a transformation of two or more bond-forming reactions under identical reaction conditions, in which the latter transformations take place at the functionalities obtained in the former bond forming reactions’.[11] Domino reactions are the ultimate expression of synthetic orchestration and execution. Sir Robert Robinson performed one of the earliest domino reactions, in his truly innovative – and now classic – synthesis of tropinone (Scheme 1a).[12] Domino reactions may be intra- or intermolecular, or a mixture of both, and can include many processes, such as polymerisation reactions (Scheme 1b),[13] unimolecular ‘cascades’ (Scheme 1c),[14] and multiple component reactions (Scheme 1d).[15] In some respects, certain polymerisation processes can be seen as extreme incarnations of domino reactions, in that many thousands of bonds are formed in a single operation, in the conversion of a simple monomer (usually an alkene) into a large organic structure. The advent of ‘living’ polymerisation techniques[16] has allowed exquisite control over the structure and molecular weight distribution of polymers. Since non-polymeric structures are the focus of this review, we direct the interested reader to key reviews on polymer formation.[17]
Consecutive reactions, unlike domino reactions, require some kind of change in reaction conditions to complete the bond forming sequence. Such changes may include, for example, successive additions of reagents or an increase in temperature. A good example of consecutive reactions can be found in the total synthesis of oligosaccharides by Huang and coworkers.[18] Up to four glycosides can be united in a one-pot sequence of consecutive couplings (Scheme 1e). The one-pot process circumvents costly purifications, and achieves a comparable outcome to automated solid phase synthesis.[19]
Consecutive reactions arise through reagent/reactant incompatibilities and the need to conduct certain sequences in a consecutive manner can be viewed as a weakness of current synthetic methods (i.e. the necessary reagents have not yet been invented to allow the process to be conducted in a true domino sense). Nevertheless, from a practical standpoint, the order and timing of reagent additions is more a tactical issue than a strategic one, and many extremely impressive consecutive-type processes have been invented. For this reason, in this review we sometimes relax the strict definition of domino reactions to include striking, one-pot consecutive sequences. To maintain a tight focus, we highlight strategies and tactics rather than techniques devised for consecutive reactions, e.g. flow chemistry, solid phase synthesis, automated synthesis, etc.
A strategy complementary to domino and consecutive processes involves two or more discrete reactive sites in a substrate undergoing independent reactions. The two or more reactions in question can be equivalent or non-equivalent, with the former being the much more commonly encountered. Independent reactions are distinct from domino and consecutive reactions in that a bond-forming event is not contingent upon the previous one (Fig. 2). A particularly prominent subclass of independent reactions is two-directional synthesis, as used in Johnson et al.’s classic squalene synthesis (Scheme 1f).[20] In the synthesis of chlorothricolide, Roush and Sciotti reported a rare example of two different chemical reactions, promoted by the same reaction conditions, occurring at discrete sites in the one substrate (Scheme 1g).[21] ‘Tandem’ is a term often used to describe reactions of this type.
|
Finally, it should be noted that some reactions, such as cycloadditions (e.g. the Diels–Alder reaction[22]), and metal-catalysed cycloisomerisations (e.g. the Pauson–Khand reaction[23]) form two or more covalent bonds, and for this reason enjoy a privileged status in synthetic chemistry. Cases where these reactions are performed alone formally constitute multi-bond forming processes, but are not covered in this review. Instead, we consider cases where these and other transformations are strung together, using one of the three strategies/tactics above, to lead to even more breathtakingly rapid increases in structural complexity.
The remainder of this review is divided into two broad sections: the former on domino reactions[24] and the latter on independent reactions. The larger section on domino reactions is subdivided into those involving one, two, and more than two skeletal components.
One-Component Domino Reactions
The development of the Stork–Eschenmoser hypothesis in the 1950s[25] led to many synthetic chemists daring to attempt to emulate nature’s highly selective polycyclisation reactions to assemble terpenoid frameworks. Pioneers in this area include Goldsmith[26] and van Tamelen[27] but perhaps the most significant contributions were made by Johnson.[28] Johnson et al. spectacularly demonstrated the viability of a biomimetic polycyclisation approach to steroids with a landmark total synthesis of progesterone.[14] This late-stage, acid-promoted domino reaction (Scheme 1c) formed four new bonds, three new rings, and five new stereocentres with a high degree of relative stereocontrol, allowing the synthesis of progesterone to be completed in just three more steps. This famous work is the predecessor of many different, one-step approaches to polycyclic frameworks.[29] Notable and varied contributions that cannot be highlighted in detail in such a short review include those from the groups of Corey,[30] Grubbs,[31] Keay,[32] Negishi,[33] Overman,[34] Oppolzer,[35] Trost,[36] Vollhardt,[37] Wang,[38] and Wulff.[39] Recently, the groups of Isihara and Snyder have made excellent progress towards the mimicry of nature’s halonium-induced cyclisations.[40]
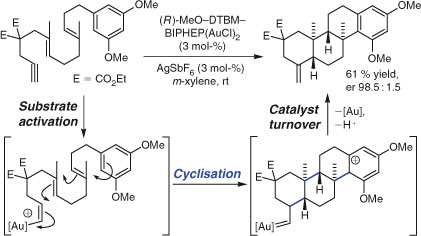
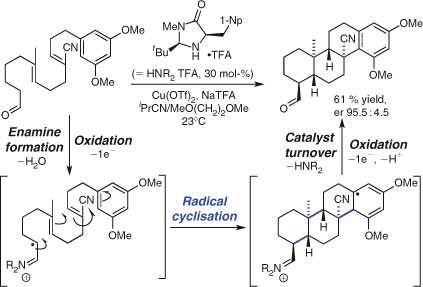
Synthesis through cationic polycyclisation has remained an active area of research. Important recent developments include the realisation of efficient catalytic enantioselective methods, which build on the pioneering work of Yamamoto and coworkers.[41] Thus, Toste and coworkers deployed an efficient, high-turnover chiral AuI complex-catalysed polycyclisation reaction to synthesise a variety of enantioenriched bi-, tri-, and tetra-cyclic frameworks (Scheme 2).[42] The approach involves the chemoselective (soft chiral Lewis acid) activation of an alkyne, which triggers polycyclisation by an initial 6-exo-dig ring closure. Formation of up to three C–C bonds proceeded in good yields and high diastereo- and enantioselectivities. Related examples of cationic, metal-catalysed, enantioselective polycyclisations have been reported by the groups of Corey,[43] Loh,[44] and Gagné,[45] and Jacobsen and coworkers have reported a complementary organocatalytic technique.[46]
|
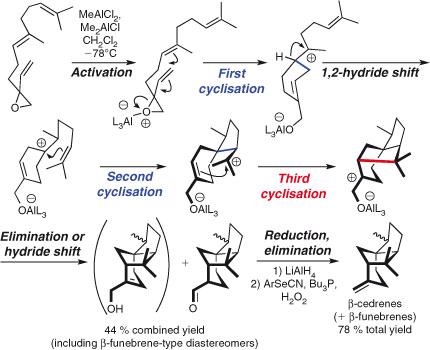
While the head-to-tail cyclisations of terpenes have been mimicked in many laboratories, tail-to-head polycyclisations have, until most recently, proven elusive. In their recent total syntheses of β-funebrenes, β-cedrenes, cumacrene, and the dunnienoic acids, Shenvi and Pronin imitated the anion-sequestering ability of an enzyme to effect tail-to-head cyclisations in the laboratory, resulting in hitherto unsynthesised, strained natural products (Scheme 3).[47] While this new multi-bond forming strategy lacks stereocontrol, selectivity will no doubt improve as further work is conducted in the area.
Radical polycyclisation has long been recognised as a complementary approach to cationic cyclisations, sometimes leading to structures with different stereochemistries.[48] The groups of Breslow,[49] and later Julia,[50] demonstrated the viability of radical cyclisations using simple polyene substrates and organic radical initiators that were incorporated into the product.[51] Different methods have been pioneered by the likes of Demuth and coworkers[52] and Pattenden and coworkers.[53,54] Oxidative metal-mediated radical polycyclisations[55] have also seen significant application, with notable examples from the groups of Snider,[56] Zoretic,[57] Barrero,[58] and Gonzáles.[59] Historically, asymmetric syntheses by these methods invariably featured chiral substrates.[60] Recently, MacMillan and coworkers have disclosed radical polycyclisations forming up to six new rings, using their previously developed catalytic enantioselective organo-SOMO strategy (SOMO, singly occupied molecular orbital).[61] These reactions proceeded with the formation of up to six new C–C bonds and 11 contiguous stereocentres (five quaternary), in good overall yields and high enantioselectivities (Scheme 4).[62]
|
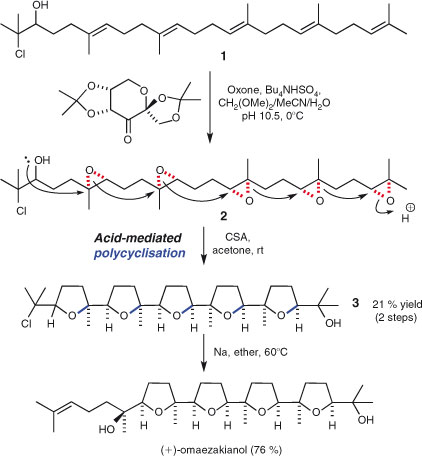
The rather obvious benefits of domino reactions must be weighed up against the (sometimes significant) synthetic effort required to prepare the precursors. Furthermore, high-yielding polycyclisations typically require substrates with built in ‘error-correcting’ groups for (de)stabilisation of intermediates and (de)activation of participating groups. Some syntheses, however, are striking in their brevity since they avoid these lengthy precursor assemblies. The synthesis of polyether natural products has provided some classic examples in this regard.[63] A superb example is Corey and coworkers' recent and remarkably short, enantioselective synthesis of (+)-omaezakianol (Scheme 5).[64a] A five-fold asymmetric epoxidation of chlorohydrin 1 generates penta-epoxide 2, which, upon treatment with camphorsulfonic acid, undergoes a spectacular substrate-controlled five-fold polycyclisation sequence to generate pentacycle 3. Just one more step furnishes the natural product, in only six steps from squalene.
Cycloadditions, powerful multi-bond forming processes in themselves, have been put to extremely good use in polycyclisation reactions.[65] Nicolaou et al.’s application of the Black biosynthetic hypothesis[66] in the synthesis of endiandric acids, using a hydrogenation/double-electrocyclisation/Diels–Alder sequence, is a classic.[67] Wender and Howbert’s metaphotocycloaddition route to hirsutene,[68] the more recent syntheses of hirsutellone B by Nicolaou et al.[69] and Sorensen and coworkers,[70] the double Diels–Alder approaches to FR182877 by Sorenson and coworkers,[71] Evans and Starr,[72] and most recently Nakada and coworkers,[73] and Denmark et al.’s [4+2]/[3+2] domino approach towards daphnilactone B are also outstanding examples.[74] Generation of a reactive ylide from a carbenoid, a strategy pioneered by Padwa from the 1980s, with enantioselective methods emerging from the laboratory of Hodgson,[75] has also proven an effective strategy in the generation of several bonds and significant structural complexity in one step.[76,77]
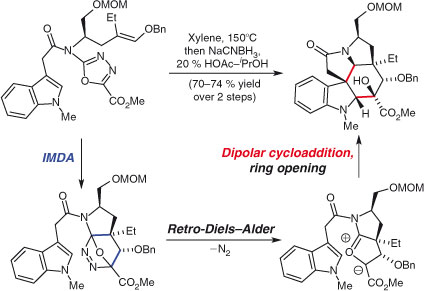
The research group of Boger has championed a complementary method of ylide formation to that of Padwa’s in an uncatalysed, pericyclic intramolecular sequence featuring a hetero-Diels–Alder, retro-Diels–Alder, and dipolar cycloaddition. Two very recent applications of this strategy are in the asymmetric syntheses of vindoline and vindorosine,[78] and (+)-fendleridine and (+)-1-acetylaspidoalbidine.[79] Each features a similar domino sequence of pericyclic reactions, generating, in the case of vindorosine (Scheme 6), three rings, four carbon–carbon bonds, and six stereocentres, in 55 % yield after reduction. The remarkable synthesis of the complex alkaloid was finished in a further nine steps.
