

Comparison of microbial diversity in rumen and small intestine of Xinong Saanen dairy goats using 16S rRNA gene high-throughput sequencing
Cong Li A , Yanan Geng A , Ping Wang A , Huaiping Shi A and Jun Luo A B
A , Yanan Geng A , Ping Wang A , Huaiping Shi A and Jun Luo A B
A Key Laboratory of Animal Genetics, Breeding and Reproduction of Shaanxi Province, College of Animal Science and Technology, Northwest A&F University, No. 22 Xinong Road, Yangling, Shaanxi 712100, China.
B Corresponding author. Email: luojun@nwsuaf.edu.cn
Animal Production Science - https://doi.org/10.1071/AN20459
Submitted: 13 October 2020 Accepted: 12 July 2021 Published online: 17 August 2021
Journal Compilation © CSIRO 2021 Open Access CC BY
Abstract
Context: Gastrointestinal microorganisms play an important role in ruminant digestion and metabolism, immune regulation and disease prevention and control. Different parts of the digestive tract have different functions and microbial community structures.
Aims: This study aims to explore the microbial diversity in the rumen and the small intestine of Xinong Saanen dairy goats.
Methods: Rumen fluid and jejunum fluid from three Xinong Saanen dairy bucks with the average slaughter weight of 33.93 ± 0.68 kg were collected and analysed for microbial diversity, by using 16S rRNA gene high-throughput sequencing.
Key results: In total, 1118 operational taxonomic units (OTUs) were identified, with 1020 OTUs and 649 OTUs being clustered to rumen and jejunum samples respectively. Alpha-diversity indices were significantly (P < 0.05) different between rumen and jejunum, as indicated by the fact that the rumen microbial community diversity, richness and uniformity/evenness were higher than those of jejunum. At the phylum level, the dominant phyla in the rumen were Bacteroidetes (66.7%) and Firmicutes (25.1%), accounting for 91.8% of the rumen microorganisms. The dominant phylum in the jejunum was Firmicutes, accounting for 73.0% of the jejunum microorganisms. At the genus level, the dominant bacteria in the rumen were Prevotella_1, norank_f_Bacteroidales_BS11_gut_group, Rikenellaceae_RC9_gut_group, Christensenellaceae_R-7_group and Family_XIII_AD3011_group, whereas the dominant bacteria in the jejunum were Omboutsia, Aeriscardovia, Intestinibacter, unclassified_f_Peptostreptococcaceae and unclassified_f_Bifidobacteriaceae. Clusters of Orthologous Groups (COG) and Kyoto Encyclopedia of Genes and Genomes (KEGG) results showed that the major functions of microorganisms in the rumen and jejunum were carbohydrate metabolism, amino acid metabolism, nucleotide metabolism, membrane transport and translation. Interestingly, fructose and mannose metabolism and peptidoglycan biosynthesis were abundant in the rumen, while homologous recombination and nucleotide excision repair were abundant in the jejunum.
Conclusions: Our study clarified the differences in microbial diversity and community structure between the rumen and the jejunum in Xinong Saanen dairy goats. Prevotella was the most predominant genus in the rumen, compared with Romboutsia, Bifidobacterium as well as Peptostreptococcaceae genera, which were the predominant genera in the jejunum.
Implications: In combination with the functional prediction of microorganisms and the metabolic characteristics of different parts of the digestive tract in ruminants, our findings provided information for further exploring the relationship among genes, species and functions of microorganisms and their hosts’ nutritional and physiological functions.
Keywords: 16S rRNA gene high-throughput sequencing, dairy goats, functional prediction, microbial diversity.
Introduction
The rumen is the largest digestive organ of ruminants. In total, 70–80% of the digestible dry matter (DM) and 50% of the crude fibre in the feed are degraded in the rumen (Yang et al. 2019). Part of nutrients are absorbed by the rumen wall, whereas the remaining nutrients are absorbed in the jejunum and other sites along the digestive tract. Gastrointestinal microorganisms are the generic terms for all microorganisms that inhabit the digestive tract, including bacteria, fungi, protozoa and archaea. The microbial genome is rich in genes involved in the metabolism of carbohydrates, amino acids, methane, vitamin and short-chain fatty acids, many of which are not available in the animal body itself. In addition, the microorganisms per se and their metabolites have unique benefits to the host by participating in nutrient digestion and absorption, immune regulation and disease control and prevention (Carberry et al. 2014; Weimer 2015).
Recently, the importance of gastrointestinal microorganisms has attracted increased interest in ruminant nutrition research. Culture and non-culture methods of phosphorus lipid fatty acid spectrum analysis and polymerase chain reaction–denaturing gradient gel electrophoresis (PCR–DGGE) were common techniques generally used to explore the microbial diversity in the past decades. However, these techniques have several common problems, such as, complicated operational procedures, being tedious, having a high cost, and difficulty in discovering trace bacterium (Combes et al. 2011). With the rapid development of high-throughput sequencing technology, 16S rDNA sequencing has been developed to an important method to explore microbial diversity due to its advantages on sequencing depth, high-throughput, reliability and cost effectiveness (Wang et al. 2014). Eckburg et al. (2005) first detected the gastrointestinal microbial diversity through 16S rRNA gene high-throughput sequencing, which broke the technique limitations in rumen microbiology isolation and culture and provided more comprehensive and accurate data for individual microbial-diversity research. The rumen microbiota of sheep analysed by 16S rRNA gene high-throughput sequencing showed that the sequence similarities between most of Prevotella species (87.8%) in the rumen with the known Prevotella species were less than 97.0%, indicating that most of the Prevotella species were non-cultivable (Bekele et al. 2010). Shanks et al. (2011) found that both feeding conditions and dietary conditions affected the microbial species and community structure in the intestinal tract, by using 16S rRNA gene high-throughput sequencing. Durso et al. (2011) reported that antibiotic resistance genes and bacterial toxin genes were mainly concentrated in the Actinomyces and Proteobacteria, by analysing the microorganisms in faeces samples of beef cattle.
As a cultivated breed, Xinong Saanen dairy goat has been strictly selected and cultivated through a long period, based on the Saanen dairy goat from abroad. Due to its superior production performance, this breed is widely promoted in China. Currently, over 50% of the total dairy goats raised in China have the bloodline of Xinong Saanen dairy goats. However, studies on the gastrointestinal microbial diversity of Xinong Saanen dairy goats are very limited. Therefore, in the current study, we aimed to explore the differences in microbial species and community structures between rumen and jejunum in Xinong Saanen dairy goats through 16S rRNA gene high-throughput sequencing technology. The findings will be helpful for elucidating the relationships on nutritional and physiological functions between gastrointestinal microorganisms and their hosts in ruminants.
Materials and methods
Ethics statement
All protocols for tissue sample collection of experimental individuals were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC, Permit number 15 516) at Northwest A&F University.
Experimental animals and design
In total, six Xinong Saanen dairy bucks with an average weight of 22.43 ± 0.75 kg at 6 months of age were considered as the study population in the present study. All dairy goats were selected from Breeding Station of Xinong Saanen Dairy Goat at Northwest A&F University (34°16′N, 108°4′27.95′′E), Shaanxi province, China. Two goats were group-housed in a yard and were fed by total mixed ration twice per day at 0800 hours and 1600 hours. The experiment lasted 12 weeks, and the composition and nutrient concentrations of basal diets were shown in Table 1.

