

Chicks and single-nucleotide polymorphisms: an entrée into identifying genes conferring disease resistance in chicken
Hans H. Cheng A B E , Sean MacEachern C , Sugalesini Subramaniam B and William M. Muir DA USDA, ARS, Avian Disease and Oncology Laboratory, 3606 East Mount Hope Road, East Lansing, MI 48823, USA.
B Comparative Medicine and Integrative Biology Graduate Program, College of Veterinary Medicine, Michigan State University, East Lansing, MI 48824, USA.
C Cobb-Vantress Inc., PO Box 1030, 4703 US Highway 412 East, Siloam Springs, AR 72761, USA.
D Department of Animal Sciences, 1151 Lilly Hall, Purdue University, West Lafayette, IN 47907, USA.
E Corresponding author. Email: hans.cheng@ars.usda.gov
Animal Production Science 52(3) 151-156 https://doi.org/10.1071/AN11099
Submitted: 3 June 2011 Accepted: 12 January 2012 Published: 13 February 2012
Journal Compilation © CSIRO Publishing 2012 Open Access CC BY-NC-ND
Abstract
Marek’s disease (MD) is one of the most serious chronic infectious disease threats to the poultry industry worldwide. Selecting for increased genetic resistance to MD is a control strategy that can augment current vaccinal control measures. Although our previous efforts integrating various genomic screens successfully identified three resistance genes, the main limitation was mapping precision, which hindered our ability to identify and further evaluate high-confidence candidate genes. Towards identifying the remaining genes of this complex trait, we incorporated three additional approaches made substantially more powerful through next-generation sequencing and that exploit the growing importance of expression variation. First, we screened for allele-specific expression (ASE) in response to Marek’s disease virus (MDV) infection, which, when allelic imbalance was identified, is sufficient to indicate a cis-acting element for a specific gene. Second, sequencing of genomic regions enriched by chromatin immunoprecipitation (ChIP) combined with transcript profiling identified motifs bound and genes directly regulated by MDV Meq, a bZIP transcription factor and the viral oncogene. Finally, analysis of genomic sequences from two experimental lines divergently selected for MD genetic resistance allowed inference about regions under selection as well as potential causative polymorphisms. These new combined approaches have resulted in a large number of high-confidence genes conferring MD resistance reflecting the multigenic basis of this trait, which expands our biological knowledge and provides corresponding single-nucleotide polymorhpisms (SNPs) that can be directly evaluated for their genetic contribution towards disease resistance.
Additional keywords: gene expression, genetic resistance, Marek’s disease, next-generation sequencing, poultry.
Introduction
Modern animal agriculture has been very successful meeting the growing demands of consumers worldwide for high-quality, safe and affordable animal products. This trend is most evident for the poultry industry. For example, in the past 40 years, poultry production has grown more than five-fold, making poultry the primary meat consumed in the USA. The USDA Economic Research Service predicts that this trend will continue in the USA for the foreseeable future, with red meat (beef, pork and lamb) consumption anticipated to decrease by more than 5% in the next 10 years, while poultry consumption is projected to increase by more than 8%! This same trend is also likely to follow outside the USA, with poultry meat production having increased 436% from 1970 to 2005, compared with 186% and 57% for pork and beef, respectively (FAO database, faostat.fao.org, verified 30 January 2012).
Several major issues confront the poultry industry today. With high-density chicken rearing and reduced genetic diversity from industry consolidation (Muir et al. 2008), control of infectious diseases and preventing disease outbreaks are critical for sustaining economic viability, maintaining public confidence in poultry products and enhancing animal welfare. Among diseases, MD, a lymphoproliferative disease of poultry caused by the highly oncogenic α-herpesvirus MDV, continues to be at or near the top of the list. The concern of MD is further enhanced by the unpredictable and spontaneous vaccine breaks that can result in devastating losses to poultry farms. Annual losses worldwide by MD due to meat condemnation and reduced egg production exceed US$1 billion (Purchase 1985), which is a minimum estimate because the figure has not been revised to reflect inflation, new disease outbreaks or MDV-induced immunosuppression.