|
Two-Component Domino Reactions
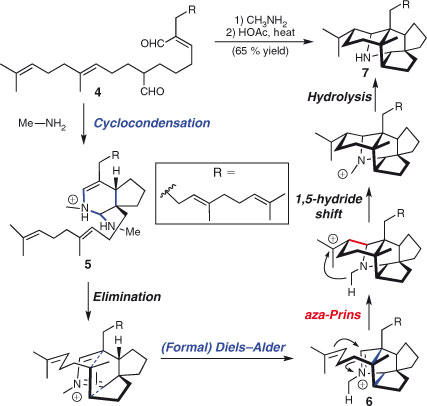
The inclusion of intermolecular processes in a domino sequence will impart convergence and hence, in principle, could lead to a more efficient synthesis. Famous examples of two component domino reactions can be found in Heathcock et al.’s landmark synthesis of daphniphyllum alkaloids (Scheme 7).[80] Thus, acyclic unsaturated dialdehyde 4 undergoes a cyclo-condensation sequence with methylamine to furnish bicyclic aza-diene 5. An intramolecular (formal) hetero-Diels–Alder cycloaddition generates tricycle 6. Aza-Prins cyclisation followed by a hydride shift yields the complex carbocyclic framework of the daphniphyllum alkaloids, from which the final product 7 is generated by iminium hydrolysis. This spectacular transformation forms six new bonds and proceeds in a remarkable 65 % yield to provide a single diastereomer of 7. That the discovery of this particular sequence came about through the use of a mislabelled reagent bottle adds further fascination to this already compelling story.[81]
Other approaches that use the union of an aldehyde and an amine to trigger a remarkable series of reactions include Corey and coworkers’ synthesis of aspidophytine,[82] an alkaloid that has also been synthesised by Mejia-Oneto and Padwa using his domino cyclisation/[3+2] cycloaddition approach.[83] Sarpong and Fischer recently performed condensation reactions of in situ-deprotected carbonyl and amine groups to initiate a domino sequence in the synthesis of (+)-complanadine A.[84] Overman and coworkers promote an aza-Cope/Mannich sequence by condensation of an amine and formaldehyde in their recent synthesis of (–)-actinophyllic acid.[85]
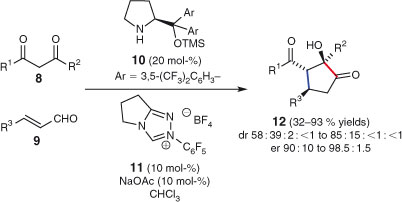
The next example of a combined intermolecular–intramolecular domino sequence highlights some advantages over ‘stop-and-go’ chemistry.[6,86] Lathrop and Rovis’ synthesis of highly substituted cyclopentanones, using a synergistic catalytic system, is presented in Scheme 8.[87] Thus, the two starting materials, β-diketone 8 and enal 9, along with chiral amine organocatalyst 10 and N-heterocyclic carbene precatalyst 11, are combined in the one flask to afford cyclopentanones 12. In contrast, when the two processes (organocatalysed Michael addition and N-heterocyclic carbene-catalysed intramolecular benzoin reaction) are carried out separately, lower yields and enantioselectivities are recorded. The problem with the stop-and-go approach is that the intermediate is prone to epimerisation and decomposition. This example also highlights the new and rapidly growing area of ‘synergistic’ domino reactions, which employ different catalysts that are tolerant of each other and possess orthogonal reactivities.[88]
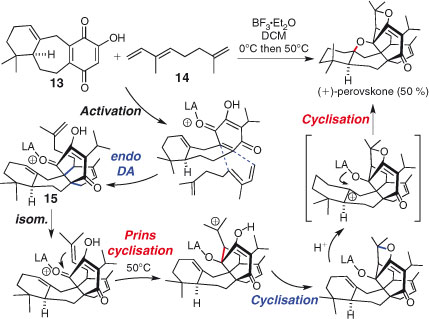
Cycloaddition reactions can also act as effective triggers for domino sequences. Recent examples include Nicolaou et al.’s synthesis of antibiotic BE-43472B, involving a stereoselective Diels–Alder/hemiacetalisation sequence[89] and Aubé and Zeng’s Diels–Alder/Schmidt domino reaction in the synthesis of stenine.[90] Majetich et al. recently disclosed a synthesis of perovskone by a remarkable, biomimetic polycyclisation triggered by an intermolecular Diels–Alder reaction (Scheme 9).[91] Treatment of dienophile 13 and diene 14 with a Lewis acid led to formation of the Diels–Alder adduct 15, which, under carefully optimised conditions, underwent alkene isomerisation to allow a triple cyclisation. This remarkable one-pot sequence led to the creation of four rings, five bonds, and six stereocentres and the synthesis of enantiopure (+)-perovskone in 50 % yield.
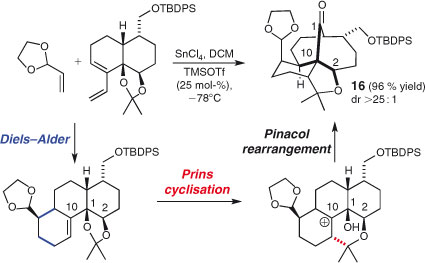
Barriault and coworkers developed an unusual, highly diastereoselective sequence involving a Gassman-type Diels–Alder reaction, Prins cyclisation, and finally a pinacol rearrangement to generate bridged/fused tetracyclic systems 16 (Scheme 10).[92] The efficiency and stereoselectivity of this process are striking, especially given the complex nature of the product.
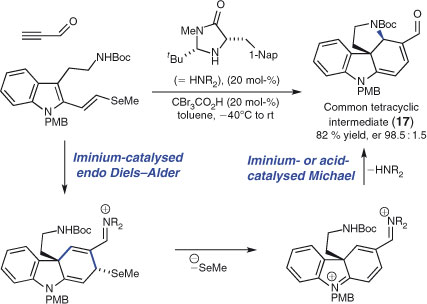
MacMillan and coworkers recently employed an impressive intermolecular–intramolecular domino sequence for the efficient synthesis of an array of alkaloid natural products (Scheme 11).[93] The approach involved the generation of key intermediate 17, from which no less than six structurally related natural products were synthesised. Tetracycle 17 was prepared by an ‘organocascade’ sequence, commencing with a catalytic enantioselective iminium-mediated Diels–Alder reaction, followed by a selenide elimination and terminated with an aza-Michael addition. This polished and cleverly designed sequence forms two C–C bonds and one C–N bond, two new rings and two stereocentres in high enantioselectivity. Importantly, it produces a structure with a wealth of possible avenues for further manipulation into the alkaloids (–)-kopsanone, (+)-vincadifformine, (–)-akuammicine, (–)-kopsinine, (+)-aspidospermidine, and (–)-strychnine.
|
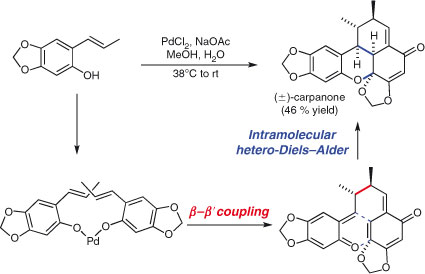
Dimerisation reactions are maximally convergent, and when performed in a domino sense can lead to some astonishingly concise syntheses of complex structures. Chapman et al.’s synthesis of (±)-carpanone features a classic domino dimerisation reaction that highlights the power of symmetry recognition.[94] Chapman et al. saw that carpanone is a dimeric natural product and performed a one-pot coupling/intramolecular hetero-Diels–Alder reaction to form three new bonds and assemble the natural product, as a single diastereomer, in 46 % yield (Scheme 12).
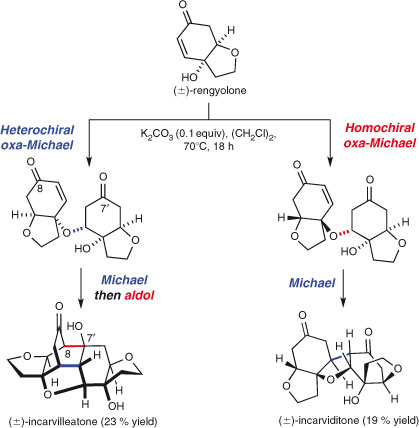
Nicolaou et al.’s syntheses of rugolosin and thiostrepton are further outstanding examples of multi-bond forming dimerisations.[95] Recently, Lawrence and coworkers synthesised the natural products (±)-incarviditone and (±)-incarvilleatone by multi-bond forming domino biomimetic dimersations of rengyolone (Scheme 13).[96] A particularly interesting feature of this synthesis is that the outcome of the reaction is determined by the relative configurations of the two reacting rengyolone molecules. A domino sequence of oxa-Michael and Michael additions between enantiomers of the same absolute configuration led to incarviditone, while a different oxa-Michael/Michael/aldol sequence between enantiomers of opposite absolute configuration led to incarvilleatone.
Diversity oriented synthesis is also benefiting from developments in two-component domino reactions. Recently, Kumar and coworkers described an interesting approach to library generation. Instead of adopting the established method, which involves carrying out the same reaction(s) simultaneously on a range of different substrates to produce a group of related structures, this group employed different cascade pathways using a common precursor.[97]
Multi-Component Reactions (MCRs)
Multi-component reactions (MCRs) enjoy an elevated position in the hierarchy of organic transformations, and have been the topic of numerous reviews.[98] Since they involve the convergent combination of three or more starting materials, at least two new bonds must be formed, which places them in the realm of cycloaddition reactions in terms of their synthetic power. The first MCRs, such as the Strecker,[99] Hantzsch,[100] and Biginelli[101] reactions, were reported in the 19th century and are still widely used. Sir Robert Robinson’s synthesis of tropinone, the forerunner of multicomponent transformations in natural product synthesis, has already been highlighted (Scheme 1a). Multicomponent reactions remain the subject of intense investigation, especially isocyanide-based MCRs such as the Ugi reaction (Scheme 1d),[102] in the context of diversity oriented synthesis.[103,104]
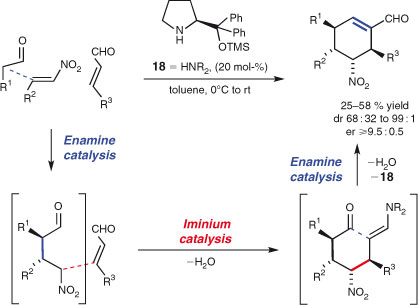
There has been much interest in enantioselective organocatalytic multi-component processes in recent times.[105] A leading example from this new wave of MCRs is Enders et al.’s enantioselective synthesis of functionalised cyclohexenes (Scheme 14).[106] The products are formed in high diastereo- and enantioselectivities by the organocatalysed union of three acyclic components. Thus, essentially a single enantiomer of a product with a new ring, four new stereocentres, and three new carbon–carbon bonds is generated from simple, readily available precursors. Notably, organocatalyst 18 mediates highly stereoselective enamine- and iminium-based processes.[107]
Through the incorporation of a consecutive intramolecular Diels–Alder reaction, the Enders group has subsequently extended this process into the one-pot synthesis of bi- and tri-cyclic frameworks, forming five new C–C bonds and up to eight stereocentres.[108]
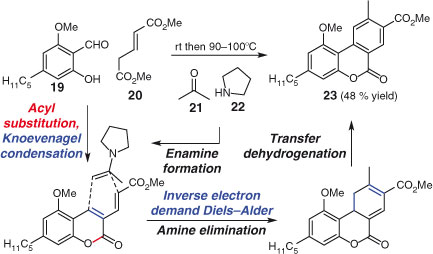
The use of cycloadditions in MCRs is a particularly effective way of generating complexity in a rapid and convergent manner.[109,110] The Knoevenagel condensation,[111] which generates a hetero- or carbo-diene, has been used to generate reactive intermediates that undergo cycloaddition.[112] Nandaluru and Bodwell have recently used such an approach in the synthesis of cannabinol (Scheme 15).[113] The successful route features a three-component reaction comprised of six mechanistic steps. Thus, starting materials 19, 20, and 21 are united by the intervention of pyrrolidine (22) as a simple amine catalyst to furnish tricycle 23 in 48 % yield. The sequence involves nucleophilic acyl substitution, intramolecular Knoevenagel condensation, an inverse electron demand Diels–Alder reaction, elimination, and finally transfer dehydrogenation. Tricycle 23 is converted into the natural product cannabinol in a further four steps.
|
The Kerr group has developed a formal [3+3] multi-component cycloaddition and deployed the process in the total synthesis of (+)-phyllantidine[114] and (+)-nakadomarin A.[115] In the synthesis of (+)-nakadomarin A, chiral cyclopropane 24 reacts with in situ formed nitrone 27, the product of condensation between hydroxylamine 25 and aldehyde 26 (Scheme 16). Oxazine 28 is formed in high yield as a single diastereomer, allowing subsequent installation of an important quaternary stereocentre from the geminal diester functionality and the completion of the total synthesis of the manzamine natural product.
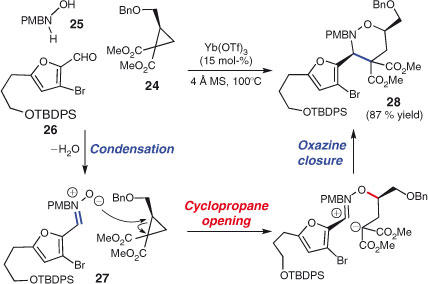
|
The Dixon group has recently targeted other manzamines with a stereoselective, multi-component domino nitro-Mannich lactamisation methodology.[116]
Despite the attraction of their inherently convergent nature, there are relatively few examples of true domino MCRs in target synthesis.[117] Significant developments in this area will no doubt be witnessed in the coming years.[118]
The Nicolaou group has used a remarkable palladium-catalysed multi-component reaction developed by Inoue et al.[119] in the synthesis of hyperolactone (Scheme 17).[120] Treatment of propargyl alcohol 29 with iodobenzene and a palladium catalyst under an atmosphere of carbon monoxide and carbon dioxide effects carbonylative[121] arylation, and subsequent formation of a carbonate sets off a sequence of additions, insertions, and eliminations to generate spirocycle 29, in 77 % yield.