|
Sample collection
At end of the 12th week, three goats were randomly selected and slaughtered, with the average slaughter weight of 33.93 ± 0.68 kg. The digestive tracts of the selected goats after slaughter were stripped, and the contents of the rumen and jejunum were collected. All contents were filtered through four layers of sterile gauze, and then, the filtrates were centrifuged at 1788.8g at 4°C. Finally, 5 mL rumen fluid and 5 mL jejunum fluid were collected and rapidly frozen in liquid nitrogen and stored at –80°C for further analysis.
Extraction of bacterial genomic DNA and PCR amplification
The bacterial genomic DNA was extracted from rumen fluid and jejunum fluid by TIANamp Stool DNA Kit (TianGen, Beijing, China), according to the manufacturer’s instructions. The quantity and quality of extracted genomic DNA were determined by NanoDropTM ND-2000c spectrophotometer (Thermo Scientific, Wilmington, USA) and 1% agarose gel electrophoresis, and the DNA was then stored at –20°C for future use.
The V3–V4 hypervariable regions of bacterial 16S rRNA gene were selected as the target fragments for PCR amplification. The primer sequences of 338F (5′-ACTCCTACGGGAGGCAGCAG-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′) were applied to amplify bacterial genomic DNA. PCR amplifications were performed in a total volume of 20 μL. The following reagents were used for amplification: 10 ng of template DNA, 0.8 μL of each primer (5 μM), 2 μL of 2.5 mM dNTPs, 4 μL of 5 × FastPfu buffer and 0.4 μL of FastPfu polymerase. The PCR procedures included a pre-denaturation at 95°C for 2 min, followed by 30 cycles of 30 s at 95°C, 30 s at 55°C for annealing, 30 s at 72°C for elongation, and a 5-min final extension at 72°C. PCR products were measured by 2% agarose gel electrophoresis (Fig. S1, available as Supplementary material to this paper) and were purified with the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA) and quantified using QuantiFluorTM-ST (Promega, USA), according to the manufacturer’s protocol.
Library construction and 16S rRNA gene sequencing
Sequencing libraries were constructed using TruSeq TM DNA Sample Prep Kit (Illumina, San Diego, USA), library quality was measured using an Agilent 4200 Bioanalyzer (Agilent Technologies, China), and library concentration was detected using a Qubit 4.0 Fluorometer (Thermofisher, USA). In addition, adapters with indices were added to the end of the PCR products for high-throughput sequencing. The Illumina MiSeq platform (Illumina) was applied and was conducted by Majorbio Bio-Pharm Technology Co. Ltd (Shanghai, China).
Processing of sequencing data and bioinformatic analysis
Raw fastq files were quality-filtered by Trimmomatic and merged by FLASH with the following criteria: (i) the reads were truncated at any site receiving an average quality score of <20 over a 50 bp sliding window; (ii) sequences with an overlap longer than 10 bp were merged according to their overlap, with mismatch no more than 2 bp; and (iii) sequences of each sample were separated according to barcodes (exactly matching) and primers (allowing 2 nucleotide mismatching), and reads containing ambiguous bases were removed (Caporaso et al. 2010).
Operational taxonomic units (OTUs) were clustered with 97% similarity cut-off by using UPARSE (ver. 7.1, http://drive5.com/uparse/; Ye et al. 2016), with a novel ‘greedy’ algorithm that performs chimera filtering and OTU clustering simultaneously. The taxonomy of each 16S rRNA gene sequence was analysed by RDP classifier algorithm (http://rdp.cme.msu.edu/; Wang et al. 2007) against the Silva (SSU123) 16S rRNA database (release 128, http://www.arb-silva.de; DeSantis et al. 2006), using confidence threshold of 70%. The alpha diversity of microorganisms was analysed using Mothur platform (ver. 1.30.2, https://www.mothur.org/wiki/Download_mothur), including Chao and Ace indices reflecting community richness, Shannon and Simpson indices reflecting community diversity, as well as Shannoneven index reflecting community evenness of microorganisms (Schloss et al. 2009). Unweighted UniFrac metric-based principal coordinate analysis was performed to compare sample distances between two groups (Lozupone and Knight 2005). For functional prediction, PICRUSt (http://picrust.github.io/picrust/) and Tax4Fun (http://tax4fun.gobics.d.e) were applied to compare with Clusters of Orthologous Groups (COG) and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases respectively (Langille et al. 2013).
Statistical analyses
Statistical analysis was performed through SPSS 23.0 software (SPSS Inc., Chicago, IL, USA). The Kruskal–Wallis test was applied to assess the significant differences in the microbial phyla and genera between the two groups. Statistical significances were declared as significant at P < 0.05 and highly significant at P < 0.01. The results are presented as means with standard errors of the mean.
Results
Analysis of sequencing depth, OTU quantity and rarefaction curve
In total, 200 514 sequences were annotated through annotating at different taxonomic levels according to the clustering of 97% similarity OTUs, which belonged to 19 phyla, 226 genera and 1118 OTUs. As shown in Table S1 (available as Supplementary material to this paper), the sequencing coverage of all samples using 16S rRNA gene high-throughput sequencing reached >99% in Xinong Saanen dairy goats. In total, 160 136 valid sequences were obtained from rumen samples, with an average of 53 379 sequences per sample and an average sequence length of 440 bp. These valid sequences were mapped to 1020 OTUs in total, of which 18 OTUs had the sequence abundances >1% and OTU779 reached the highest sequence abundance of 4.5%. For jejunum samples, 134 127 valid sequences were obtained, with an average of 44 709 sequences per sample and an average sequence length of 428 bp. In total, 649 OTUs were identified; of these, 12 OTUs had sequence abundances of >1% and OTU15 had the highest sequence abundance of 27.9%.
The rarefaction curves of rumen and jejunum in dairy goats approaching a plateau indicated that the sampling could cover the majority of the bacterial diversity (Fig. S2, available as Supplementary material to this paper). When the sequencing depth was less than 10 000 reads, the Sobs and Shannon indices in the rumen and jejunum were markedly increased as the sequencing depth progressed. However, when the sequencing depth was greater than 10 000 reads, the Sobs and Shannon indices in the rumen and jejunum had a tendency to stabilisation. Importantly, the indices of Sobs and Shannon in the rumen were always greater than those in the jejunum, indicating a greater species diversity in the rumen than in the jejunum. In addition, low requirements of sequencing depth were observed for Shannon index, namely, the accurate and stable values of Shannon index can be obtained through a low sequencing depth (Fig. S2b), whereas the value of Sobs index was gently increased as the sequencing depth increased (Fig. S2a).