The main control strategy for MD is vaccination. The first USA vaccine was HVT, a related herpesvirus of turkey, introduced in the late 1960s (Okazaki et al. 1970; Witter et al. 1970). Since then, additional vaccines with better efficacy have been introduced in response to field strains that are more pathogenic. While these vaccines are very effective in preventing MD and tumour formation, they are not sterilising, and thus, do not prevent infection or shedding of pathogenic MDV (Witter 2001). Because both vaccine viruses and pathogenic MDV coexist in MD-vaccinated flocks, it is highly possible that these conditions are in part responsible for the evolution of strains with increasing virulence (Witter 1997). On the basis of pathogenicity shifts, it has been suggested that a new MD vaccine is useful for ~10 years (Kreager 1996). Thus, in the long term, controls measures alternative to vaccines are needed to successfully control MD incidence.
The field of genomics offers one of the more exciting avenues for controlling MD and other diseases. While still in its formative years, by identifying quantitative trait loci (QTL) and genes that control heritable traits of agricultural importance, it is possible to select for birds with superior agricultural traits such as improved disease resistance via marker-assisted selection (MAS). Other positive attributes commonly cited for MAS include greater speed and accuracy than for traditional breeding. Furthermore, for infectious diseases, MAS would eliminate the exposure risk to elite flocks associated with handling a hazardous pathogen. The release of the chicken genome sequence (Hillier et al. 2004) and ongoing improvements only increase the power of this discipline.
As shown in Fig. 1, previously, we combined (1) at the DNA level, QTL scans to identify genomic regions that influenced MD incidence, (2) at the RNA level, transcript profiling to identify genes that were differentially expressed between MD resistance and susceptible lines following MDV infection, and (3) at the protein level, virus-host protein–protein interaction screens that identified high-confidence genes associated with resistance to MD (Cheng et al. 2008). The rationale for using more than one approach is that the strengths of each system can be combined to yield results of higher confidence. Another justification is that given the large volume of data produced by genomics, each method provides an additional screen to limit the number of targets to verify and characterise in future experiments. Efforts to experimentally characterise growth hormone (Liu et al. 2001), stem-cell antigen 2 (Mao et al. 2010), and major histocompatibility complex class II β chain (Niikura et al. 2007) validated their corresponding genes as influencing genetic resistance to MD.
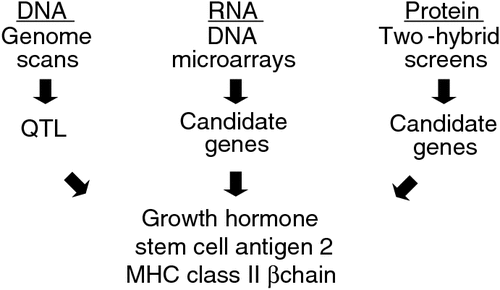
|
Despite this success, we were able to explain only a fraction of the total variation accounting for genetic resistance to MD. The main limitation has been our mapping resolution when using linkage or association analyses. In other words, genetic mapping cannot resolve down to a single-gene level, which is desired for further validation and biological characterisation. This limitation is still present in genome-wide association studies that have tens of thousands of individuals with up to 1 million genotypes per individual, as evidenced by commentaries that address the ‘missing heritability’ (Maher 2008). Because this limitation is shared with virtually every complex trait, and the likelihood that even whole genome sequences will not provide a resolution, we have been incorporating new strategies shown in Fig. 2 that largely overcome this obstacle.
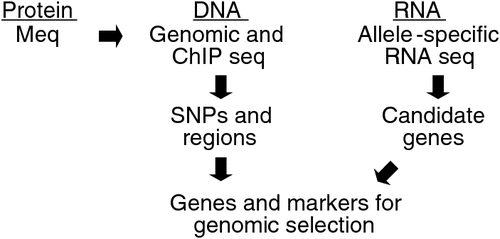
|
Allele-specific expression in response to MDV infection
First inspired by the very modest differences in protein sequence divergence between humans and chimpanzees (King and Wilson 1975), and later confirmed by whole genome sequence assemblies, it became logical to conclude that non-synonymous substitutions were not sufficient to explain trait variation both within and among species. Thus, it was hypothesised that differences in gene expression (when, where, and how much) are a major contributor of phenotypic variation (Knight 2005; Wray 2007; Pastinen 2010). Unfortunately, in stark contrast to protein coding variation, very little is known about the extent of variation in gene regulatory elements within populations. To make matters more difficult, even when sequence variation is known, it is extremely difficult to predict what polymorphisms will influence gene expression or, more importantly, which traits. Consequently, until one surveys for variation in gene expression and associated trait differences, and identifies the underlying sequences that account for these differences, our understanding of a genome, even those with a genome sequence similar to that of the chicken, will be limited.