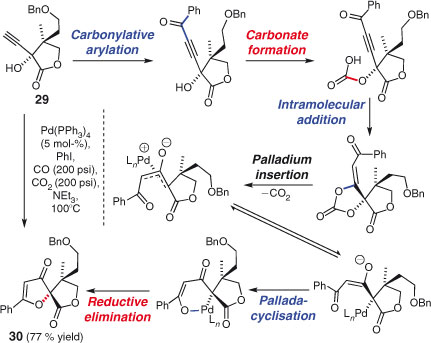
|
Other groups have focussed on improving well known MCRs and applying them in synthesis. The Ugi reaction, for example, while often considered unsuited to stereoselective target synthesis, has been employed in several recent total syntheses.[122] Ruitjer and coworkers used two classic MCRs – an Ugi reaction and a Passerini reaction – in the convergent synthesis of the antiviral drug telaprevir (Scheme 18).[123] The synthesis is also three times shorter than the original reported procedure.[124]
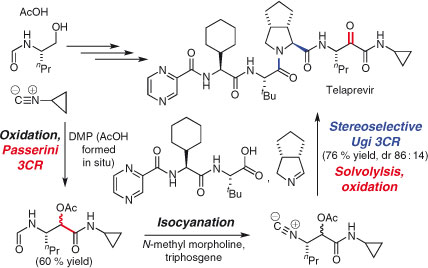
|
Many chemists have developed carefully designed ‘one-pot’ consecutive sequences that unite multiple components. Unlike the MCRs discussed so far, these are not domino reactions, because reagents are added or conditions are changed sequentially in order to avoid undesired processes. When executed well, such a tactic is still extremely meritorious, since intermediates are not isolated and many components can be combined in a convergent manner leading to a remarkable increase in structural complexity.[125]
The Hayashi group has adopted this approach to great effect. Recently, the group completed a notable one-pot synthesis of dipeptidyl peptidase IV inhibitor, ABT-341 (Scheme 19).[126] The carefully optimised one-pot sequence consists of six steps and furnishes the pure product as a single diastereomer in 63 % yield. The first, enantioselective step is an organocatalysed Michael addition between 31 and 32, proceeding with an enantiomeric ratio of 98.5 : 1.5. Careful removal of excess acetaldehyde and solvent leaves behind the product 34 and the organocatalyst 33, the latter being inert during the rest of the sequence. A two-component domino Michael addition/intramolecular Horner–Wadsworth–Emmons reaction establishes the cyclohexene ring but with incorrect stereochemistry, which is rectified in the next step. Hydrolysis of the ester of 35 is followed by amide formation and subsequent reduction of the nitro group to the amine completes the synthesis. This work builds upon the group’s earlier, equally remarkable three-pot synthesis of influenza drug (–)-oseltamivir (Tamiflu).[127]
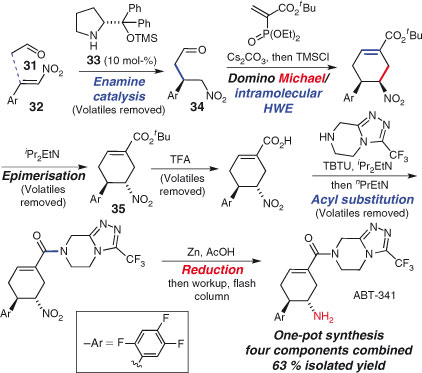
|
The MacMillan group has demonstrated the successive use of a metal catalyst and two organocatalysts in a one-pot transformation, in the enantioselective synthesis of (–)-aromadendranediol.[128] Specifically, consecutive use of the Grubbs II catalyst, imidazolidinone 36b, and proline brought about a three component union through olefin metathesis and iminium and enamine catalysis (Scheme 20).[128,129]
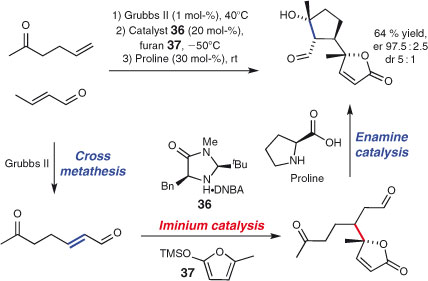
|
In the search for a library of potentially bioactive compounds, Waldmann and coworkers recently developed a three-component, one pot sequence that proceeds by a striking 12-step mechanism (Scheme 21).[130] The sequence involves two intermolecular Michael additions and various ring-closing and ring-opening steps. Overall yields of up to 80 % show control over the selectivity of this process, which involves three separate additions of reagents. Considering the impressive length of this mechanistic sequence, the fact that only two new rings and three new bonds are formed in the process is surprising.

Independent Reactions
All examples discussed so far involve multi-bond forming events that are linked by clear-cut chronology. Specifically, the first bond-forming event sets up the functionality for the second, which sets up the third, etc. The multi-bond forming processes discussed in the present section involve bonds that are formed in no particular order. Several sub-genres of this grouping – which we choose to name ‘independent reactions’ – exist but the field is dominated by contributions in the multi-directional synthesis arena. The synthesis of squalene provides an instructive example. Throughout their pioneering work on the synthesis of squalene, Johnson and coworkers implemented a two-directional chain extension strategy (Scheme 1f).[20] Since each step is a two-fold functionalisation, bonds are made in a ‘two for the price of one’ fashion, hence the structure is generated twice as fast as the more familiar unidirectional approach. The benefits of such a strategy continue to be exploited in the synthesis of C2-symmetric targets.[131]
Since few natural products are symmetrical, one might conclude that multi-directional synthesis is very limited in application. This is not the case! Careful structure analysis can often reveal some measure of ‘hidden’ symmetry in a target. If a symmetrical precursor to a non-symmetrical target is accessed then evidently, a desymmetrisation event will be necessary. Schreiber was a pioneer of the two-directional chain extension/terminus differentiation strategy[132] and his group has employed the approach in magnificent syntheses of a range of unsymmetrical targets, including hikizimycin[133] and mycoticins A and B.[134] This strategy remains popular, with recent examples including those from the laboratories of Zhang,[135] Metz,[136] Molinski,[137] Blakemore,[138] Nelson,[139] and Vogel.[140] Stockman and coworkers have reported the application of the two-directional approach in both target and diversity oriented synthesis.[141] A skillful recent example is their formal total synthesis of halichlorine, which involved two-directional chain extension of ethyl formate (40) using addition, oxidation, and cross-metathesis reactions (Scheme 22).[142] Chain termini were differentiated by a two-component, domino cyclisation sequence to generate 41. Further manipulation intercepted an intermediate from Clive and coworkers’s earlier approach,[143] thereby completing a formal total synthesis.
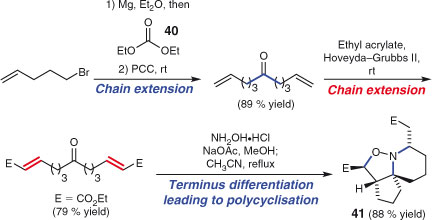
|
Multi-directional chain extension is neither limited to acyclic chains nor to symmetrical precursors. In their impressive synthesis of hibarimicinone,[144] Shair and coworkers carried out a two-fold annulation process upon pseudo-symmetric biaryl precursor 42, which was generated by desymmetrising an earlier, symmetric intermediate (Scheme 23). The same enone 43 reacts at the two distinct ‘termini’ of 42. Thus, independent domino Michael–Claisen sequences yield octacycle 44 in a single operation that forms four key bonds in the target and promotes the completion of a particularly elegant synthesis.[145]
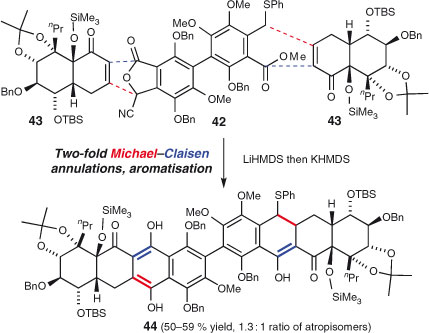
|
Diels and Alder’s first report of the transformation now known as the Diels–Alder reaction involves independent cycloadditions to the two alkenes of benzoquinone by two molecules of cyclopentadiene.[22,146] Another interesting three component reaction using independent dipolar cycloadditions to benzoquinone (45) was recently disclosed by Waldmann and coworkers (Scheme 24).[147] Enantioselective [3+2] cycloadditions are deployed in a two-directional sense to unite three components, forming up to eight stereocentres. Judicious modification of reaction conditions allowed the control of both chemo- and regioselectivity, which promises access to a wide range of related structures.
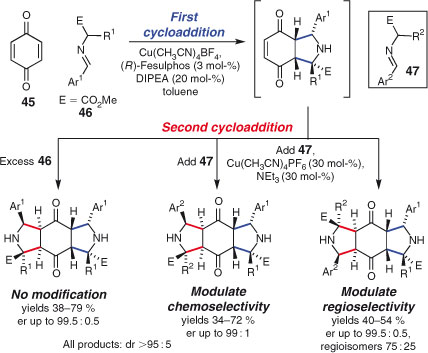
|
With the development of selective and functional group-tolerant olefin and ene-yne metathesis reaction conditions,[148] sequences of such processes for multi-bond formation have emerged. The laboratories of Clark,[149,150] Zhang,[151] and Nakata[152] have exploited two-fold ring-closing metathesis (RCM) strategies to independently and simultaneously form two rings of polyether natural products. These approaches are pre-dated by seminal work by the groups of Mioskowski,[153] Nicolaou,[154] and Martin.[155] Very recently, Selfridge and Feldman utilised a double metathesis on a symmetrical substrate to obtain the linked bicyclic core of ent-lomaiviticin.[156] Two-directional RCM reactions can also be performed as part of a domino sequence, for instance in the ring-opening metathesis/double-RCM sequence employed in Phillips and Pfeiffer’s elegant synthesis of (+)-cyanthiwigin U (Scheme 25),[157] and the metathesis sequence employed in Nelson and coworkers’ recent synthesis of molecular ‘libraries’.[158] Two-fold ene-yne metathesis, pioneered by the group of Grubbs in the 1990s,[159] has also proven to be an efficient strategy for the synthesis of structures containing up to four new rings.[160,161] Some of the most recent examples feature in the work reported by the groups of Yang,[162] Fukuyama,[163] Metz,[164] Oguri,[165] Movassaghi,[166] Mulzer,[167] and Lee.[168]
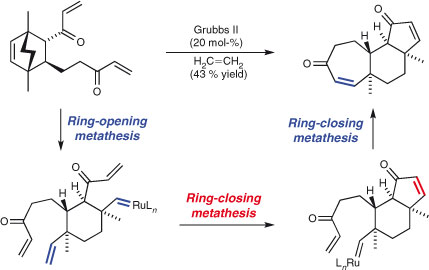
|
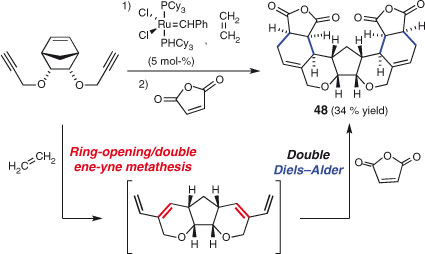
Olefin metathesis steps have also been combined with other transformations in independent reaction sequences.[11,160c,169] A notable example is the work of North and Banti, who exploited a one-pot, two-directional, double ene-yne metathesis/double Diels–Alder sequence (Scheme 26).[170] The reaction forms a meso compound (48) with six new rings, eight new stereocentres, and eight new covalent bonds. This example demonstrates how a two-directional strategy can further amplify the complexity generating effect of an already powerful consecutive sequence.
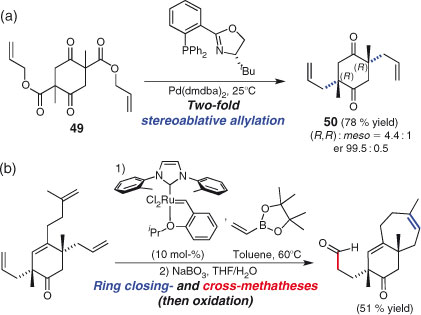
The route to cyanthiwigin F by Stoltz and Enquist is a sophisticated application of the two-fold strategy.[171] A two-directional, catalytic asymmetric approach is used to target a chiral C2-symmetric intermediate (Scheme 27a). Thus, a mixture of the three stereoisomers of allyl ester 49 (namely the two C2-symmetric enantiomers and the one achiral meso diastereomer) was converted, by the formation of two new C–C bonds in an enantioselective double Tsuji–Trost allylation,[172] into a highly enriched sample of the chiral diastereomer 50. Syntheses of other members of the cyanthiwigin natural product family utilised the same powerful ‘stereoablative’ method.[173] The advanced synthetic technology on show in this reaction illustrates the level to which two-directional methods have evolved over the past three decades. This synthesis also features a pair of independent metathesis reactions carried out in a one-pot manner (Scheme 27b): one a ring-closing and the other a cross-metathesis process.
|
Two-fold synthesis clearly offers a powerful method for quickly forming bonds, by doubling the amount of complexity generated with each operation. Many examples of even higher order independent ‘n-fold’ transformations exist, although seldom in the synthesis of naturally occurring materials.[174] The authors’ group has pioneered the synthesis and exploration of the reactivity of a fundamental class of unsaturated hydrocarbons, the dendralenes.[175] As polyalkenes, dendralenes are prime substrates for multi-bond forming processes. The ivyanes, polycyclic helically chiral molecules that exhibit extraordinarily high heats of combustion, are generated in one step from dendralenes by exhaustive cyclopropanation sequences.[176] In the case of [8]ivyane, the synthesis from [8]dendralene (51) proceeds by an eight-fold cyclopropanation to form 16 new carbon–carbon bonds (Scheme 28).