Alpha diversity analysis of microbial communities
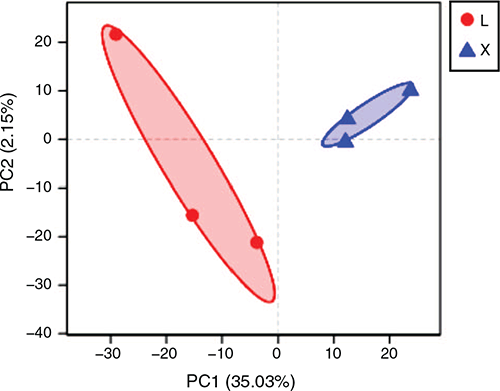
Alpha diversity analysis reflects the abundance and diversity of microbial communities. The principal-coordinate analysis showed that the rumen samples were separated from the jejunum samples (Fig. 1). The Chao and Ace indices reflect microbial community richness, the Shannon index reflects microbial community diversity, while the Shannoneven index reflects microbial community evenness. All of these indices were significantly (P < 0.05) higher in the rumen than in the jejunum (Table 2). The Simpson index reflecting microbial community diversity was pronouncedly lower in the rumen than in the jejunum (P < 0.05; Table 2). The noticeably higher richness, diversity and evenness were observed in the rumen than in the jejunum.
|

|
Microbial community structure analysis based on phylum level
Analysis of microbial community structure in the rumen and jejunum at the phylum level showed that the microorganisms in both rumen and jejunum were annotated to 17 phyla (Fig. 2a). Among these, 15 phyla were found both in the rumen and jejunum, while two phyla were unique in that their relative abundances were less than 0.01%. The subsequent analyses were performed to target the microbial species with relative abundances greater than 1% in the rumen and jejunum (Fig. 2b). In total, five phyla with relative abundances greater than 1% were identified in the rumen, and were listed as the relative abundances from high to low, including Bacteroidetes, Firmicutes, Fibrobacteres, Spirochaetes and Actinobacteria. Among these, Bacteroidetes and Firmicutes accounted for 66.7% and 25.1% respectively, of the rumen microorganisms. For jejunum, three phyla with relative abundances greater than 1% were found, and were listed from high to low relative abundances as follows: Firmicutes, Actinobacteria and Tenericutes. Among these, Firmicutes accounted for 73.0% of the jejunum microorganisms.

|
Microbial community structure analysis based on genus level
At the genus level, the microorganisms in the rumen were annotated to 176 genera, including 35 unique genera, while 191 genera, containing 50 unique genera, were annotated in the jejunum (Fig. 3a). In total, 141 genera were found both in the rumen and the jejunum. Analysis of microbial species with relative abundances >1% in the rumen and jejunum showed that 20 genera in total were identified in the rumen (Fig. 3b). Of these, the top five genera with high relative abundances accounted for 56.7% of the rumen microorganisms, including Prevotella_1, norank_f_Bacteroidales_BS11_gut_group, Rikenellaceae_RC9_gut_group, Christensenellaceae_R-7_group and Family_XIII_AD3011_group. In addition, 21.4% of unidentified microorganisms were observed in the rumen. In the jejunum, 17 genera with relative abundances greater than 1% were found. Of these, Romboutsia, Aeriscardovia, Intestinibacter, unclassified_f_Peptostreptococcaceae and unclassified_f_Bifidobacteriaceae were the top five genera with high relative abundances, accounting for 59.6% of the jejunum microorganisms. The unidentified microorganisms account for 16.4% of the jejunum microorganisms.

|
Functional prediction by COG database
Comparison with the standardised OTUs corresponding to the greengenes and COG database, the top five enriched COG terms with high abundances in the rumen were amino acid transport and metabolism, cell wall/membrane/envelope biogenesis, carbohydrate transport and metabolism, translation, ribosomal structure and biogenesis as well as chromatin structure and dynamics (Fig. 4). In jejunum, the top five enriched COG terms with high abundances were amino acid transport and metabolism, transcription, carbohydrate transport and metabolism, replication, recombination and repair as well as general function prediction only (Fig. 4).

|
Functional prediction by KEGG database
Multiple KEGG pathways were dramatically enriched for standardised OTUs corresponding to the green genes. At the first level, six categories of KEGG pathways (relative abundances in rumen and jejunum) were enriched in the rumen and jejunum respectively, including metabolism (63.7%, 53.5%), genetic information processing (16.8%, 23.2%), environmental information processing (12.3%, 17.5%), cellular processes (3.8%, 2.7%), human diseases (1.8%, 1.8%), organismal systems (1.6%, 1.2%) (Fig. 5). Among these, the highest-percentage category was the metabolism pathways.

|
At the second level, 41 categories of KEGG pathways, in total, were enriched in the rumen and jejunum (Fig. 5). The top five enriched KEGG categories with high abundances in the rumen were carbohydrate metabolism (17.1%), amino acid metabolism (10.6%), metabolism of cofactors and vitamin (7.4%), nucleotide metabolism (7.3%) as well as membrane transport (7.1%). However, the top five enriched KEGG categories in jejunum with high abundances were membrane transport (14.3%), carbohydrate metabolism (13.1%), translation (11.1%), amino acid metabolism (9.1%) as well as nucleotide metabolism (8.3%).
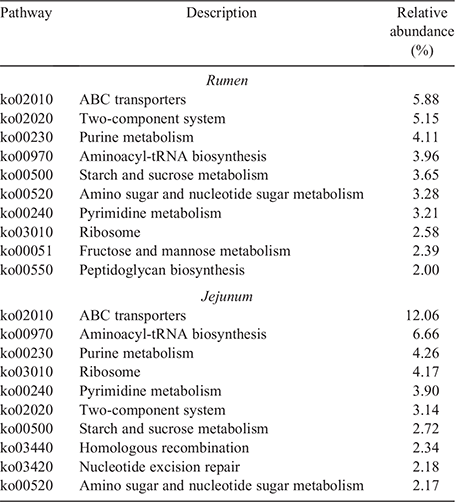
In total, 272 KEGG pathways were enriched in the rumen and jejunum at the third level. The top 10 enriched KEGG pathways with high abundances in the rumen and jejunum are presented in Table 3. Of these, eight pathways were found both in the rumen and jejunum, including amino sugar and nucleotide sugar metabolism, starch and sucrose metabolism, aminoacyl-tRNA biosynthesis, pyrimidine metabolism, purine metabolism, ribosome, ABC transporters as well as a two-component system. Two unique KEGG pathways, including fructose and mannose metabolism as well as peptidoglycan biosynthesis were found in the rumen. Nucleotide excision repair and homologous recombination were the two unique KEGG pathways observed in the jejunum.
|
Discussion
Microbial diversity in the rumen and jejunum of Xinong Saanen dairy goats
The amount of 16S rRNA sequencing data is affected by the sequencing technology and the sample size. Previous studies in Holstein, Jersey cow, Brown Swiss dairy cows have shown that the accurate and reliable results can be obtained if the sequencing depth is greater than 10 000 reads for each sample (Myer et al. 2015; Paz et al. 2016; Zubiria et al. 2019). In this study, the number of reads in individual samples of rumen and jejunum greater than 40 000, and the flattened rarefaction curves indicated that we obtained sufficient sequencing amounts. The greater number of valid sequences and OTU representative sequences in the rumen than in the jejunum suggested that the microbial abundances in the rumen were higher than in the jejunum. Alpha diversity indices reflected the richness, evenness, diversity and coverage of the microbial communities. The richness and diversity of microbial communities in the rumen were lower than those in the caecum in bovine through alpha diversity-index analysis (Myer et al. 2015). The richness and diversity of microbial communities evaluated in yak have also shown that the abundances observed in the ileum were higher than those in the duodenum, jejunum and caecum (Zhang et al. 2020). In the present study, the Chao and Ace indices reflecting richness, the Shannon and Simpson indices reflecting diversity as well as the Shannoneven index reflecting evenness of microbial communities were evaluated to compare the differences in microbial diversity and communities between the rumen and jejunum in dairy goats. The larger index values of Chao, Ace and Shannon, and the smaller index values of Simpson indicated that the more abundant and diverse species were observed in the samples (Qian et al. 2015). Our results showed that all alpha diversity indices were significantly different between rumen and jejunum, and higher diversity, richness as well as evenness of microbial communities were observed in the rumen than in the jejunum in dairy goats. This response was consistent with the reports on the higher microbial diversity associated with the digestion and utilisation of cellulose in the rumen (Patra and Yu 2015).