One technique to identify variation in gene expression is to screen for ASE. The concept is very simple, which also greatly adds to the power and attraction of this approach. For all genes of interest, the relative expression levels of the two alleles are compared within a RNA sample derived from an individual that is heterozygous for a measurable polymorphism such as a coding SNP (cSNP). When allelic imbalance or differential expression is observed, then a polymorphic cis-acting element must be present for that gene ‘since allelic variation is by definition reflective of cis-acting influence’ (Stamatoyannopoulos 2004). The primary strength of the ASE approach is that extraneous (‘trans-acting’) effects are eliminated, because the two alleles being compared are present in the same diploid cell. Thus, allelic imbalance immediately identifies a gene with an allele as being under the influence of a polymorphic cis-acting regulatory element and, therefore, within the transcriptional regulatory region in which the gene in question resides. This does not mean that trans-acting factors do not influence or modulate the expression of a specific allele, but does mean that when unequal expression of alleles is observed, it is sufficient to indicate a cis-acting or genetic element. More importantly, because genetic factors that influence transcriptional regulation in cis are generally in close proximity to the gene itself, identification of a cis-acting regulatory element essentially identifies a specific gene or locus that is most likely to contain the polymorphism leading to the allelic expression imbalance. Alternatively, another mechanism for allelic imbalance in the absence of a polymorphism is imprinting which parent-of-origin determines allelic expression; however, imprinting has not been observed in chickens (O’Neill et al. 2000), presumably due to the absence of specific DNA methyltransferases (Yokomine et al. 2006).
We have conducted a genome-wide ASE screen for chicken non-major histocompatibility complex genes that respond to MDV infection, with preliminary results reported (MacEachern et al. 2011). The experiment was performed in the following three steps: (1) generation of the samples, (2) transcriptome sequencing of limited samples, followed by analysis to identify cSNPs and, when found, preliminary evidence for ASE in response to MDV infection, and (3) validation of results by screening all samples with selected cSNPs.
To generate the RNA samples, highly inbred experimental Line 6 (MD resistant) and Line 7 (MD susceptible) birds were intermated to maximise the number of genes heterozygous for each cSNP. Reciprocal matings were conducted to allow for possible maternal or imprinting influences on gene expression. The progeny from each reciprocal cross were split, with half uninfected and the other half infected with MDV (2000 plaque forming units, JM strain) at 2 weeks of age. Twelve F1 birds from each treatment group were killed at 1, 4, 7, 11, 13 and 15 days post infection (dpi), splenic tissue was recovered and the RNA isolated. Genomic DNA from 12 F1 birds for each mating direction was also recovered to act as assay controls, i.e. allelic ratio of each gene is known to be 1 : 1.
ASE assays require a cSNP to monitor the abundance of each expressed allele. To get a cost-efficient, genome-wide and unbiased survey of cSNP(s) within all of the expressed genes, and an indication of ASE, two replicate RNA pools consisting of six uninfected or six MDV-infected F1 birds at a single time point (4 dpi) and each reciprocal mating were sequenced using a next-generation sequencer (Illumina GA, Illumina, San Diego, CA, USA). The reads were aligned to the chicken genome (Version 2.1) with MAQ (Version 0.50; http://maq.sourceforge.net/maq-man.shtml, verified 30 January 2012). FastQ files were initially parsed, and all adaptor sequences were removed as were poor-quality reads (quality scores <40). Alignments were parsed and transcripts examined for the presence of cSNPs that differentiated alleles in the two inbred lines. After filtering, 14–17+ million reads were mapped per treatment group and 22 655 high-quality cSNPs were identified. As exemplified in Fig. 3, our main interest was to identify cSNPs (and genes) where the allelic expression ratios were altered as a result of MDV infection. Statistical analysis revealed that 5360 (23.6%) cSNPs in 3773 genes exhibited a statistically significant allelic imbalance in response to MDV infection.