|
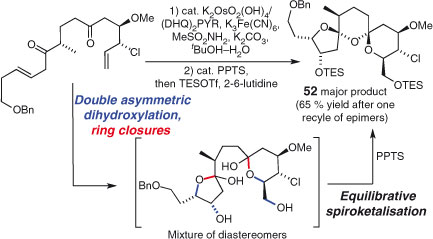
Dendralenes are by no means the only polyenes that have found use in n-fold reactions. Sharpless and coworkers’ six-fold dihydroxylation of squalene from 1993 is a landmark example of the enantiopurity amplification that results from a reagent-controlled, multi-bond forming reaction.[177–179] The breathtakingly short approaches to polyether natural products (+)-omaezakianol (Scheme 5)[64] and glabrescol[180] by Corey and coworkers feature stereoselective five- and four-fold epoxidations, respectively. Paterson et al.’s synthesis of the DEF tricyclic spiroketal fragment of spirastrellolide A (Scheme 29)[181] is another example of ingenious synthetic design using independent reactions combined with domino sequences. Application of a two-fold stereoselective dihydroxylation sets up the requisite functionality for a double acetalisation to tricycle 52, installing no less than six C–O bonds, five stereocentres, and three rings in one pot after exposure to mild acid.
|
The ‘independent reactions’ strategy is complementary to the domino/consecutive strategy and, importantly, the two approaches are not incompatible. Undoubtedly, important avenues for future exploration will involve the design of syntheses that exploit the two approaches simultaneously (Fig. 3). Importantly, to avoid the accumulation of unwanted side products, each discrete bond-forming event must be highly selective.
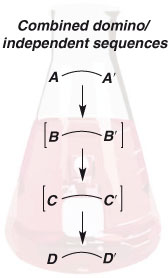
|
Conclusion
In 1992, Clayton Heathcock wrote: ‘Although our approaches to problems have matured, we need even more mature strategies of synthesis. There is no reason that organic chemists should not be able to surpass nature’s virtuosity in the synthesis of complex organic structures. In fact, we are still very far from this goal in most cases.’[182] The authors of the present review believe that Heathcock’s statement is as relevant today as it was 20 years ago.
Multi-bond forming processes and the impressive synthetic achievements that they allow are being reported at an accelerating pace. In this review, we have attempted to sample both pioneering work and recent contributions that exhibit the most sophisticated current methods. We believe that, while future contributions will report increasingly improved methods and applications, the strategies and tactics employed will be variations or combinations of the general themes defined in the present review.
How can practitioners of the science of synthesis move the discipline forward? The key to unleashing the true potential of synthetic organic chemistry lies in the development of better chemical transformations. Specifically, we need reagent and catalyst systems that exhibit broad substrate scope but also extraordinarily high selectivity. We invite the reader to ask the following question when analysing any synthetic reaction sequence: how can consecutive but discrete reactions be united into a single synthetic operation? If we strive to answer this simple question by amalgamating separate reactions into the same process, we are compelled to improve our toolkit of selective and compatible transformations. The more effort that we expend in devising such processes, the less time we will subsequently spend doing target synthesis in the laboratory.
* Dedication: In memory of Athel Beckwith.
Acknowledgments
The authors gratefully acknowledge the financial support of the Australian Research Council.
References
[1] (a) The official website of the Nobel Prize, nobelprize.org, lists (a) 2010 Nobel Prize to Heck, Negishi, and Suzuki “for palladium-catalysed cross couplings in organic synthesis”.(b) 2005 Nobel Prize to Chauvin, Grubbs, and Schrock “for the development of the metathesis method in organic synthesis”.
[2] K. C. Nicolaou, J. S. Chen, Classics in Total Synthesis III 2011 (Wiley-VCH: Weinheim).
[3] M. Razzak, J. K. De Brabander, Nat. Chem. Biol. 2011, 7, 865.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsVOisL3O&md5=a0c08bffea5bdd1c31be632c63a190acCAS |
[4] We are referring to limits imposed by the laws of nature here.
[5] (a) The ideal synthesis is an idea initially articulated by Hendrickson and then developed and championed by Wender. See: J. B. Hendrickson, J. Am. Chem. Soc. 1975, 97, 5784.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE2MXlvFajs7Y%3D&md5=cb04a2f89de7e014b1f08d43eba2a772CAS |
(b) P. A. Wender, M. P. Croatt, B. Witulski, Tetrahedron 2006, 62, 7505.
| Crossref | GoogleScholarGoogle Scholar |
(c) P. A. Wender, B. L. Miller, Nature 2009, 460, 197.
| Crossref | GoogleScholarGoogle Scholar |
(d) See also: T. Gaich, P. S. Baran, J. Org. Chem. 2010, 75, 4657.
| Crossref | GoogleScholarGoogle Scholar |
[6] We do not wish to underplay the importance of total synthesis. There is pressing need for total synthesis to continue to pioneer new methods and strategies as stepping stones towards the ‘ideal synthesis’. For a discussion of the problem of total synthesis within the context of a specific natural product/medicinal agent, see: A. M. Walji, D. W. C. MacMillan, Synlett 2007, 1477.
| 1:CAS:528:DC%2BD2sXotVynu7Y%3D&md5=26c9a1a81d89d8b4cb029d911de50142CAS |
[7] H. Ledford, Nature 2010, 468, 608.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsFSku7bP&md5=3d6ca89e6f84da4cc4ca64f3830242b2CAS |
[8] We eschew the word ‘multiple’ in favour of ‘multi’, in order to prevent any possible association with unsaturated bonds.
[9] L. F. Tietze, U. Beifuss, Angew. Chem. Int. Ed. Engl. 1993, 32, 131.
| Crossref | GoogleScholarGoogle Scholar |
[10] Ł. Albrecht, H. Jiang, K. A. Jørgensen, Angew. Chem. Int. Ed. 2011, 50, 8492.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXpvFahsbs%3D&md5=8f15eea47a5791a484234f9675aae0caCAS |
[11] L. F. Tietze, G. Brasche, K. M. Gericke, Domino Reactions in Organic Synthesis, 1st ed., 2006 (Wiley-VCH: Weinheim).
[12] R. Robinson, J. Chem. Soc. Trans. 1917, 111, 762.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaC2sXhsFShsw%3D%3D&md5=486885b969be9af76f1025d5a2b9e24bCAS |
[13] J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo, S. H. Thang, Macromolecules 1998, 31, 5559.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXkvF2gs7k%3D&md5=b6c833121ee15ed9473a3f1c9c1c7141CAS |
[14] W. S. Johnson, M. B. Gravestock, B. E. McCarry, J. Am. Chem. Soc. 1971, 93, 4332.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE3MXltFSms74%3D&md5=6bd9593200198c5164daa28f152271b1CAS |
[15] A. Endo, A. Yanagisawa, M. Abe, S. Tohma, T. Kan, T. Fukuyama, J. Am. Chem. Soc. 2002, 124, 6552.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38Xjs1aksLY%3D&md5=848d05cb97b96707689582b0265fb9f3CAS |
[16] Controlled and Living Polymerizations: From Mechanisms to Applications, (Eds. K. Matyjaszewski, A. H. E. Müller) 2009 (Wiley-VCH: Weinheim).
[17] (a) For reviews on polymerisation, see: G. Moad, E. Rizzardo, S. H. Thang, Aust. J. Chem. 2012, 65, 985.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhtlWmu7%2FE&md5=aa37bfea601cd67a23853d2d03121563CAS |
(b) Synthesis of Polymers (Eds. D. A. Schluter, C. Hawker, J. Sakamoto) 2012 (Wiley-VCH: Weinheim).
[18] Z. Wang, L. Zhou, K. El-Boubbou, X.-S. Ye, X. Huang, J. Org. Chem. 2007, 72, 6409.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXnvFGntLk%3D&md5=f031e2794b2ae61be22d5089634d4d57CAS |
[19] O. J. Plante, E. R. Palmacci, P. H. Seeberger, Science 2001, 291, 1523.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXhsVeru7w%3D&md5=d3277b86be6dcc4b22853d33d6c53849CAS |
[20] W. S. Johnson, L. Werthemann, W. R. Bartlett, T. J. Brocksom, T.-T. Li, D. J. Faulkner, M. R. Petersen, J. Am. Chem. Soc. 1970, 92, 741.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE3cXnvFamtA%3D%3D&md5=e70dba8b9ffadcfecf7341d2aac466ddCAS |
[21] W. R. Roush, R. J. Sciotti, J. Am. Chem. Soc. 1998, 120, 7411.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXkvF2gsr0%3D&md5=2473ba1a79d97836dc6ae2ef058f8036CAS |
[22] O. Diels, K. Alder, Justus Liebigs Ann. Chem. 1928, 460, 98.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaB1cXpsFyn&md5=9259d4442a2fea355efa743fa7997eb0CAS |
[23] I. U. Khand, G. R. Knox, P. L. Pauson, W. E. Watts, M. I. Foreman, J. Chem. Soc., Perkin Trans. 1 1973, 977.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE3sXhsF2jtro%3D&md5=abeecd497b59cf32470da6b718a2ffefCAS |
[24] With the occasional exception involving consecutive reactions, as noted.
[25] (a) G. Stork, A. W. Burgstahler, J. Am. Chem. Soc. 1955, 77, 5068.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaG28XksVCnsQ%3D%3D&md5=0aa168df4c2a549b47723c3fcb0e785eCAS |
(b) A. Eschenmoser, L. Ruzicka, O. Jeger, D. Arigoni, Helv. Chim. Acta 1955, 38, 1890.
| Crossref | GoogleScholarGoogle Scholar |
(c) For an English translation and commentary, see: A. Eschenmoser, D. Arigoni, Helv. Chim. Acta 2005, 88, 3011.For an English translation and commentary, see:
| Crossref | GoogleScholarGoogle Scholar |
(d) P. A. Stadler, A. Eschenmoser, H. Schinz, G. Stork, Helv. Chim. Acta 1957, 40, 2191.
| Crossref | GoogleScholarGoogle Scholar |
[26] (a) Key papers include: D. J. Goldsmith, J. Am. Chem. Soc. 1962, 84, 3913.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaF3sXjtlGnsg%3D%3D&md5=4b4dd9e37b2b3b93d76a08ac0fa963d7CAS |
(b) D. J. Goldsmith, C. F. Phillips, J. Am. Chem. Soc. 1969, 91, 5862.
| Crossref | GoogleScholarGoogle Scholar |
[27] (a) Key papers include: E. E. van Tamelen, J. Willet, M. Schwartz, R. Nadeau, J. Am. Chem. Soc. 1966, 88, 5937.
| 1:CAS:528:DyaF2sXmsFKmuw%3D%3D&md5=9c4a62d9e04e4f5a819e5119b3bfef8eCAS |
(b) E. E. Van Tamelen, J. P. McCormick, J. Am. Chem. Soc. 1969, 91, 1847.
| Crossref | GoogleScholarGoogle Scholar |
(c) E. E. Van Tamelen, T. M. Leiden, J. Am. Chem. Soc. 1982, 104, 2061.
| Crossref | GoogleScholarGoogle Scholar |
(d) E. E. Van Tamelen, J. R. Hwu, J. Am. Chem. Soc. 1983, 105, 2490.
| Crossref | GoogleScholarGoogle Scholar |
(e) For an early review, see: E. E. Van Tamelen, Acc. Chem. Res. 1968, 1, 111.
| Crossref | GoogleScholarGoogle Scholar |
[28] (a) A summary of Johnson’s contributions can be found in: (a) W. S. Johnson, A Fifty-Year Love Affair with Organic Chemistry 1998 (American Chemical Society: Washington, DC).
(b) For an earlier review, see: W. S. Johnson, Bioorg. Chem. 1976, 5, 51.
| Crossref | GoogleScholarGoogle Scholar |
[29] (a) For an excellent review of pioneering biomimetic polycyclisation reactions towards terpenoid structures, see: R. A. Yoder, J. N. Johnston, Chem. Rev. 2005, 105, 4730.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXht1Gms7bM&md5=5e346c341ce2c19e6980c34bcf555982CAS |
(b) A more recent review: M. Kotora, F. Hessler, B. Eignerová, Eur. J. Org. Chem. 2012, 29.
| Crossref | GoogleScholarGoogle Scholar |
[30] (a) Key early papers include: E. J. Corey, J. Lee, J. Am. Chem. Soc. 1993, 115, 8873.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2cXhtVOnsr8%3D&md5=5e716524535d3338c4a730beffe5425eCAS |
(b) E. J. Corey, J. Lee, D. R. Liu, Tetrahedron Lett. 1994, 35, 9149.
| Crossref | GoogleScholarGoogle Scholar |
(c) E. J. Corey, S. Lin, J. Am. Chem. Soc. 1996, 118, 8765.
| Crossref | GoogleScholarGoogle Scholar |
(d) E. J. Corey, G. Luo, L. S. Lin, J. Am. Chem. Soc. 1997, 119, 9927.
| Crossref | GoogleScholarGoogle Scholar |
(e) For an impressive recent example, see: K. Surendra, E. J. Corey, J. Am. Chem. Soc. 2009, 131, 13928.
| Crossref | GoogleScholarGoogle Scholar |
[31] W. J. Zuercher, M. Scholl, R. H. Grubbs, J. Org. Chem. 1998, 63, 4291.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXjslKntrs%3D&md5=50d1e4cb1968adc5caceb119600676c1CAS |
[32] S. P. Maddaford, N. G. Andersen, W. A. Cristofoli, B. A. Keay, J. Am. Chem. Soc. 1996, 118, 10766.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK28Xmt1Gmt78%3D&md5=fad63fcab0541a29aff3f1b763e25f53CAS |
[33] Y. Zhang, G. Z. Wu, G. Agnel, E. I. Negishi, J. Am. Chem. Soc. 1990, 112, 8590.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3cXmtlSqsbY%3D&md5=33ebe86493b3474cdbee567487d5dfc0CAS |
[34] (a) M. Bogenstätter, A. Limberg, L. E. Overman, A. L. Tomasi, J. Am. Chem. Soc. 1999, 121, 12206.