Microbial community structure in the rumen and jejunum of Xinong Saanen dairy goats
As the dominant flora in the gastrointestinal tracts of mammals, Firmicutes and Bacteroides are helpful for animals to digest plant-derived feed (Ley et al. 2008; Singh et al. 2012; de Oliveira et al. 2013). Bacteroidetes species have function in promoting the utilisation of carbohydrates in gastrointestinal tracts (Spence et al. 2006), especially as the main degraders of non-fibrous carbohydrates (Russell and Diez-Gonzalez 1997). Firmicutes species play vital roles in the degradation of fibres and cellulose (Brulc et al. 2009; Thoetkiattikul et al. 2013). Thus, high concentrations of Firmicutes and Bacteroides are the typical characteristics of gastrointestinal microbial communities in herbivorous animals (Yang et al. 2019). Consistently, our findings in Xinong Saanen dairy goats also confirmed that Firmicutes and Bacteroides account for 91.8% of bacterium and are the dominant flora in the rumen. Our results are also in accordance with those of previous studies (Brulc et al. 2009; Kim et al. 2011; Guo et al. 2020), where Firmicutes and Bacteroides were dominant flora in the rumen. Additionally, our findings showed that Firmicutes, Actinomycetes and Tenericutes were identified as the dominant flora/phyla in the jejunum, of which Firmicutes accounted for 73.0% of jejunal microorganisms. Such responses are in agreement with earlier observations in goats (Mao et al. 2013) and cattle (Shanks et al. 2011). Meanwhile, other research (Di Rienzi et al. 2013; Wang et al. 2017; Li et al. 2019) has shown that Proteobacteria and Cyanophyta are highly abundant in the small intestine of ruminants, which may be caused by different colonisation of microorganisms in the jejunum of different ruminants. Our results are the first to illuminate the differences in microbial community structures between the rumen and jejunum in Xinong Saanen dairy goats.
Prevotella is the most dominant genus of rumen bacterium in ruminants (Bekele et al. 2010). The main function of Prevotella is to degrade non-fibre components in the feed. As the main proteolytic bacteria in the rumen, Prevotella species also produce a large number of complex enzymes to accelerate the degradation of starch (Avgustin et al. 1997; Purushe et al. 2010). Similar to previous research in goats (Wang et al. 2018; Li et al. 2019), the findings in the present study showed that Prevotella, Bacteroidales, Rikenellaceae and Christensenellaceae were the predominant genera in the rumen of Xinong Saanen dairy goats. Rikenellaceae was also reported to be involved in degrading structural carbohydrates (Zened et al. 2013).
The predominant genera in the jejunum of mammals are Lactobacillus and Bifidobacterium (Mitsuoka 1996), which exhibit a variety of beneficial impacts on the host as probiotics (LeBlanc et al. 2013; Wang et al. 2013; Dylag et al. 2014). In addition, many indispensable nutrients, such as amino acids and vitamin B can be synthesised by Bifidobacterium (Swedlow and Danuser 2012). In the present study, Romboutsia, Bifidobacterium, Aeriscardovia, Intestinibacter, and an unclassified genus of Peptostreptococcaceae were identified as the predominant genera in the jejunum. Among these, the last three genera belonged to the family Peptostreptococcaceae. Our results are in close agreement with those of previous research (Zhang et al. 2020), where Romboutsia was the dominant genus and Peptostreptococcaceae was the dominant family in the jejunum of yak. Peptostreptococcaceae as be the dominant family was also consistent with observations in cattle (Rudi et al. 2012).
Functional prediction of microorganisms in the rumen and jejunum of Xinong Saanen dairy goats
The interaction of gastrointestinal microorganisms with their hosts plays a crucial role in regulating host metabolism, immune function and nutrition utilisation (Zhang et al. 2018). Previous studies have reported that microbial communities varied in different intestinal segments in camel (He et al. 2018), sheep (Zhang et al. 2018), chicken (Wen et al. 2019) and cashmere goat (Li et al. 2019). The study on yak and cashmere goat showed significant differences of microorganisms between the small intestine and the large intestine (Li et al. 2019; Zhang et al. 2020). Therefore, it is speculated that different microbial communities may be the reason for the functional differences in different digestive-tract parts. In the present study, functional prediction of microorganisms through COG and KEGG databases showed that pathways with the highest abundances are enriched in metabolism both in rumen and jejunum of Xinong Saanen dairy goats. It mainly included amino acid metabolism, nucleotide metabolism, carbohydrate metabolism, among others. In addition, the pathways of membrane transport and translation were also highly abundant. These results are in agreement with observations in India buffalo (Singh et al. 2014). All findings indicated that microorganisms in both rumen and jejunum have significant impacts on nutrient digestion and metabolism in ruminants. The nutrients in the feed were decomposed into nutrients directly used by ruminants, or converted into other easily digestible forms through microorganisms, and then absorbed and utilised in a transmembrane transport manner. For example, volatile fatty acids, such as acetic acid, propionic acid and butyric acid, were produced from the fibre and hemi-fibre in feed under the action of fibre hydrolases, which were transported by the monocarboxylic acid transporter and absorbed by the rumen wall to provide energy for the body (Moschen et al. 2012). In addition, the microorganisms had the function of decomposing the nitrogen-containing substances in the feed, and then converting them into microbial mycoprotein or amino acids, which were absorbed in the small intestine to improve the nitrogen-utilisation rate of ruminants.
Except for the common KEGG pathways shared by the rumen and jejunum, it is noteworthy that two unique KEGG pathways were identified in the rumen and jejunal fluid in Xinong Saanen dairy goats. The two KEGG pathways uniquely identified in the rumen were closely related to biosynthesis and metabolism of carbohydrate, including fructose and mannose metabolism and peptidoglycan biosynthesis. This phenomenon is probably related to the digestive characteristics of different parts of the digestive tract of ruminants. As is known, rumen is the main digestive organ of ruminants, in which a large number of microorganisms live in for fibre fermentation. Then, the stable pH environment and energy source are needed for this process in the rumen. As a soluble carbohydrate, fructose ferments faster in the rumen (Sniffen et al. 1992), produces more butyric acid and the hydrolysed propionic acids are absorbed faster, than for starch (DeFrain et al. 2004; Vallimont et al. 2004; Oba et al. 2010). Thus, fructose provides the energy for rapid propagation of fermentation of microorganisms in the rumen. In addition, partially replacing starch with soluble carbohydrate in ruminant diets, rumen pH would be increased to prevent rumen acidosis (Guo et al. 2013; Baurhoo and Mustafa 2014), and simultaneously, production performances would be improved, such as enhancing milk yields and milk fat percentage, improving the components of milk fatty acids (Martel et al. 2011; DeVries and Gill 2012). Mannose had a competitive inhibitory relationship with glucose in the process of cell transport (Thorens and Mueckler 2010), which interfered with the ability of bacteria to metabolise glucose and reduced the contents of lactic acids. It also plays an important role in maintaining a stable pH environment in the rumen. Peptidoglycan, as the main component of bacterial cell wall, was heavily synthesised in the rumen, which might be related to the grinding effects of rumen on feed. Moreover, peptidoglycan can absorb toxic substances in the environment due to its strong adhesion ability, which not only improves the antioxidant and anti-inflammatory capabilities of the cells (Murphey et al. 2008), but is also used as an activator of the immune system, with antitumour and immunomodulatory effects (He et al. 2017).