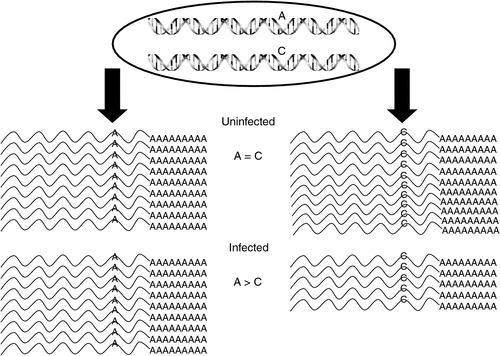
|
To economically validate and extend the results, 1536 selected SNPs were screened on RNA samples from all F1 birds by using Illumina GoldenGate arrays (Illumina, San Diego, CA, USA). We performed an analysis of variance for ASE in each cSNP, sex, reciprocal cross and infection status across all dpi and tested the interaction of each factor with infection. The results show that expression of each cSNP varied greatly, and that infection, dpi and cross were all factors that significantly affect rates of expression. No significant interactions were detected for sex × infection and cross × infection; therefore, there is little evidence of maternal or epigenetic effects in response to MDV infection. However, a significant interaction was detected for dpi × infection and dpi × cross × infection that indicates that infection status has a significant impact on the expression levels in these genes. The key result is that 1184 (96%) of the 1233 cSNPs showed evidence for ASE in response to MDV infection. Thus, extrapolating to the 5360 SNPs exhibiting ASE, we have identified potentially 3773 genes between our two lines that may account for their differences in genetic resistance to MD.
ChIP seq for genes directly regulated by MDV Meq
Genetic resistance to MD is characterised by the lack of tumours or nerve enlargements following exposure to MDV. MDV Meq is a bZIP transcription factor and the likely MDV oncogene (Jones et al. 1992; Lupiani et al. 2004), suggesting that one pathway for resistance is the inability of Meq to regulate the transcription of specific genes in individuals resistant to MD, thereby failing to initiate transformation. Therefore, it is of interest to define DNA-binding sites and the genes that are directly regulated by Meq. Previous work with ChIP has shown that Meq binds to specific sites on the MDV genome that are dependent on whether Meq is a homodimer or a heterodimer with c-Jun (Levy et al. 2003). Also, different forms of Meq are expressed and, in general, the full-length form is highly expressed during viral latency and in MD tumours, while the Meq-vIL8 variant, which shows no transactivation ability, is expressed at low levels during lytic replication. Recent advancements in sequencing and bioinformatics analyses can identify and define the sequences in the chicken genome bound by Meq alone or in combination with c-Jun, the preferential dimerisation partner for Meq. The identification of DNA-binding sites combined with DNA microarray analyses that profile genes regulated by Meq may reveal positional candidate genes that confer genetic resistance to MD.
Thus far, using the DF1 cell line (Himly et al. 1998) and DF1 stably transfected and expressing Meq, ChIP seq analysis using Bowtie (Langmead et al. 2009) and QuEST (Valouev et al. 2008) have identified 22 334 and 19 360 genomic regions that bind Meq and c-Jun, respectively. Examining only the highest-scoring 1000 peaks, there are 3031 and 2092 genes within 2 Kb of the Meq and c-Jun binding sites, respectively, with 602 of the peaks in common. Motif analysis for the Meq-binding sites have confirmed existing motifs (TGACA/GTCA and ACACA) (Levy et al. 2003) as well as identified several potential new ones. In parallel, the same cells were processed on Affymetrix DNA microarrays to reveal differentially expressed genes. Integrating the ChIP seq results with microarray analysis revealed 351 genes that were within 2 Kb of the highest-scoring Meq-binding peaks, as well as being differentially expressed. Pathway analysis has suggested enrichment of genes in the mitogen-activated protein kinase and WNT signalling pathways for MDV-induced transformation, both of which are key pathways for detecting extracellular ligands, regulating cellular growth and carcinogenesis.
Thus, these results have provided genomic sequences and genes that are directly regulated by MDV Meq. Of the 351 candidate genes directly regulated by Meq, 318 exhibit ASE in response to MDV infection, suggesting that binding and transcription regulation by Meq is a probable molecular mechanism. If true, it is of particular interest to see whether the ‘Meq-regulated’ genes are more likely to influence MD than are the other genes exhibiting ASE in response to MDV infection.
Genomic resequencing to identify regions under selection
The genomes of Lines 6 (MD resistant) and 7 were sequenced with paired end reads of fragments averaging 2.5 Kb between the ends. After quality trimming, the reads were aligned (chicken build 2.1) by using ABI Bioscope (Life Technologies Corp., Carlsbad, CA, USA) and SNPs were called using Samtools pileup (Li et al. 2009). Because these lines are from the same base population, the impacts of bidirectional selection on the genome were found by limiting the SNPs to those fixed for alternative alleles between lines. Next, haplotypes were formed where successive chromosomal fixations occurred in the same direction. The strength of selection is reflected in chromosome haplotype length (CHL), i.e. the hitchhiking effect (Maynard Smith and Haigh 1974). CHLs of ~60 Kb in length, successively fixed in the same direction segmented by CHL of decreasing length where recombination events occurred before complete fixation, have been observed. Although these results are still preliminary, ~25% of the ASE SNPs are in common with genes detected in this manner. Given that there are ~23 000 genes in the poultry genome, of which >16% show ASE, this association between ASE and regions showing CHL is highly significant.