| Crossref | GoogleScholarGoogle Scholar |
(b) M. M. Abelman, L. E. Overman, J. Am. Chem. Soc. 1988, 110, 2328.
| Crossref | GoogleScholarGoogle Scholar |
(c) N. E. Carpenter, D. J. Kucera, L. E. Overman, J. Org. Chem. 1989, 54, 5846.
| Crossref | GoogleScholarGoogle Scholar |
(d) For a review, see: L. E. Overman, M. M. Abelman, D. J. Kucera, V. D. Tran, D. J. Ricca, Pure Appl. Chem. 1992, 64, 1813.
| Crossref | GoogleScholarGoogle Scholar |
[35] W. Oppolzer, R. J. DeVita, J. Org. Chem. 1991, 56, 6256.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3MXmtlWktrs%3D&md5=0f2d5319350e3fa7428ef286f2c14d5cCAS |
[36] (a) B. M. Trost, Y. Shi, J. Am. Chem. Soc. 1991, 113, 701.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3MXotFSluw%3D%3D&md5=56c4d2b1c66d9a311e50d4aa34ccdd3bCAS |
(b) B. M. Trost, Y. Shi, J. Am. Chem. Soc. 1992, 114, 791.
| Crossref | GoogleScholarGoogle Scholar |
[37] (a) R. L. Funk, K. P. C. Vollhardt, J. Am. Chem. Soc. 1980, 102, 5253.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL3cXltFCrs74%3D&md5=40ebecb232131c0306c0723a2daa4538CAS |
(b) S. H. Lecker, N. H. Nguyen, K. P. C. Vollhardt, J. Am. Chem. Soc. 1986, 108, 856.
| Crossref | GoogleScholarGoogle Scholar |
(c) Recent variations on this theme include: M. Petit, C. Aubert, M. Malacria, Org. Lett. 2004, 6, 3937.
| Crossref | GoogleScholarGoogle Scholar |
(d) U. Groth, N. Richter, A. Kalogerakis, Synlett 2006, 905.
| Crossref | GoogleScholarGoogle Scholar |
[38] Y. W. Andemichael, Y. Huang, K. K. Wang, J. Org. Chem. 1993, 58, 1651.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3sXhvFajurw%3D&md5=38fdca821e01dc8ad090d4ad8f7bb065CAS |
[39] J. M. Bao, V. Dragisich, S. Wenglowsky, W. D. Wulff, J. Am. Chem. Soc. 1991, 113, 9873.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3MXmsl2ms7c%3D&md5=c3d77adaaaf3dd3c219bc5879a431a45CAS |
[40] (a) A. Sakakura, A. Ukai, K. Ishihara, Nature 2007, 445, 900.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhvFGit7Y%3D&md5=18a4f5f3dca3a7db7f51a59961a631cbCAS |
(b) S. A. Snyder, D. S. Treitler, A. P. Brucks, J. Am. Chem. Soc. 2010, 132, 14303.
| Crossref | GoogleScholarGoogle Scholar |
[41] (a) H. Ishibashi, K. Ishihara, H. Yamamoto, J. Am. Chem. Soc. 2004, 126, 11122.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXmslSqt78%3D&md5=fe043d0ecb3ea369dbd326881b9d0d44CAS |
(b) K. Ishihara, S. Nakamura, H. Yamamoto, J. Am. Chem. Soc. 1999, 121, 4906.
| Crossref | GoogleScholarGoogle Scholar |
[42] S. G. Sethofer, T. Mayer, F. D. Toste, J. Am. Chem. Soc. 2010, 132, 8276.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXmvFaktrg%3D&md5=810c16b0f14383700eff538b7b66a2a3CAS |
[43] (a) K. Surendra, W. W. Qiu, E. J. Corey, J. Am. Chem. Soc. 2011, 133, 9724.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXntVenurY%3D&md5=6b28e33abc448f4e40335b2cb66b6d82CAS |
(b) K. Surendra, E. J. Corey, J. Am. Chem. Soc. 2012, 134, 11992.
| Crossref | GoogleScholarGoogle Scholar |
[44] Y.-J. Zhao, B. Li, L.-J. S. Tan, Z.-L. Shen, T.-P. Loh, J. Am. Chem. Soc. 2010, 132, 10242.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXos1ersLk%3D&md5=8e0ec41e333e8d629010de4af993940cCAS |
[45] C. A. Mullen, A. N. Campbell, M. R. Gagné, Angew. Chem. Int. Ed. 2008, 47, 6011.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXpslagtbc%3D&md5=76a67c68aa44151ab0c0df1a3e887713CAS |
[46] R. R. Knowles, S. Lin, E. N. Jacobsen, J. Am. Chem. Soc. 2010, 132, 5030.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXktFekuro%3D&md5=409dd4fd532c321686a4d0854929b307CAS |
[47] S. V. Pronin, R. A. Shenvi, Nat. Chem. 2012, 4, 915.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xhtlylsb7O&md5=702b91cb8213b779bdbbfe22d376458fCAS |
[48] (a) For recent reviews on radical polycyclisations, see: J. Justicia, L. Alvarez de Cienfuegos, A. G. Campana, D. Miguel, V. Jakoby, A. Gansauer, J. M. Cuerva, Chem. Soc. Rev. 2011, 40, 3525.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXns12nu78%3D&md5=8990e92637c94e40d10289e9394a5751CAS |
(b) A. Baralle, A. Baroudi, M. Daniel, L. Fensterbank, J. Goddard, E. Lacôte, M. Larraufie, G. Maestri, M. Malacria, C. Ollivier, in Encyclopedia of Radicals in Chemistry, Biology and Materials (Eds. C. Chatgilialoglu, A. Studer), Ch. 27, pp. 729–765 (Wiley: Chichester).
[49] R. Breslow, S. S. Olin, J. T. Groves, Tetrahedron Lett. 1968, 9, 1837.
| Crossref | GoogleScholarGoogle Scholar |
[50] J. Y. Lallemand, M. Julia, D. Mansuy, Tetrahedron Lett. 1973, 14, 4461.
| Crossref | GoogleScholarGoogle Scholar |
[51] Professor Athel Beckwith also made significant contributions to the understanding of the selectivity of radical addition ring closures, including an early radical polycyclisation reaction to form the triquinane framework. A. L. J. Beckwith, C. H. Schiesser, Tetrahedron 1985, 41, 3925.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL28XkvFKks78%3D&md5=89177426f8814b9e30ece94001789b14CAS |
[52] (a) U. Hoffmann, Y. Gao, B. Pandey, S. Klinge, K. D. Warzecha, C. Krueger, H. D. Roth, M. Demuth, J. Am. Chem. Soc. 1993, 115, 10358.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2cXoslKg&md5=9a955bc42321f3651199cc6e2dc91066CAS |
(b) X. Xing, M. Demuth, Eur. J. Org. Chem. 2001, 537.
| Crossref | GoogleScholarGoogle Scholar |
[53] (a) Acyl selenides as starting materials: L. Chen, G. B. Gill, G. Pattenden, Tetrahedron Lett. 1994, 35, 2593.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2cXlsFOqtrc%3D&md5=6ff75bc5b19eb577bc801d06afa3f93fCAS |
(b) S. Handa, P. S. Nair, G. Pattenden, Helv. Chim. Acta 2000, 83, 2629.
| Crossref | GoogleScholarGoogle Scholar |
[54] (a) Macrocyclisation/transannular sequences: for an initial important contribution from Porter, see: N. A. Porter, V. H. T. Chang, D. R. Magnin, B. T. Wright, J. Am. Chem. Soc. 1988, 110, 3554.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1cXit1Wgs7k%3D&md5=4a8f88b8e3a382f9b3251645fafb10f4CAS |
(b) For a review on early developments: S. Handa, G. Pattenden, Contemp. Org. Synth. 1997, 4, 196.
| Crossref | GoogleScholarGoogle Scholar |
(c) For recent applications, see: G. Pattenden, M. A. Gonzalez, S. McCulloch, A. Walter, S. J. Woodhead, Proc. Natl. Acad. Sci. USA 2004, 101, 12024.
| Crossref | GoogleScholarGoogle Scholar |
(d) G. Pattenden, D. A. Stoker, J. M. Winne, Tetrahedron 2009, 65, 5767.
| Crossref | GoogleScholarGoogle Scholar |
[55] (a) For reviews, see: A. F. Barrero, J. F. Quílez del Moral, E. M. Sánchez, J. F. Arteaga, Eur. J. Org. Chem. 2006, 1627.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XjsV2jsLY%3D&md5=a0253bd09a218dd7f2694d0b4cfffa68CAS |
(b) B. B. Snider, Chem. Rev. 1996, 96, 339.
| Crossref | GoogleScholarGoogle Scholar |
[56] B. B. Snider, J. Y. Kiselgof, B. M. Foxman, J. Org. Chem. 1998, 63, 7945.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXmsVaksLo%3D&md5=a6b56c06c725d7d5e302644d90658112CAS |
[57] P. A. Zoretic, Z. Shen, M. Wang, A. A. Ribeiro, Tetrahedron Lett. 1995, 36, 2925.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXlsVSqtb0%3D&md5=169979824610560a38a6eefc677e4048CAS |
[58] A recent example: V. Domingo, J. F. Arteaga, J. L. L. Perez, R. Pelaez, J. F. Q. del Moral, A. F. Barrero, J. Org. Chem. 2012, 77, 341.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsFKktrnI&md5=0b254607c56c33363f8aa3bdd7354596CAS |
[59] M. A. González, S. Molina-Navarro, J. Org. Chem. 2007, 72, 7462.
| Crossref | GoogleScholarGoogle Scholar |
[60] (a) For examples, see: Q. Zhang, R. M. Mohan, L. Cook, S. Kazanis, D. Peisach, B. M. Foxman, B. B. Snider, J. Org. Chem. 1993, 58, 7640.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2cXivF2jsrY%3D&md5=6781df9899193b6d4a1d9a29fb335c8cCAS |
(b) C. Heinemann, M. Demuth, J. Am. Chem. Soc. 1997, 119, 1129.
| Crossref | GoogleScholarGoogle Scholar |
(c) D. Yang, X.-Y. Ye, S. Gu, M. Xu, J. Am. Chem. Soc. 1999, 121, 5579.
| Crossref | GoogleScholarGoogle Scholar |
[61] T. D. Beeson, A. Mastracchio, J.-B. Hong, K. Ashton, D. W. C. MacMillan, Science 2007, 316, 582.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXksFeksLY%3D&md5=9f2373dd65bd8c19c5dccc22cc70692cCAS |
[62] S. Rendler, D. W. C. MacMillan, J. Am. Chem. Soc. 2010, 132, 5027.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXjvVejsbw%3D&md5=20398528e679dbf8b5d81bc877ec7f3bCAS |
[63] (a) For reviews, see: I. Vilotijevic, T. F. Jamison, Angew. Chem. Int. Ed. 2009, 48, 5250.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXosV2hsb0%3D&md5=4dbc6bcdc7723d0b3bdc974099986098CAS |
(b) F. E. McDonald, R. Tong, J. C. Valentine, F. Bravo, Pure Appl. Chem. 2007, 79, 281.
| Crossref | GoogleScholarGoogle Scholar |
(c) For recent examples, see: A. R. Van Dyke, T. F. Jamison, Angew. Chem. Int. Ed. 2009, 48, 4430.
| Crossref | GoogleScholarGoogle Scholar |
(d) I. Vilotijevic, T. F. Jamison, Science 2007, 317, 1189.
| Crossref | GoogleScholarGoogle Scholar |
(e) Y. Morimoto, H. Yata, Y. Nishikawa, Angew. Chem. Int. Ed. 2007, 46, 6481.
| Crossref | GoogleScholarGoogle Scholar |
(f) R. Tong, F. E. McDonald, Angew. Chem. Int. Ed. 2008, 47, 4377.
| Crossref | GoogleScholarGoogle Scholar |
(g) J. Tanuwidjaja, S.-S. Ng, T. F. Jamison, J. Am. Chem. Soc. 2009, 131, 12084.
| Crossref | GoogleScholarGoogle Scholar |
(h) M. A. Boone, R. Tong, F. E. McDonald, S. Lense, R. Cao, K. I. Hardcastle, J. Am. Chem. Soc. 2010, 132, 5300.
| Crossref | GoogleScholarGoogle Scholar |
(i) J. J. Topczewski, J. G. Kodet, D. F. Wiemer, J. Org. Chem. 2011, 76, 909.
| Crossref | GoogleScholarGoogle Scholar |
[64] (a) Z. Xiong, R. Busch, E. J. Corey, Org. Lett. 2010, 12, 1512.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXjtVyhtLo%3D&md5=67c6910582eb61f446ac350124ab2023CAS |
(b) L. K. Geisler, S. Nguyen, C. J. Forsyth, Org. Lett. 2004, 6, 4159.
| Crossref | GoogleScholarGoogle Scholar |
(c) For a similar, but less spectacular, synthesis, see: Y. Morimoto, T. Okita, H. Kambara, Angew. Chem. Int. Ed. 2009, 48, 2538.