For jejunum, the two unique KEGG pathways were related to DNA repair, including homologous recombination and nucleotide excision repair. Nucleotide excision repair is responsible for repairing a variety of localised DNA damage, including from radiation, protein-DNA crosslinking or chemical drugs (Sakthivel and Hariharan 2017). Homologous recombination is the exchange of two homologous DNA molecules under the catalysis of recombinase. The bacterial genome even can be assembled through homologous recombination of long DNA fragments in microorganisms. It has been reported that the homologous recombination ability of Saccharomyces cerevisiae has been used to achieve the replacement of synthetic chromosome fragments with wild chromosome fragments (Mitchell et al. 2017; Richardson et al. 2017; Xie et al. 2017). Our findings showed a strong DNA repair function of the microorganisms in the jejunum, which was probably related to heavily damaged microbial DNA in the jejunum because of the low-pH environment of the jejunum. In addition, when the diet reached the jejunum through the mouth of the ruminant, DNA was damaged due to a large number of rumen microorganisms being exposed to sudden pH changes in the environment. The microorganisms without acid tolerance were thoroughly decomposed into nucleotides and then absorbed and re-used by other microorganisms or jejunum. Acid-resistant microorganisms colonised the jejunum made their functions in digesting nutrients.
Conclusions
In the present study, the differences of microbial diversity and community structure between the rumen and jejunum were evaluated to understand their different functions in digestion, absorption and utilisation of diets in dairy goats. Our findings showed that there was higher diversity, richness and evenness of microbial communities in the rumen than in the jejunum in Xinong Saanen dairy goats. The predominant phyla were Bacteroidetes and Firmicutes in the rumen, while Firmicutes was predominant in the jejunum. Prevotella was the most predominant genus in the rumen, compared with Romboutsia, Bifidobacterium as well as Peptostreptococcaceae genera, which were the predominant genera in the jejunum. The main functions of rumen and jejunum were enriched in carbohydrate metabolism, amino acid metabolism, nucleotide metabolism, membrane transport and translation. The uniquely identified pathways in the rumen were closely related with biosynthesis and metabolism of carbohydrates, while DNA repair pathways as uniquely identified in the jejunum showed the strong DNA repair functions of the microorganisms in the jejunum. Our findings have laid the foundation for exploring microorganisms of rumen and small intestine in ruminants, and also provided evidence for further elucidating the microbial functional genes closely related to some important nutritional and physiological functions.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgement
This work was jointly supported by the Key Research and Development Plan of Shaanxi Province, China (2018ZDCXL-NY-01-05), Agricultural Science and Technology Innovation Transformation Project of Shaanxi Province, China (NYKJ-2019-YL14) and the Innovation Support Plan of Shaanxi Province (2021TD-31).
References
Avgustin G, Wallace RJ, Flint HJ (1997) Phenotypic diversity among ruminal isolates of Prevotella ruminicola: proposal of Prevotella brevis sp. nov., Prevotella bryantii sp. nov., and Prevotella albensis sp. nov. and redefinition of Prevotella ruminicola. International Journal of Systematic Bacteriology 47, 284–288.| Phenotypic diversity among ruminal isolates of Prevotella ruminicola: proposal of Prevotella brevis sp. nov., Prevotella bryantii sp. nov., and Prevotella albensis sp. nov. and redefinition of Prevotella ruminicola.Crossref | GoogleScholarGoogle Scholar | 9103611PubMed |
Baurhoo B, Mustafa A (2014) Short communication: effects of molasses supplementation on performance of lactating cows fed high-alfalfa silage diets. Journal of Dairy Science 97, 1072–1076.
| Short communication: effects of molasses supplementation on performance of lactating cows fed high-alfalfa silage diets.Crossref | GoogleScholarGoogle Scholar | 24315324PubMed |
Bekele AZ, Koike S, Kobayashi Y (2010) Genetic diversity and diet specificity of ruminal Prevotella revealed by 16S rRNA gene-based analysis. FEMS Microbiology Letters 305, 49–57.
| Genetic diversity and diet specificity of ruminal Prevotella revealed by 16S rRNA gene-based analysis.Crossref | GoogleScholarGoogle Scholar | 20158525PubMed |
Brulc JM, Antonopoulos DA, Miller ME, Wilson MK, Yannarell AC, Dinsdale EA, Edwards RE, Frank ED, Emerson JB, Wacklin P, Coutinho PM, Henrissat B, Nelson KE, White BA (2009) Gene-centric metagenomics of the fiber-adherent bovine rumen microbiome reveals forage specific glycoside hydrolases. Proceedings of the National Academy of Sciences of the United States of America 106, 1948–1953.
| Gene-centric metagenomics of the fiber-adherent bovine rumen microbiome reveals forage specific glycoside hydrolases.Crossref | GoogleScholarGoogle Scholar | 19181843PubMed |
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R (2010) QIIME allows analysis of high-throughput community sequencing data. Nature Methods 7, 335–336.
| QIIME allows analysis of high-throughput community sequencing data.Crossref | GoogleScholarGoogle Scholar | 20383131PubMed |
Carberry CA, Kenny DA, Kelly AK, Waters SM (2014) Quantitative analysis of ruminal methanogenic microbial populations in beef cattle divergent in phenotypic residual feed intake (RFI) offered contrasting diets. Journal of Animal Science and Biotechnology 5, 41
| Quantitative analysis of ruminal methanogenic microbial populations in beef cattle divergent in phenotypic residual feed intake (RFI) offered contrasting diets.Crossref | GoogleScholarGoogle Scholar | 25276350PubMed |
Combes S, Michelland RJ, Monteils V, Cauquil L, Soulie V, Tran NU, Gidenne T, Fortun-Lamothe L (2011) Postnatal development of the rabbit caecal microbiota composition and activity. FEMS Microbiology Ecology 77, 680–689.
| Postnatal development of the rabbit caecal microbiota composition and activity.Crossref | GoogleScholarGoogle Scholar | 21658088PubMed |
de Oliveira MN, Jewell KA, Freitas FS, Benjamin LA, Totola MR, Borges AC, Moraes CA, Suen G (2013) Characterizing the microbiota across the gastrointestinal tract of a Brazilian Nelore steer. Veterinary Microbiology 164, 307–314.
| Characterizing the microbiota across the gastrointestinal tract of a Brazilian Nelore steer.Crossref | GoogleScholarGoogle Scholar | 23490556PubMed |
DeFrain JM, Hippen AR, Kalscheur KF, Jardon PW (2004) Feeding glycerol to transition dairy cows: effects on blood metabolites and lactation performance. Journal of Dairy Science 87, 4195–4206.
| Feeding glycerol to transition dairy cows: effects on blood metabolites and lactation performance.Crossref | GoogleScholarGoogle Scholar | 15545383PubMed |
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL (2006) Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Applied and Environmental Microbiology 72, 5069–5072.
| Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB.Crossref | GoogleScholarGoogle Scholar | 16820507PubMed |
DeVries TJ, Gill RM (2012) Adding liquid feed to a total mixed ration reduces feed sorting behavior and improves productivity of lactating dairy cows. Journal of Dairy Science 95, 2648–2655.
| Adding liquid feed to a total mixed ration reduces feed sorting behavior and improves productivity of lactating dairy cows.Crossref | GoogleScholarGoogle Scholar | 22541492PubMed |
Di Rienzi SC, Sharon I, Wrighton KC, Koren O, Hug LA, Thomas BC, Goodrich JK, Bell JT, Spector TD, Banfield JF, Ley RE (2013) The human gut and groundwater harbor non-photosynthetic bacteria belonging to a new candidate phylum sibling to Cyanobacteria. eLife 2, e01102
| The human gut and groundwater harbor non-photosynthetic bacteria belonging to a new candidate phylum sibling to Cyanobacteria.Crossref | GoogleScholarGoogle Scholar | 24137540PubMed |
Durso LM, Harhay GP, Bono JL, Smith TP (2011) Virulence-associated and antibiotic resistance genes of microbial populations in cattle feces analyzed using a metagenomic approach. Journal of Microbiological Methods 84, 278–282.
| Virulence-associated and antibiotic resistance genes of microbial populations in cattle feces analyzed using a metagenomic approach.Crossref | GoogleScholarGoogle Scholar | 21167876PubMed |
Dylag K, Hubalewska-Mazgaj M, Surmiak M, Szmyd J, Brzozowski T (2014) Probiotics in the mechanism of protection against gut inflammation and therapy of gastrointestinal disorders. Current Pharmaceutical Design 20, 1149–1155.
| Probiotics in the mechanism of protection against gut inflammation and therapy of gastrointestinal disorders.Crossref | GoogleScholarGoogle Scholar | 23755726PubMed |
Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA (2005) Diversity of the human intestinal microbial flora. Science 308, 1635–1638.
| Diversity of the human intestinal microbial flora.Crossref | GoogleScholarGoogle Scholar | 15831718PubMed |
Guo Y, Xu X, Zou Y, Yang Z, Li S, Cao Z (2013) Changes in feed intake, nutrient digestion, plasma metabolites, and oxidative stress parameters in dairy cows with subacute ruminal acidosis and its regulation with pelleted beet pulp. Journal of Animal Science and Biotechnology 4, 31
| Changes in feed intake, nutrient digestion, plasma metabolites, and oxidative stress parameters in dairy cows with subacute ruminal acidosis and its regulation with pelleted beet pulp.Crossref | GoogleScholarGoogle Scholar | 23947764PubMed |
Guo J, Li P, Liu S, Miao B, Zeng B, Jiang Y, Li L, Wang L, Chen Y, Zhang H (2020) Characterization of the Rumen Microbiota and Volatile Fatty Acid Profiles of Weaned Goat Kids under Shrub–Grassland Grazing and Indoor Feeding. Animals 10, 176
| Characterization of the Rumen Microbiota and Volatile Fatty Acid Profiles of Weaned Goat Kids under Shrub–Grassland Grazing and Indoor Feeding.Crossref | GoogleScholarGoogle Scholar |
He J, Wu Z, Pan D, Guo Y, Zeng X (2017) Effect of selenylation modification on antitumor activity of peptidoglycan from Lactobacillus acidophilus. Carbohydrate Polymers 165, 344–350.
| Effect of selenylation modification on antitumor activity of peptidoglycan from Lactobacillus acidophilus.Crossref | GoogleScholarGoogle Scholar | 28363558PubMed |
He J, Yi L, Hai L, Ming L, Gao W, Ji R (2018) Characterizing the bacterial microbiota in different gastrointestinal tract segments of the Bactrian camel. Scientific Reports 8, 654
| Characterizing the bacterial microbiota in different gastrointestinal tract segments of the Bactrian camel.Crossref | GoogleScholarGoogle Scholar | 29330494PubMed |
Kim M, Morrison M, Yu Z (2011) Status of the phylogenetic diversity census of ruminal microbiomes. FEMS Microbiology Ecology 76, 49–63.
| Status of the phylogenetic diversity census of ruminal microbiomes.Crossref | GoogleScholarGoogle Scholar | 21223325PubMed |
Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Thurber RLV, Knight R, Beiko RG, Huttenhower C (2013) Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nature Biotechnology 31, 814
| Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences.Crossref | GoogleScholarGoogle Scholar |
LeBlanc JG, Milani C, de Giori GS, Sesma F, van Sinderen D, Ventura M (2013) Bacteria as vitamin suppliers to their host: a gut microbiota perspective. Current Opinion in Biotechnology 24, 160–168.
| Bacteria as vitamin suppliers to their host: a gut microbiota perspective.Crossref | GoogleScholarGoogle Scholar | 22940212PubMed |
Ley RE, Lozupone CA, Hamady M, Knight R, Gordon JI (2008) Worlds within worlds: evolution of the vertebrate gut microbiota. Nature Reviews. Microbiology 6, 776–788.
| Worlds within worlds: evolution of the vertebrate gut microbiota.Crossref | GoogleScholarGoogle Scholar | 18794915PubMed |
Li B, Zhang K, Li C, Wang X, Chen Y, Yang Y (2019) Characterization and Comparison of Microbiota in the Gastrointestinal Tracts of the Goat (Capra hircus) During Preweaning Development. Frontiers in Microbiology 10, 2125
| Characterization and Comparison of Microbiota in the Gastrointestinal Tracts of the Goat (Capra hircus) During Preweaning Development.Crossref | GoogleScholarGoogle Scholar | 31572331PubMed |
Lozupone C, Knight R (2005) UniFrac: a new phylogenetic method for comparing microbial communities. Applied and Environmental Microbiology 71, 8228–8235.
| UniFrac: a new phylogenetic method for comparing microbial communities.Crossref | GoogleScholarGoogle Scholar | 16332807PubMed |
Mao S, Huo W, Zhu W (2013) Use of pyrosequencing to characterize the microbiota in the ileum of goats fed with increasing proportion of dietary grain. Current Microbiology 67, 341–350.
| Use of pyrosequencing to characterize the microbiota in the ileum of goats fed with increasing proportion of dietary grain.Crossref | GoogleScholarGoogle Scholar | 23636494PubMed |
Martel CA, Titgemeyer EC, Mamedova LK, Bradford BJ (2011) Dietary molasses increases ruminal pH and enhances ruminal biohydrogenation during milk fat depression. Journal of Dairy Science 94, 3995–4004.
| Dietary molasses increases ruminal pH and enhances ruminal biohydrogenation during milk fat depression.Crossref | GoogleScholarGoogle Scholar | 21787935PubMed |
Mitchell LA, Wang A, Stracquadanio G, Kuang Z, Wang X, Yang K, Richardson S, Martin JA, Zhao Y, Walker R, Luo Y, Dai H, Dong K, Tang Z, Yang Y, Cai Y, Heguy A, Ueberheide B, Fenyo D, Dai J, Bader JS, Boeke JD (2017) Synthesis, debugging, and effects of synthetic chromosome consolidation: synVI and beyond. Science 355, eaaf4831
Mitsuoka T (1996) Intestinal flora and human health. Asia Pacific Journal of Clinical Nutrition 5, 2–9.