| Crossref | GoogleScholarGoogle Scholar |
(d) For similar recent cascade cyclisations to form polyether natural products, see: P. Yang, P.-F. Li, J. Qu, L.-F. Tang, Org. Lett. 2012, 14, 3932.
| Crossref | GoogleScholarGoogle Scholar |
(e) J. A. Marshall, R. K. Hann, J. Org. Chem. 2008, 73, 6753.
| Crossref | GoogleScholarGoogle Scholar |
[65] J. Poulin, C. M. Grise-Bard, L. Barriault, Chem. Soc. Rev. 2009, 38, 3092.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXht12ht73E&md5=5ca8a0ab82037af0724ab2596f2a007bCAS |
[66] W. M. Bandaranayake, J. E. Banfield, D. St. C. Black, J. Chem. Soc., Chem. Commun. 1980, 902.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL3MXht1ynsA%3D%3D&md5=3d4cc11e8f91791d68637433203c1d71CAS |
[67] (a) K. C. Nicolaou, N. A. Petasis, R. E. Zipkin, J. Uenishi, J. Am. Chem. Soc. 1982, 104, 5555.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL38XlsVClt7o%3D&md5=6e0ca0fe0f75d406bf5eda32c27cfe08CAS |
(b) K. C. Nicolaou, N. A. Petasis, J. Uenishi, R. E. Zipkin, J. Am. Chem. Soc. 1982, 104, 5557.
| Crossref | GoogleScholarGoogle Scholar |
(c) K. C. Nicolaou, R. E. Zipkin, N. A. Petasis, J. Am. Chem. Soc. 1982, 104, 5558.
| Crossref | GoogleScholarGoogle Scholar |
(d) K. C. Nicolaou, N. A. Petasis, R. E. Zipkin, J. Am. Chem. Soc. 1982, 104, 5560.
| Crossref | GoogleScholarGoogle Scholar |
[68] P. A. Wender, J. J. Howbert, Tetrahedron Lett. 1982, 23, 3983.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL3sXovFSgsw%3D%3D&md5=eff4df96afbeaef50377c77e787d6331CAS |
[69] K. C. Nicolaou, D. Sarlah, T. R. Wu, W. Zhan, Angew. Chem. Int. Ed. 2009, 48, 6870.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtVOrtrzE&md5=4eaf4ee7fcdad6cd19aed12ec9914c94CAS |
[70] S. D. Tilley, K. P. Reber, E. J. Sorensen, Org. Lett. 2009, 11, 701.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXisVSqtA%3D%3D&md5=8ac6c425a8a78263c4d4e2f313b4bf9fCAS |
[71] D. A. Vosburg, C. D. Vanderwal, E. J. Sorensen, J. Am. Chem. Soc. 2002, 124, 4552.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XisVeksrg%3D&md5=82cf81a2497ec6f2a7b0e6e03eac0ab1CAS |
[72] D. A. Evans, J. T. Starr, Angew. Chem. 2002, 114, 1865.
| Crossref | GoogleScholarGoogle Scholar |
[73] N. Tanaka, T. Suzuki, T. Matsumura, Y. Hosoya, M. Nakada, Angew. Chem. Int. Ed. 2009, 48, 2580.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXktlOrtbc%3D&md5=586a7d3b84412b77ab5d188efeac326aCAS |
[74] S. E. Denmark, R. Y. Baiazitov, S. T. Nguyen, Tetrahedron 2009, 65, 6535.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXoslGltr8%3D&md5=3dbc61cc99e3717021d14e0b2570784cCAS |
[75] D. M. Hodgson, A. H. Labande, F. Y. T. M. Pierard, M. Á. Expósito Castro, J. Org. Chem. 2003, 68, 6153.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXltV2isrg%3D&md5=28acb2207cb80ebc870faf816c5b1643CAS |
[76] (a) A. Padwa, S. P. Carter, H. Nimmesgern, J. Org. Chem. 1986, 51, 1157.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL28XitVOisrw%3D&md5=d2eed99cbbbb9232a334dc3dcce6fd26CAS |
(b) A. Padwa, Chem. Soc. Rev. 2009, 38, 3072.
| Crossref | GoogleScholarGoogle Scholar |
(c) A. Padwa, Tetrahedron 2011, 67, 8057.
| Crossref | GoogleScholarGoogle Scholar |
[77] Carbenoids have also found use in different domino polycyclisation strategies, such as that recently employed to synthesise (±)-minfiensine and (±)-vincorine. M. Zhang, X. Huang, L. Shen, Y. Qin, J. Am. Chem. Soc. 2009, 131, 6013.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXjslelsLY%3D&md5=316039baf2b12beeee2fe03cdf8a2c6fCAS |
[78] Y. Sasaki, D. Kato, D. L. Boger, J. Am. Chem. Soc. 2010, 132, 13533.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtV2kur3M&md5=13891d25f685a7d488677a48479d2d20CAS |
[79] E. L. Campbell, A. M. Zuhl, C. M. Liu, D. L. Boger, J. Am. Chem. Soc. 2010, 132, 3009.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhvFKisrc%3D&md5=ef06ad4d0800b85b254a596296f76198CAS |
[80] C. H. Heathcock, M. M. Hansen, R. B. Ruggeri, J. C. Kath, J. Org. Chem. 1992, 57, 2544.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38Xkt1Krt78%3D&md5=e2cb0f5fc5a640ead459da18d23f2ff5CAS |
[81] C. H. Heathcock, Proc. Natl. Acad. Sci. USA 1996, 93, 14323.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK28XnsVKlu7s%3D&md5=86622e62fb894384982f252794cb0372CAS |
[82] F. He, Y. Bo, J. D. Altom, E. J. Corey, J. Am. Chem. Soc. 1999, 121, 6771.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXktVahsro%3D&md5=973cc2f96f6a2ee073981252659bdd5eCAS |
[83] J. M. Mejia-Oneto, A. Padwa, Org. Lett. 2006, 8, 3275.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XmtVShsb0%3D&md5=c5a9f9ce9007a9163da5889b0ffcdf37CAS |
[84] D. F. Fischer, R. Sarpong, J. Am. Chem. Soc. 2010, 132, 5926.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXksF2lu7s%3D&md5=32ba39dbf23e3f8b039a80a0d2b80b08CAS |
[85] C. L. Martin, L. E. Overman, J. M. Rohde, J. Am. Chem. Soc. 2010, 132, 4894.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXjtVyjsrk%3D&md5=c6a4385a10d26cbac8a84ec901ce51eaCAS |
[86] T. Hudlicky, J. W. Reed, The Way of Synthesis, 1st ed. 2007 (Wiley-VCH: Weinheim).
[87] S. P. Lathrop, T. Rovis, J. Am. Chem. Soc. 2009, 131, 13628.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtV2nsLzM&md5=a4f0c0a051aaa0ab660ef2287870ee35CAS |
[88] For a classification and review of these types of transformation, see: D. E. Fogg, E. N. dos Santos, Coord. Chem. Rev. 2004, 248, 2365.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXhtVagsLzK&md5=eae8d52a3c25dd70872320301554bbabCAS |
[89] K. C. Nicolaou, Y. H. Lim, J. Becker, Angew. Chem. Int. Ed. 2009, 48, 3444.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXltFGhtro%3D&md5=1baf1f26ad920ec99dc20b25e5a3451bCAS |
[90] Y. Zeng, J. Aubé, J. Am. Chem. Soc. 2005, 127, 15712.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXhtFCltLbI&md5=f5d32ac49ed6db8ccb390e13b8cc671cCAS |
[91] G. Majetich, Y. Zhang, X. Tian, J. E. Britton, Y. Li, R. Phillips, Tetrahedron 2011, 67, 10129.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsFertbjI&md5=8381cd5c351f7ad9039b1c46e9c0e08fCAS |
[92] R. M. A. Lavigne, M. Riou, M. L. Girardin, L. Morency, L. Barriault, Org. Lett. 2005, 7, 5921.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXht1Gisr%2FF&md5=983b0f9a30d6f18e49c21c05e4628c66CAS |
[93] S. B. Jones, B. Simmons, A. Mastracchio, D. W. C. MacMillan, Nature 2011, 475, 183.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXovFehu74%3D&md5=e2ca333dec5611a2a0b01c5269b1be09CAS |
[94] O. L. Chapman, M. R. Engel, J. P. Springer, J. C. Clardy, J. Am. Chem. Soc. 1971, 93, 6696.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE38XisFWnsw%3D%3D&md5=0aaac19f54da6cbfcd24e7482ad5fabaCAS |
[95] (a) Rugulosin: K. C. Nicolaou, Y. H. Lim, C. D. Papageorgiou, J. L. Piper, Angew. Chem. 2005, 117, 8131.
| Crossref | GoogleScholarGoogle Scholar |
(b) Thiostrepton: K. C. Nicolaou, M. Zak, B. S. Safina, A. A. Estrada, S. H. Lee, M. Nevalainen, J. Am. Chem. Soc. 2005, 127, 11176.
| Crossref | GoogleScholarGoogle Scholar |
(c) K. C. Nicolaou, B. S. Safina, M. Zak, S. H. Lee, M. Nevalainen, M. Bella, A. A. Estrada, C. Funke, F. J. Zécri, S. Bulat, J. Am. Chem. Soc. 2005, 127, 11159.
| Crossref | GoogleScholarGoogle Scholar |
[96] P. D. Brown, A. C. Willis, M. S. Sherburn, A. L. Lawrence, Org. Lett. 2012, 14, 4537.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xht1yhsLnM&md5=062b1a2049048798d4a7caccf67b700dCAS |
[97] W. Liu, V. Khedkar, B. Baskar, M. Schuermann, K. Kumar, Angew. Chem. Int. Ed. 2011, 50, 6900.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXnsF2ksrY%3D&md5=67afdfe2bb2251be3ea837bf3c777f84CAS |
[98] (a) C. de Graaff, E. Ruijter, R. V. A. Orru, Chem. Soc. Rev. 2012, 41, 3969.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xmt1yksLk%3D&md5=bf09083a85ce7563ed3182742f8d9dbcCAS |
(b) S. S. van Berkel, B. G. M. Bogels, M. A. Wijdeven, B. Westermann, F. P. J. T. Rutjes, Eur. J. Org. Chem. 2012, 3543.
| Crossref | GoogleScholarGoogle Scholar |
(c) E. Ruijter, R. Scheffelaar, R. V. A. Orru, Angew. Chem. Int. Ed. 2011, 50, 6234.
| Crossref | GoogleScholarGoogle Scholar |
(d) D. Enders, C. Grondal, M. R. M. Huettl, Angew. Chem. Int. Ed. 2007, 46, 1570.
| Crossref | GoogleScholarGoogle Scholar |
(e) D. J. Ramon, M. Yus, Angew. Chem. Int. Ed. 2005, 44, 1602.
| Crossref | GoogleScholarGoogle Scholar |
[99] A. Strecker, Justus Liebigs Ann. Chem. 1850, 75, 27.
| Crossref | GoogleScholarGoogle Scholar |
[100] A. Hantzsch, Ber. Dtsch. Chem. Ges. 1881, 14, 1637.
| Crossref | GoogleScholarGoogle Scholar |
[101] P. Biginelli, Ber. Dtsch. Chem. Ges. 1891, 24, 1317.
| Crossref | GoogleScholarGoogle Scholar |
[102] A. Endo, A. Yanagisawa, M. Abe, S. Tohma, T. Kan, T. Fukuyama, J. Am. Chem. Soc. 2002, 124, 6552.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38Xjs1aksLY%3D&md5=848d05cb97b96707689582b0265fb9f3CAS |
[103] A. Dömling, Chem. Rev. 2006, 106, 17.
| Crossref | GoogleScholarGoogle Scholar |
[104] (a) Combining MCRs with other processes leads to some even more effective multi-bond forming processes. Some recent notable examples include: A. Znabet, J. Zonneveld, E. Janssen, F. J. J. De Kanter, M. Helliwell, N. J. Turner, E. Ruijter, R. V. A. Orru, Chem. Commun. 2010, 46, 7706.
| 1:CAS:528:DC%2BC3cXht1Omtr%2FK&md5=bb120488cd20a7935dbc0ab8652f49efCAS |
(b) N. Elders, D. van der Born, L. J. D. Hendrickx, B. J. J. Timmer, A. Krause, E. Janssen, F. J. J. de Kanter, E. Ruijter, R. V. A. Orru, Angew. Chem. Int. Ed. 2009, 48, 5856.
(c) H. Mizoguchi, H. Oguri, K. Tsuge, H. Oikawa, Org. Lett. 2009, 11, 3016.
(d) N. Kumagai, G. Muncipinto, S. L. Schreiber, Angew. Chem. Int. Ed. 2006, 45, 3635.