Moschen I, Broer A, Galic S, Lang F, Broer S (2012) Significance of short chain fatty acid transport by members of the monocarboxylate transporter family (MCT). Neurochemical Research 37, 2562–2568.
| Significance of short chain fatty acid transport by members of the monocarboxylate transporter family (MCT).Crossref | GoogleScholarGoogle Scholar | 22878645PubMed |
Murphey ED, Fang G, Sherwood ER (2008) Pretreatment with the Gram-positive bacterial cell wall molecule peptidoglycan improves bacterial clearance and decreases inflammation and mortality in mice challenged with Staphylococcus aureus. Critical Care Medicine 36, 3067–3073.
| Pretreatment with the Gram-positive bacterial cell wall molecule peptidoglycan improves bacterial clearance and decreases inflammation and mortality in mice challenged with Staphylococcus aureus.Crossref | GoogleScholarGoogle Scholar | 18824898PubMed |
Myer PR, Wells JE, Smith TP, Kuehn LA, Freetly HC (2015) Cecum microbial communities from steers differing in feed efficiency. Journal of Animal Science 93, 5327–5340.
| Cecum microbial communities from steers differing in feed efficiency.Crossref | GoogleScholarGoogle Scholar | 26641052PubMed |
Oba M, Penner GB, Whyte TD, Wierenga K (2010) Effects of feeding triticale dried distillers grains plus solubles as a nitrogen source on productivity of lactating dairy cows. Journal of Dairy Science 93, 2044–2052.
| Effects of feeding triticale dried distillers grains plus solubles as a nitrogen source on productivity of lactating dairy cows.Crossref | GoogleScholarGoogle Scholar | 20412919PubMed |
Patra AK, Yu Z (2015) Essential oils affect populations of some rumen bacteria in vitro as revealed by microarray (RumenBactArray) analysis. Frontiers in Microbiology 6, 297
| Essential oils affect populations of some rumen bacteria in vitro as revealed by microarray (RumenBactArray) analysis.Crossref | GoogleScholarGoogle Scholar | 25914694PubMed |
Paz HA, Anderson CL, Muller MJ, Kononoff PJ, Fernando SC (2016) Rumen Bacterial Community Composition in Holstein and Jersey Cows Is Different under Same Dietary Condition and Is Not Affected by Sampling Method. Frontiers in Microbiology 7, 1206
| Rumen Bacterial Community Composition in Holstein and Jersey Cows Is Different under Same Dietary Condition and Is Not Affected by Sampling Method.Crossref | GoogleScholarGoogle Scholar | 27536291PubMed |
Purushe J, Fouts DE, Morrison M, White BA, Mackie RI, North American Consortium for Rumen Coutinho PM, Henrissat B, Nelson KE (2010) Comparative genome analysis of Prevotella ruminicola and Prevotella bryantii: insights into their environmental niche. Microbial Ecology 60, 721–729.
| Comparative genome analysis of Prevotella ruminicola and Prevotella bryantii: insights into their environmental niche.Crossref | GoogleScholarGoogle Scholar | 20585943PubMed |
Qian H, Lu H, Ding H, Lavoie M, Li Y, Liu W, Fu Z (2015) Analyzing Arabidopsis thaliana root proteome provides insights into the molecular bases of enantioselective imazethapyr toxicity. Scientific Reports 5, 11975
| Analyzing Arabidopsis thaliana root proteome provides insights into the molecular bases of enantioselective imazethapyr toxicity.Crossref | GoogleScholarGoogle Scholar | 26153126PubMed |
Richardson SM, Mitchell LA, Stracquadanio G, Yang K, Dymond JS, DiCarlo JE, Lee D, Huang CL, Chandrasegaran S, Cai Y, Boeke JD, Bader JS (2017) Design of a synthetic yeast genome. Science 355, 1040–1044.
| Design of a synthetic yeast genome.Crossref | GoogleScholarGoogle Scholar | 28280199PubMed |
Rudi K, Moen B, Sekelja M, Frisli T, Lee MR (2012) An eight-year investigation of bovine livestock fecal microbiota. Veterinary Microbiology 160, 369–377.
| An eight-year investigation of bovine livestock fecal microbiota.Crossref | GoogleScholarGoogle Scholar | 22749759PubMed |
Russell JB, Diez-Gonzalez F (1997) The effects of fermentation acids on bacterial growth. Advances in Microbial Physiology 39, 205–234.
| The effects of fermentation acids on bacterial growth.Crossref | GoogleScholarGoogle Scholar |
Sakthivel KM, Hariharan S (2017) Regulatory players of DNA damage repair mechanisms: role in Cancer Chemoresistance. Biomedicine and Pharmacotherapy 93, 1238–1245.
| Regulatory players of DNA damage repair mechanisms: role in Cancer Chemoresistance.Crossref | GoogleScholarGoogle Scholar | 28738540PubMed |
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and Environmental Microbiology 75, 7537–7541.
| Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities.Crossref | GoogleScholarGoogle Scholar | 19801464PubMed |
Shanks OC, Kelty CA, Archibeque S, Jenkins M, Newton RJ, McLellan SL, Huse SM, Sogin ML (2011) Community structures of fecal bacteria in cattle from different animal feeding operations. Applied and Environmental Microbiology 77, 2992–3001.
| Community structures of fecal bacteria in cattle from different animal feeding operations.Crossref | GoogleScholarGoogle Scholar | 21378055PubMed |
Singh KM, Ahir VB, Tripathi AK, Ramani UV, Sajnani M, Koringa PG, Jakhesara S, Pandya PR, Rank DN, Murty DS, Kothari RK, Joshi CG (2012) Metagenomic analysis of Surti buffalo (Bubalus bubalis) rumen: a preliminary study. Molecular Biology Reports 39, 4841–4848.
| Metagenomic analysis of Surti buffalo (Bubalus bubalis) rumen: a preliminary study.Crossref | GoogleScholarGoogle Scholar | 21947953PubMed |
Singh KM, Reddy B, Patel D, Patel AK, Parmar N, Patel A, Patel JB, Joshi CG (2014) High potential source for biomass degradation enzyme discovery and environmental aspects revealed through metagenomics of Indian buffalo rumen. BioMed Research International 2014, 267189
| High potential source for biomass degradation enzyme discovery and environmental aspects revealed through metagenomics of Indian buffalo rumen.Crossref | GoogleScholarGoogle Scholar | 25136572PubMed |
Sniffen CJ, O’Connor JD, Van Soest PJ, Fox DG, Russell JB (1992) A net carbohydrate and protein system for evaluating cattle diets: II. Carbohydrate and protein availability. Journal of Animal Science 70, 3562–3577.
| A net carbohydrate and protein system for evaluating cattle diets: II. Carbohydrate and protein availability.Crossref | GoogleScholarGoogle Scholar | 1459919PubMed |
Spence C, Wells WG, Smith CJ (2006) Characterization of the primary starch utilization operon in the obligate anaerobe Bacteroides fragilis: Regulation by carbon source and oxygen. Journal of Bacteriology 188, 4663–4672.
| Characterization of the primary starch utilization operon in the obligate anaerobe Bacteroides fragilis: Regulation by carbon source and oxygen.Crossref | GoogleScholarGoogle Scholar | 16788175PubMed |
Swedlow J, Danuser G (2012) Scale integration: the structure and dynamics of macromolecular assemblies, cells and tissues. Current Opinion in Cell Biology 24, 1–3.
| Scale integration: the structure and dynamics of macromolecular assemblies, cells and tissues.Crossref | GoogleScholarGoogle Scholar | 22342264PubMed |
Thoetkiattikul H, Mhuantong W, Laothanachareon T, Tangphatsornruang S, Pattarajinda V, Eurwilaichitr L, Champreda V (2013) Comparative analysis of microbial profiles in cow rumen fed with different dietary fiber by tagged 16S rRNA gene pyrosequencing. Current Microbiology 67, 130–137.
| Comparative analysis of microbial profiles in cow rumen fed with different dietary fiber by tagged 16S rRNA gene pyrosequencing.Crossref | GoogleScholarGoogle Scholar | 23471692PubMed |
Thorens B, Mueckler M (2010) Glucose transporters in the 21st Century. American Journal of Physiology. Endocrinology and Metabolism 298, E141–E145.
| Glucose transporters in the 21st Century.Crossref | GoogleScholarGoogle Scholar | 20009031PubMed |
Vallimont JE, Bargo F, Cassidy TW, Luchini ND, Broderick GA, Varga GA (2004) Effects of replacing dietary starch with sucrose on ruminal fermentation and nitrogen metabolism in continuous culture. Journal of Dairy Science 87, 4221–4229.
| Effects of replacing dietary starch with sucrose on ruminal fermentation and nitrogen metabolism in continuous culture.Crossref | GoogleScholarGoogle Scholar | 15545386PubMed |
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and Environmental Microbiology 73, 5261–5267.
| Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy.Crossref | GoogleScholarGoogle Scholar | 17586664PubMed |
Wang LQ, Zhao F, Liu F, Meng XC (2013) Live/dead state is not the factor influencing adhesion ability of Bifidobacterium animalis KLDS2.0603. Journal of Microbiology 51, 584–589.
| Live/dead state is not the factor influencing adhesion ability of Bifidobacterium animalis KLDS2.0603.Crossref | GoogleScholarGoogle Scholar |
Wang J, Wang F, Chu L, Wang H, Zhong Z, Liu Z, Gao J, Duan H (2014) High genetic diversity and novelty in eukaryotic plankton assemblages inhabiting saline lakes in the Qaidam basin. PLoS One 9, e112812
| High genetic diversity and novelty in eukaryotic plankton assemblages inhabiting saline lakes in the Qaidam basin.Crossref | GoogleScholarGoogle Scholar | 25549339PubMed |
Wang J, Fan H, Han Y, Zhao J, Zhou Z (2017) Characterization of the microbial communities along the gastrointestinal tract of sheep by 454 pyrosequencing analysis. Asian-Australasian Journal of Animal Sciences 30, 100–110.
| Characterization of the microbial communities along the gastrointestinal tract of sheep by 454 pyrosequencing analysis.Crossref | GoogleScholarGoogle Scholar | 27383798PubMed |
Wang Y, Zhang H, Zhu L, Xu Y, Liu N, Sun X, Hu L, Huang H, Wei K, Zhu R (2018) Dynamic Distribution of Gut Microbiota in Goats at Different Ages and Health States. Frontiers in Microbiology 9, 2509
| Dynamic Distribution of Gut Microbiota in Goats at Different Ages and Health States.Crossref | GoogleScholarGoogle Scholar | 30405569PubMed |
Weimer PJ (2015) Redundancy, resilience, and host specificity of the ruminal microbiota: implications for engineering improved ruminal fermentations. Frontiers in Microbiology 6, 296
| Redundancy, resilience, and host specificity of the ruminal microbiota: implications for engineering improved ruminal fermentations.Crossref | GoogleScholarGoogle Scholar | 25914693PubMed |
Wen C, Yan W, Sun C, Ji C, Zhou Q, Zhang D, Zheng J, Yang N (2019) The gut microbiota is largely independent of host genetics in regulating fat deposition in chickens. The ISME Journal 13, 1422–1436.
| The gut microbiota is largely independent of host genetics in regulating fat deposition in chickens.Crossref | GoogleScholarGoogle Scholar | 30728470PubMed |
Xie ZX, Li BZ, Mitchell LA, Wu Y, Qi X, Jin Z, Jia B, Wang X, Zeng BX, Liu HM, Wu XL, Feng Q, Zhang WZ, Liu W, Ding MZ, Li X, Zhao GR, Qiao JJ, Cheng JS, Zhao M, Kuang Z, Wang X, Martin JA, Stracquadanio G, Yang K, Bai X, Zhao J, Hu ML, Lin QH, Zhang WQ, Shen MH, Chen S, Su W, Wang EX, Guo R, Zhai F, Guo XJ, Du HX, Zhu JQ, Song TQ, Dai JJ, Li FF, Jiang GZ, Han SL, Liu SY, Yu ZC, Yang XN, Chen K, Hu C, Li DS, Jia N, Liu Y, Wang LT, Wang S, Wei XT, Fu MQ, Qu LM, Xin SY, Liu T, Tian KR, Li XN, Zhang JH, Song LX, Liu JG, Lv JF, Xu H, Tao R, Wang Y, Zhang TT, Deng YX, Wang YR, Li T, Ye GX, Xu XR, Xia ZB, Zhang W, Yang SL, Liu YL, Ding WQ, Liu ZN, Zhu JQ, Liu NZ, Walker R, Luo Y, Wang Y, Shen Y, Yang H, Cai Y, Ma PS, Zhang CT, Bader JS, Boeke JD, Yuan YJ (2017) ‘Perfect’ designer chromosome V and behavior of a ring derivative Science 355, eaaf4704
Yang Y, Yin Y, Chen X, Chen C, Xia Y, Qi H, Baker PN, Zhang H, Han TL (2019) Evaluating different extraction solvents for GC–MS based metabolomic analysis of the fecal metabolome of adult and baby giant pandas. Scientific Reports 9, 12017.
Ye H, Liu J, Feng P, Zhu W, Mao S (2016) Grain-rich diets altered the colonic fermentation and mucosa-associated bacterial communities and induced mucosal injuries in goats. Scientific Reports 6, 20329
| Grain-rich diets altered the colonic fermentation and mucosa-associated bacterial communities and induced mucosal injuries in goats.Crossref | GoogleScholarGoogle Scholar | 26841945PubMed |
Zened A, Combes S, Cauquil L, Mariette J, Klopp C, Bouchez O, Troegeler-Meynadier A, Enjalbert F (2013) Microbial ecology of the rumen evaluated by 454 GS FLX pyrosequencing is affected by starch and oil supplementation of diets. FEMS Microbiology Ecology 83, 504–514.
| Microbial ecology of the rumen evaluated by 454 GS FLX pyrosequencing is affected by starch and oil supplementation of diets.Crossref | GoogleScholarGoogle Scholar | 22974422PubMed |
Zhang H, Shao M, Huang H, Wang S, Ma L, Wang H, Hu L, Wei K, Zhu R (2018) The Dynamic Distribution of Small-Tail Han Sheep Microbiota across Different Intestinal Segments. Frontiers in Microbiology 9, 32
| The Dynamic Distribution of Small-Tail Han Sheep Microbiota across Different Intestinal Segments.Crossref | GoogleScholarGoogle Scholar | 29445360PubMed |
Zhang L, Jiang X, Li A, Waqas M, Gao X, Li K, Xie G, Zhang J, Mehmood K, Zhao S, Wangdui B, Li J (2020) Characterization of the microbial community structure in intestinal segments of yak (Bos grunniens). Anaerobe 61, 102115
| Characterization of the microbial community structure in intestinal segments of yak (Bos grunniens).Crossref | GoogleScholarGoogle Scholar | 31711887PubMed |
Zubiria I, Garcia-Rodriguez A, Atxaerandio R, Ruiz R, Benhissi H, Mandaluniz N, Lavin JL, Abecia L, Goiri I (2019) Effect of Feeding Cold-Pressed Sunflower Cake on Ruminal Fermentation, Lipid Metabolism and Bacterial Community in Dairy Cows. Animals 9, 755
| Effect of Feeding Cold-Pressed Sunflower Cake on Ruminal Fermentation, Lipid Metabolism and Bacterial Community in Dairy Cows.Crossref | GoogleScholarGoogle Scholar |