(e) Synthesis using more than one MCR: O. Pando, S. Stark, A. Denkert, A. Porzel, R. Preusentanz, L. A. Wessjohann, J. Am. Chem. Soc. 2011, 133, 7692.
| Crossref | GoogleScholarGoogle Scholar |
[105] (a) For recent reviews on organocatalytic multi-bond forming processes, see: C. M. Marson, Chem. Soc. Rev. 2012, 41, 7712.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xhs1ahtbrK&md5=548ddfbc81612d5ef561a12a22515c92CAS |
(b) C. Grondal, M. Jeanty, D. Enders, Nat. Chem. 2010, 2, 167.
| Crossref | GoogleScholarGoogle Scholar |
(c) H. Pellissier, Adv. Synth. Catal. 2012, 354, 237.
| Crossref | GoogleScholarGoogle Scholar |
[106] D. Enders, M. R. M. Huttl, C. Grondal, G. Raabe, Nature 2006, 441, 861.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XlvVGltrg%3D&md5=a9f2f3becaddbb486c16d813a4fc0584CAS |
[107] (a) For reviews on asymmetric multicatalytic processes, see: J. Zhou, Chem. Asian J. 2010, 5, 422.
| 1:CAS:528:DC%2BC3cXktVSmsb8%3D&md5=e0b219e8c3b5e704b174ff93f340ae2fCAS |
(b) R. C. Wende, P. R. Schreiner, Green Chem. 2012, 14, 1821.
(c) See also ref [88]
(d) For an interesting recent development in multifunctional catalyst design, see: Y. Chi, S. T. Scroggins, J. M. J. Fréchet, J. Am. Chem. Soc. 2008, 130, 6322.
| Crossref | GoogleScholarGoogle Scholar |
[108] D. Enders, M. R. M. Hüttl, G. Raabe, J. W. Bats, Adv. Synth. Catal. 2008, 350, 267.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXlt1Wit7w%3D&md5=b2103cefb8172b6c7673415445964c64CAS |
[109] (a) For some interesting novel examples featuring dipolar cycloadditions, see: P. Garner, H. U. Kaniskan, C. M. Keyari, L. Weerasinghe, J. Org. Chem. 2011, 76, 5283.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmvFChs78%3D&md5=e5e1f01a5922acb6c612590582c229c7CAS |
(b) P. Garner, L. Weerasinghe, W. J. Youngs, B. Wright, D. Wilson, D. Jacobs, Org. Lett. 2012, 14, 1326.
| Crossref | GoogleScholarGoogle Scholar |
[110] For a combination of metal cycloisomerisation and a Diels–Alder reaction, see: C. F. Bender, F. K. Yoshimoto, C. L. Paradise, J. K. D. Brabander, J. Am. Chem. Soc. 2009, 131, 11350.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXovFKltrY%3D&md5=25296557377d995ece7f01bec5c0d07eCAS |
[111] E. Knoevenagel, Ber. Dtsch. Chem. Ges. 1898, 31, 2596.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaD28XpsF2isw%3D%3D&md5=152d8a692304de05b647f6d0c9e107a7CAS |
[112] (a) For examples, see (and references therein): L. F. Tietze, Y. Zhou, Angew. Chem. Int. Ed. 1999, 38, 2045.
| 1:CAS:528:DyaK1MXkslSht7c%3D&md5=ffadd7965ee323e4d4492a0f413cb93aCAS |
(b) L. F. Tietze, N. Böhnke, S. Dietz, Org. Lett. 2009, 11, 2948.
(c) A. L. Lawrence, R. M. Adlington, J. E. Baldwin, V. Lee, J. A. Kershaw, A. L. Thompson, Org. Lett. 2010, 12, 1676.
(d) Y. Gao, G.-Q. Wang, K. Wei, P. Hai, F. Wang, J.-K. Liu, Org. Lett. 2012, 14, 5936.
(e) L. F. Tietze, J. Heterocycl. Chem. 1990, 27, 47.
[113] P. R. Nandaluru, G. J. Bodwell, Org. Lett. 2012, 14, 310.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhs1aqu7bI&md5=4c65e027d4e0ef64e5cd57ae298013d7CAS |
[114] C. A. Carson, M. A. Kerr, Angew. Chem. Int. Ed. 2006, 45, 6560.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhtFWgu7fL&md5=5d70a2905357f960c9e1dd96bc171ed4CAS |
[115] I. S. Young, M. A. Kerr, J. Am. Chem. Soc. 2007, 129, 1465.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXlt1Cqtg%3D%3D&md5=d23920b2d486bde5d0f99da26cb1af90CAS |
[116] P. Jakubec, A. Hawkins, W. Felzmann, D. J. Dixon, J. Am. Chem. Soc. 2012, 134, 17482.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhsVKmtbrK&md5=73f0ec05b039f6533d969cf592d9bc06CAS |
[117] For an excellent review, see: B. B. Touré, D. G. Hall, Chem. Rev. 2009, 109, 4439.
| Crossref | GoogleScholarGoogle Scholar |
[118] (a) Recent discoveries and new applications are paving the way for future work. For some recent targeted syntheses involving a key MCR, see: T. Ritthiwigrom, S. G. Pyne, Org. Lett. 2008, 10, 2769.
| 1:CAS:528:DC%2BD1cXms1GisLw%3D&md5=88036d24db2f4112aaa4f70a3c136f74CAS |
(b) T. Ritthiwigrom, A. C. Willis, S. G. Pyne, J. Org. Chem. 2009, 75, 815.
(c) L. V. Frolova, N. M. Evdokimov, K. Hayden, I. Malik, S. Rogelj, A. Kornienko, I. V. Magedov, Org. Lett. 2011, 13, 1118.
[119] Y. Inoue, K. Ohuchi, I. F. Yen, S. Imaizumi, Bull. Chem. Soc. Jpn. 1989, 62, 3518.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3cXhsFOqtrY%3D&md5=6d02d0891f8d1363ab8a8d864611b877CAS |
[120] K. C. Nicolaou, D. Sarlah, D. M. Shaw, Angew. Chem. Int. Ed. 2007, 46, 4708.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXntlOluro%3D&md5=260dd1e9a26874adadf6c36f680aae76CAS |
[121] For a review on other MCRs involving CO, see: M. D. Mihovilovic, P. Stanetty, Angew. Chem. Int. Ed. 2007, 46, 3612.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXmtVSqu7c%3D&md5=34b4d4bcb2c0d8c6c2fa5cf8fc0f21aeCAS |
[122] (a) T. Tanino, S. Ichikawa, M. Shiro, A. Matsuda, J. Org. Chem. 2010, 75, 1366.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhslShsrs%3D&md5=023e8fde1564a0ed30e5130c3ba9907eCAS |
(b) W. Wang, S. Joyner, K. A. S. Khoury, A. Doemling, Org. Biomol. Chem. 2010, 8, 529.
| Crossref | GoogleScholarGoogle Scholar |
(c) E. Avilés, A. D. Rodriguez, Org. Lett. 2010, 12, 5290.
| Crossref | GoogleScholarGoogle Scholar |
(d) J. Isaacson, Y. Kobayashi, Angew. Chem. Int. Ed. 2009, 48, 1845.
| Crossref | GoogleScholarGoogle Scholar |
(e) S. Takiguchi, T. Iizuka, Y.-s. Kumakura, K. Murasaki, N. Ban, K. Higuchi, T. Kawasaki, J. Org. Chem. 2010, 75, 1126.
| Crossref | GoogleScholarGoogle Scholar |
(f) T. Iizuka, S. Takiguchi, Y.-s. Kumakura, N. Tsukioka, K. Higuchi, T. Kawasaki, Tetrahedron Lett. 2010, 51, 6003.
| Crossref | GoogleScholarGoogle Scholar |
[123] A. Znabet, M. M. Polak, E. Janssen, F. J. J. de Kanter, N. J. Turner, R. V. A. Orru, E. Ruijter, Chem. Commun. 2010, 46, 7918.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtlantrvL&md5=61facfc1055bfd2bf76c38144838bb90CAS |
[124] (a) Y. Yip, F. Victor, J. Lamar, R. Johnson, Q. M. Wang, D. Barket, J. Glass, L. Jin, L. Liu, D. Venable, M. Wakulchik, C. Xie, B. Heinz, E. Villarreal, J. Colacino, N. Yumibe, M. Tebbe, J. Munroe, S.-H. Chen, Bioorg. Med. Chem. Lett. 2004, 14, 251.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXpvVWnurk%3D&md5=80fea0cc59a2ea64a3557a41776666b8CAS |
(b) J. A. Monn, M. J. Valli, J. Org. Chem. 1994, 59, 2773.
| Crossref | GoogleScholarGoogle Scholar |
[125] (a) For some recent impressive ‘one-pot’ examples, see: N. Chernyak, V. Gevorgyan, Angew. Chem. Int. Ed. 2010, 49, 2743.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXktFGrsrs%3D&md5=c56d066716f72fce1b46ee5f85bbfd6eCAS |
(b) B.-C. Hong, P. Kotame, C.-W. Tsai, J.-H. Liao, Org. Lett. 2010, 12, 776.
| Crossref | GoogleScholarGoogle Scholar |
(c) T. Sakaguchi, S. Kobayashi, S. Katsumura, Org. Biomol. Chem. 2011, 9, 257.
| Crossref | GoogleScholarGoogle Scholar |
(d) For a recent application of the ‘one-pot’ strategy applied in industry, see: F. Xu, M. Zacuto, N. Yoshikawa, R. Desmond, S. Hoerrner, T. Itoh, M. Journet, G. R. Humphrey, C. Cowden, N. Strotman, P. Devine, J. Org. Chem. 2010, 75, 7829.
| Crossref | GoogleScholarGoogle Scholar |
[126] H. Ishikawa, M. Honma, Y. Hayashi, Angew. Chem. Int. Ed. 2011, 50, 2824.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXivVGhtLs%3D&md5=4cf81ba07f093be1ea8e38810bea1d24CAS |
[127] H. Ishikawa, T. Suzuki, Y. Hayashi, Angew. Chem. Int. Ed. 2009, 48, 1304.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXitFymsbc%3D&md5=0d9ff9c726269ae5e23490b399664c11CAS |
[128] B. Simmons, A. M. Walji, D. W. C. MacMillan, Angew. Chem. Int. Ed. 2009, 48, 4349.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXms12isbg%3D&md5=1b7683101fa47ca2c4795cee8895d3ebCAS |
[129] For another recent example of the combination of organocatalysis and metal catalysis, see: W. Sun, G. Zhu, L. Hong, R. Wang, Chem.–Eur. J. 2011, 17, 13958.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsVyru7%2FI&md5=4d9e686254d6609202871e0f502dfdabCAS |
[130] H. Dückert, V. Pries, V. Khedkar, S. Menninger, H. Bruss, A. W. Bird, Z. Maliga, A. Brockmeyer, P. Janning, A. Hyman, S. Grimme, M. Schuermann, H. Preut, K. Huebel, S. Ziegler, K. Kumar, H. Waldmann, Nat. Chem. Biol. 2012, 8, 179.
| Crossref | GoogleScholarGoogle Scholar |
[131] (a) For some recent examples of the use of two-directional synthesis of symmetrical targets, see: R. Barth, J. Mulzer, Angew. Chem. 2007, 119, 5893.
| Crossref | GoogleScholarGoogle Scholar |
(b) K. M. G. O’Connell, H. S. G. Beckmann, L. Laraia, H. T. Horsley, A. Bender, A. R. Venkitaraman, D. R. Spring, Org. Biomol. Chem. 2012, 10, 7545.
| Crossref | GoogleScholarGoogle Scholar |
(c) K. C. Nicolaou, S. Totokotsopoulos, D. Giguere, Y.-P. Sun, D. Sarlah, J. Am. Chem. Soc. 2011, 133, 8150.
| Crossref | GoogleScholarGoogle Scholar |
(d) Y. Morimoto, M. Takaishi, N. Adachi, T. Okita, H. Yata, Org. Biomol. Chem. 2006, 4, 3220.
| Crossref | GoogleScholarGoogle Scholar |
[132] (a) Terminus differentiation techniques have been neatly classified by Schreiber: C. S. Poss, S. L. Schreiber, Acc. Chem. Res. 1994, 27, 9.
| 1:CAS:528:DyaK2cXns1KgtQ%3D%3D&md5=8a7047582a7a3c7059e8204dfa0638ccCAS |
(b) For a review, see: S. R. Magnuson, Tetrahedron 1995, 51, 2167.
| Crossref | GoogleScholarGoogle Scholar |
[133] N. Ikemoto, S. L. Schreiber, J. Am. Chem. Soc. 1992, 114, 2524.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38XhsFahtbs%3D&md5=ef9bfe3de5e9648598b6c1cfdc2a4a77CAS |
[134] C. S. Poss, S. D. Rychnovsky, S. L. Schreiber, J. Am. Chem. Soc. 1993, 115, 3360.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3sXkt1Ortbg%3D&md5=c3fe03377238f69b6ba82276f06c1d93CAS |
[135] Y. Zhang, C. C. Arpin, A. J. Cullen, M. J. Mitton-Fry, T. Sammakia, J. Org. Chem. 2011, 76, 7641.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtFOqu7%2FJ&md5=577f9c420131cc2eb35eb88f9c910537CAS |
[136] Y. Wang, O. Kataeva, P. Metz, Adv. Synth. Catal. 2009, 351, 2075.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtF2hu7fM&md5=31faadfe543276da0b4ff4d651d82f96CAS |
[137] E. W. Rogers, T. F. Molinski, J. Org. Chem. 2009, 74, 7660.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtFWqt7rK&md5=3d74ec7962862bed71757f624f3a7fc6CAS |
[138] N. R. Norcross, J. P. Melbardis, M. F. Solera, M. A. Sephton, C. Kilner, L. N. Zakharov, P. C. Astles, S. L. Warriner, P. R. Blakemore, J. Org. Chem. 2008, 73, 7939.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtFeqs7rF&md5=b3c0cd51577e2a94f02f7c45c785524eCAS |
[139] J. M. Holland, M. Lewis, A. Nelson, J. Org. Chem. 2003, 68, 747.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XpvVantLk%3D&md5=05dee97027d4c69cc1eb0125950ec34eCAS |
[140] C. J. Exner, M. Turks, F. Fonquerne, P. Vogel, Chem. – Eur. J. 2011, 17, 4246.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjvVShsbo%3D&md5=c699e7342c1d7289ab5b43058143ad74CAS |
[141] (a) D. Robbins, A. F. Newton, C. Gignoux, J.-C. Legeay, A. Sinclair, M. Rejzek, C. A. Laxon, S. K. Yalamanchili, W. Lewis, M. A. O’Connell, R. A. Stockman, Chem. Sci. 2011, 2, 2232.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXht12isrrP&md5=efef1d47a4a2a1085286921a5912ff04CAS |
(b) A. F. Newton, S. J. Roe, J.-C. Legeay, P. Aggarwal, C. Gignoux, N. J. Birch, R. Nixon, M.-L. Alcaraz, R. A. Stockman, Org. Biomol. Chem. 2009, 7, 2274.
| Crossref | GoogleScholarGoogle Scholar |
(c) P. Aggarwal, G. Procopiou, D. Robbins, G. Harbottle, W. Lewis, R. A. Stockman, Synlett 2012, 423.
[142] C. Gignoux, A. F. Newton, A. Barthelme, W. Lewis, M.-L. Alcaraz, R. A. Stockman, Org. Biomol. Chem. 2012, 10, 67.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsFKmtbjF&md5=a1e352186eda1994b240848e631c3e83CAS |
[143] D. Liu, H. P. Acharya, M. Yu, J. Wang, V. S. C. Yeh, S. Kang, C. Chiruta, S. M. Jachak, D. L. J. Clive, J. Org. Chem. 2009, 74, 7417.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtFWqurnP&md5=4c23ec7e61cf30651cfadc9e87f695f7CAS |
[144] B. B. Liau, B. C. Milgram, M. D. Shair, J. Am. Chem. Soc. 2012, 134, 16765.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xhtlekt7jJ&md5=1ebe727d9d7f8b345a03e8dd84d8550aCAS |
[145] For another recent example of two-fold reaction sequences on pseudo-symmetrical substrates, see: E. Airiau, T. Spangenberg, N. Girard, B. Breit, A. Mann, Org. Lett. 2010, 12, 528.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhs1WjsLfO&md5=69ec30e6ae0adae274341820b88112dcCAS |
[146] J. A. Berson, Tetrahedron 1992, 48, 3.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38XhtVGiurg%3D&md5=a94d8733e81344b26fccc1c3feb116b5CAS |
[147] M. Potowski, M. Schurmann, H. Preut, A. P. Antonchick, H. Waldmann, Nat. Chem. Biol. 2012, 8, 428.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XktVaksrY%3D&md5=926917bd8023964f54ad820bcfedea84CAS |
[148] A. H. Hoveyda, A. R. Zhugralin, Nature 2007, 450, 243.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXht1yntrvN&md5=1e795bfcf02807427c711e020e349276CAS |
[149] J. S. Clark, M. C. Kimber, J. Robertson, C. S. P. McErlean, C. Wilson, Angew. Chem. Int. Ed. 2005, 44, 6157.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXhtFWisLbP&md5=1c49ddf727c839928307aa7bfa1a0ec3CAS |
[150] J. S. Clark, O. Hamelin, Angew. Chem. Int. Ed. 2000, 39, 372.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXns1GhsA%3D%3D&md5=4cbe5c0e5a65e0a7017463fddf116a1cCAS |
[151] Y. Zhang, J. D. Rainier, Org. Lett. 2009, 11, 237.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhsVCgu73F&md5=177d0c90bcb2c55cc4866c78752146e0CAS |
[152] G. Matsuo, K. Kawamura, N. Hori, H. Matsukura, T. Nakata, J. Am. Chem. Soc. 2004, 126, 14374.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXos1Wju7o%3D&md5=ba59e4622d49408603413faa637fe8a8CAS |
[153] C. Baylon, M.-P. Heck, C. Mioskowski, J. Org. Chem. 1999, 64, 3354.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXitFSgsrk%3D&md5=7f600995e20286e6d2bb589b2871eb6fCAS |
[154] K. C. Nicolaou, D. G. McGarry, P. K. Sommers, J. Am. Chem. Soc. 1990, 112, 3696.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3cXitlGjsbY%3D&md5=e5e10b7d20b06ebf6f487ec09a3d651dCAS |
[155] (a) J. L. Ravelo, A. Regueiro, J. D. Martin, Tetrahedron Lett. 1992, 33, 3389.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38XltFWgtL0%3D&md5=2ff332de9b8f4e43a0341af23c3b14e2CAS |
(b) For a recent example from the same laboratory, see: B. Cheng, J. D. Sunderhaus, S. F. Martin, Org. Lett. 2010, 12, 3622.
| Crossref | GoogleScholarGoogle Scholar |
[156] K. S. Feldman, B. K. Selfridge, Org. Lett. 2012, 14, 5484.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhsFamtrjK&md5=d107161c778a2a614d468ff94e4ef18aCAS |
[157] M. W. B. Pfeiffer, A. J. Phillips, J. Am. Chem. Soc. 2005, 127, 5334.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXis1ens7g%3D&md5=ddd061885729ac09eae16b663749d992CAS |
[158] D. Morton, S. Leach, C. Cordier, S. Warriner, A. Nelson, Angew. Chem. Int. Ed. 2009, 48, 104.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXosFGmsQ%3D%3D&md5=56891319323fb1dcecfaf8b93de4eb42CAS |
[159] (a) S.-H. Kim, N. Bowden, R. H. Grubbs, J. Am. Chem. Soc. 1994, 116, 10801.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXhslyisbg%3D&md5=e2834f261abb723d0c95f7a2a533ef24CAS |
(b) S.-H. Kim, W. J. Zuercher, N. B. Bowden, R. H. Grubbs, J. Org. Chem. 1996, 61, 1073.
| Crossref | GoogleScholarGoogle Scholar |
[160] W. J. Zuercher, M. Scholl, R. H. Grubbs, J. Org. Chem. 1998, 63, 4291.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXjslKntrs%3D&md5=50d1e4cb1968adc5caceb119600676c1CAS |
[161] (a) For reviews, see: S. T. Diver, A. J. Giessert, Chem. Rev. 2004, 104, 1317.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXhvFGgs7g%3D&md5=0a686e925c5a6700a3ae3274b1ec0f3eCAS |
(b) M. Mori, Adv. Synth. Catal. 2007, 349, 121.
| Crossref | GoogleScholarGoogle Scholar |
(c) J. Li, D. Lee, Eur. J. Org. Chem. 2011, 4269.
| Crossref | GoogleScholarGoogle Scholar |
[162] L. Chang, H. Jiang, J. Fu, B. Liu, C.-c. Li, Z. Yang, J. Org. Chem. 2012, 77, 3609.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xjs1Oit78%3D&md5=3e47192005533773d6911350e3c5c8e6CAS |
[163] Y. Adachi, N. Kamei, S. Yokoshima, T. Fukuyama, Org. Lett. 2011, 13, 4446.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXpsFSrtrc%3D&md5=bdcf50ce95755a2e53478954db1646c9CAS |
[164] M. Schubert, P. Metz, Angew. Chem. Int. Ed. 2011, 50, 2954.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjt1yqu7s%3D&md5=aa57e242dba9f6a50e264e252268598fCAS |
[165] H. Oguri, T. Hiruma, Y. Yamagishi, H. Oikawa, A. Ishiyama, K. Otoguro, H. Yamada, S. Ōmura, J. Am. Chem. Soc. 2011, 133, 7096.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjsVGjsL8%3D&md5=4d839334ec35326873939833b25ea4bcCAS |
[166] D. S. Siegel, G. Piizzi, G. Piersanti, M. Movassaghi, J. Org. Chem. 2009, 74, 9292.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVOhtr%2FE&md5=4f535f0b4dbfaa92e5bd94bb9b98a761CAS |
[167] J. Ramharter, H. Weinstabl, J. Mulzer, J. Am. Chem. Soc. 2010, 132, 14338.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXht1WmtrjN&md5=58e6cda9f9de8c5d6c12fdd0ca52e9ccCAS |
[168] S. Mukherjee, D. Lee, Org. Lett. 2009, 11, 2916.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXntVSku7k%3D&md5=7a26e80198acd6fcc01b96d74cac6808CAS |
[169] (a) Metathesis also features in some impressive, enantioselective domino sequences, for reviews see: S. Kress, S. Blechert, Chem. Soc. Rev. 2012, 41, 4389.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XnsF2qs7g%3D&md5=ac6c9ab3c851459e3c93250a8e10846fCAS |
(b) A. H. Hoveyda, S. J. Malcolmson, S. J. Meek, A. R. Zhugralin, Angew. Chem. Int. Ed. 2010, 49, 34.
| Crossref | GoogleScholarGoogle Scholar |
[170] (a) D. Banti, M. North, Tetrahedron Lett. 2002, 43, 1561.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38Xpsl2quw%3D%3D&md5=a503156701c06cabb986aa78daadd17eCAS |
(b) D. Banti, M. North, Adv. Synth. Catal. 2002, 344, 694.
| Crossref | GoogleScholarGoogle Scholar |
(c) For an even more recent example of the combined metathesis-Diels–Alder approach, see: A. Aljarilla, M. C. Murcia, A. G. Csákÿ, J. Plumet, Eur. J. Org. Chem. 2009, 822.
| Crossref | GoogleScholarGoogle Scholar |
[171] J. A. Enquist, B. M. Stoltz, Nature 2008, 453, 1228.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXnslGksb8%3D&md5=570de207e9950ed42d1626cdb95c63a3CAS |
[172] (a) J. Tsuji, I. Minami, I. Shimizu, Tetrahedron Lett. 1983, 24, 1793.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL3sXltVCrsb4%3D&md5=7501876f9d316b0f2969cb14a75f8e98CAS |
(b) B. M. Trost, T. J. Fullerton, J. Am. Chem. Soc. 1973, 95, 292.
| Crossref | GoogleScholarGoogle Scholar |
[173] J. A. Enquist, S. C. Virgil, B. M. Stoltz, Chem. – Eur. J. 2011, 17, 9957.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXptVehtbY%3D&md5=9363a1ef2bfa7c8727aef486aae7afa8CAS |
[174] The syntheses of dendrimers provide examples of high order multi-directional synthesis. For a review, see: T. Iizuka, S. Takiguchi, Y.-s. Kumakura, N. Tsukioka, K. Higuchi, T. Kawasaki, Tetrahedron Lett. 2010, 51, 6003.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtlSmt7jM&md5=aea67c42032f3a48823ec2ad75f3c6c9CAS |
[175] H. Hopf, M. S. Sherburn, Angew. Chem. Int. Ed. 2012, 51, 2298.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xitlygsr0%3D&md5=d66e3c468bf87f476b994cc12933fed1CAS |
[176] G. Bojase, T. V. Nguyen, A. D. Payne, A. C. Willis, M. S. Sherburn, Chem. Sci. 2011, 2, 229.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmsVOjtg%3D%3D&md5=d584ea413b808284b521567ee0e66a4eCAS |
[177] G. A. Crispino, P. T. Ho, K. B. Sharpless, Science 1993, 259, 64.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3sXitFansbg%3D&md5=466da22396e56ff6b047b4b03fcbe3c3CAS |
[178] (a) From an achiral starting material, an enantiomeric ratio of approximately 105 : 1 is generated by virtue of an effect known as the Horeau principle. See W. Langenbeck, G. Triem, Z. Physik. Chem. 1936, A177, 401.
| 1:CAS:528:DyaA2sXhslyhuw%3D%3D&md5=f2a6c138793fb37060b9c901fa20e26eCAS |
(b) J. P. Vigneron, M. Dhaenens, A. Horeau, Tetrahedron 1973, 29, 1055.
| Crossref | GoogleScholarGoogle Scholar |
(c) V. Rautenstrauch, Bull. Soc. Chim. Fr. 1994, 131, 515.
(d) D. S. Glueck, Catal. Sci. Technol. 2011, 1, 1099.
| Crossref | GoogleScholarGoogle Scholar |
[179] For a recent example of this effect in independent reactions, see: S. Bell, B. Wüstenberg, S. Kaiser, F. Menges, T. Netscher, A. Pfaltz, Science 2006, 311, 642.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XptVyksg%3D%3D&md5=527005615a185a4c907605415dc71e64CAS |
[180] Z. Xiong, E. J. Corey, J. Am. Chem. Soc. 2000, 122, 9328.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXmt1WitLo%3D&md5=ee44c76765d9e741173fa04cf5006686CAS |
[181] (a) I. Paterson, E. A. Anderson, S. M. Dalby, J. H. Lim, P. Maltas, C. Moessner, Chem. Commun. 2006, 4186.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhtVCru7bP&md5=6d3348ac7210e25f8b3620ea0d9e583cCAS |
(b) A second-generation synthesis has recently been completed utilising this methodology: I. Paterson, P. Maltas, S. M. Dalby, J. H. Lim, E. A. Anderson, Angew. Chem. Int. Ed. 2012, 51, 2749.
| Crossref | GoogleScholarGoogle Scholar |
[182] C. H. Heathcock, Angew. Chem. Int. Ed. Engl. 1992, 31, 665.
| Crossref | GoogleScholarGoogle Scholar |