Conclusions
The field of genomics has and continues to be heavily influenced by new or improved technologies such as next-generation sequencing. However, the use of these technologies is not sufficient for major gains, which is why we are incorporating them in novel ways with other existing tools or methods to achieve optimal power. It is in this context that we screened for ASE in response to MDV infection. As early as 2002, Yan et al. (2002) demonstrated that variation in allelic expression could identify specific genes for complex traits. This concept was extended to genome-wide screens in the seminal work by Brem et al. (2002), where they combined DNA microarrays with genotyping to identify expression or eQTL. While appealing in their power, eQTL screens have the same limitations as inherent in QTL mapping, namely that the resolving power is normally not sufficient to identify specific genes. It was not until ASE screens that capitalised on SNP chips (e.g. Serre et al. 2008) and the next-generation sequencing (e.g. Pickrell et al. 2010) were used to monitor the expression of both alleles of a gene that genomic screens had the resolving power to identify specific genes accounting for expression variation.
ASE screens are particularly attractive to identify candidate genes for genetic resistance to infectious pathogens. This is because of the simplistic method where one just compares the cSNP ratios between samples from two states, uninfected v. infected. Our results suggest that many genes (3700+) with a genetic basis account for the differences in MD incidence between our two chicken lines. This is not surprising, given the sensitivity of our ASE screens and the growing consensus that complex traits are controlled by many genes. Our results are consistent with those found in a much larger mouse study where 41% of the 15 884 genes surveyed showed ASE (Keane et al. 2011). Most interestingly, polymorphisms associated with exons, introns and flanking regions are more likely to be large-effect QTL than those in intergenic positions, which also suggests a significant role for transcriptional regulation. However, until additional studies are conducted, it is speculative to determine the extent that gene regulation and ASE account for in complex traits.
The inclusion of ChIP seq using anti-Meq antibodies and resequencing of our divergently selected lines provides additional knowledge and power on the underlying mechanisms and polymorphisms that may account for the ASE response and, thus, genetic resistance to MD. This also helps highlight how screens at different levels synergise each other and provide more meaningful results.
In conclusion, the incorporation of new technologies that primarily survey transcriptional variation and their underlying mechanisms shows great promise in identifying the molecular and genetic basis of complex traits, such as genetic resistance to MD. In our case, the ultimate proof and validation lies in whether the cSNPs identified in our ASE screens are associated with MD incidence and can be utilised in genomic selection to improve commercial poultry lines.
Acknowledgements
We thank Laurie Molitor for excellent technical assistance, and Seth Crosby for conducting the Illumina GoldenGate assays. This project was supported in part by National Research Initiative competitive grants no. 2008-35205-18745 and 2009-35205-05035 from the USDA National Institute of Food and Agriculture Animal Genome Program.
References
Brem RB, Yvert G, Clinton R, Kruglyak L (2002) Genetic dissection of transcriptional regulation in budding yeast. Science 296, 752–755.| Genetic dissection of transcriptional regulation in budding yeast.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XjtlOnt70%3D&md5=958f465b27c51d3e9dc62bb6910177d9CAS |
Cheng H, Niikura M, Kim T, Mao W, MacLea KS, Hunt H, Dodgson J, Burnside J, Morgan R, Ouyang M, Lamont S, Dekkers J, Fulton J, Soller M, Muir W (2008) Using integrative genomics to elucidate genetic resistance to Marek’s disease in chickens. In ‘Animal genomics for animal health. Developments in biologicals. Vol. 132’. (Eds M-H Pinard, C Gay, P-P Pastoret, B Dodet) pp. 365–372. (Karger: Basel, Switzerland)
Hillier LW, Miller W, Birney E, Warren W, Hardison RC, et al International Chicken Genome Sequencing Consortium (2004) Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 432, 695–716.
| Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXhtVGmtb7M&md5=cf7c36ddc1f0793a557c6bbd830d1cf0CAS |
Himly M, Foster DN, Bottoli I, Iacovoni JS, Vogt PK (1998) The DF-1 chicken fibroblast cell line: transformation induced by diverse oncogenes and cell death resulting from infection by avian leukosis viruses. Virology 248, 295–304.
| The DF-1 chicken fibroblast cell line: transformation induced by diverse oncogenes and cell death resulting from infection by avian leukosis viruses.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXlvVOntrk%3D&md5=6f445d92bfdacffa4216a0e8e19f4f8bCAS |
Jones D, Lucy L, Liu JL, Kung HJ, Tillotson JK (1992) Marek disease virus encodes a basic-leucine zipper gene resembling the fos/jun oncogenes that is highly expressed in lymnphoblastoid tumors. Proceedings of the National Academy of Sciences, USA 89, 4042–4046.
| Marek disease virus encodes a basic-leucine zipper gene resembling the fos/jun oncogenes that is highly expressed in lymnphoblastoid tumors.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3sXitV2jtLs%3D&md5=d3857f33077f79d8d86b898de1edcb37CAS |
Keane TM, Goodstadt L, Danecek P, White MA, Wong K, et al (2011) Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 477, 289–294.
| Mouse genomic variation and its effect on phenotypes and gene regulation.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtFOlsL7N&md5=f71d7e1275c07df5b3a0c4f783be1962CAS |
King MC, Wilson AC (1975) Evolution at two levels in human and chimpanzees. Science 188, 107–116.
| Evolution at two levels in human and chimpanzees.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE2MXhs1Orur4%3D&md5=ec42178cc2413498b233dcf7eb81cfa8CAS |
Knight JC (2005) Regulatory polymorphisms underlying complex disease traits. Journal of Molecular Medicine 83, 97–109.
Kreager K (1996) Industry concerns workshop. In ‘Current research on Marek’s disease’. (Eds RF Silva, HH Cheng, PM Coussens, LF Lee, LF Velicer) pp. 509–511. (American Association of Avian Pathologists: Kennett Square, PA)
Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology 10, R25
| Ultrafast and memory-efficient alignment of short DNA sequences to the human genome.Crossref | GoogleScholarGoogle Scholar |
Levy AM, Izumiya Y, Brunovskis P, Xia L, Parcells MS, Reddy SM, Lee L, Chen HW, Kung HJ (2003) Characterization of the chromosomal binding sites and dimerization partners of the viral oncoprotein Meq in Marek’s disease virus-transformed T cells. Journal of Virology 77, 12 841–12 851.
| Characterization of the chromosomal binding sites and dimerization partners of the viral oncoprotein Meq in Marek’s disease virus-transformed T cells.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXptFOjsLc%3D&md5=dc577a2299b350b72023b6b216bbe00dCAS |
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R (2009) The sequence alignment/map format (SAM) and SAMtools. Bioinformatics 25, 2078–2079.
| The sequence alignment/map format (SAM) and SAMtools.Crossref | GoogleScholarGoogle Scholar |
Liu HC, Kung HJ, Fulton JE, Morgan RW, Cheng HH (2001) Growth hormone interacts with the Marek’s disease virus SORF2 protein and is associated with disease resistance in chicken. Proceedings of the National Academy of Sciences, USA 98, 9203–9208.
| Growth hormone interacts with the Marek’s disease virus SORF2 protein and is associated with disease resistance in chicken.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXlvFSrsrs%3D&md5=37680cbe7a8aa4f2c48b7506a13a5199CAS |
Lupiani B, Lee LF, Cui X, Gimeno I, Anderson A, Morgan RW, Silva RF, Witter RL, Reddy SM (2004) Marek’s disease virus-encoded Meq gene is involved in transformation of lymphocytes but is dispensable for replication. Proceedings of the National Academy of Sciences, USA 101, 11 815–11 820.
| Marek’s disease virus-encoded Meq gene is involved in transformation of lymphocytes but is dispensable for replication.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXntVers7o%3D&md5=73582d7f823cc86912e14e8bfa7edba5CAS |
MacEachern S, Muir WM, Crosby S, Cheng HH (2011) Genome-wide identification of allele-specific expression (ASE) in response to Marek’s disease virus infection using next generation sequencing. BMC Proceedings 5, S14
| Genome-wide identification of allele-specific expression (ASE) in response to Marek’s disease virus infection using next generation sequencing.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXnsFamtLk%3D&md5=44af39be45a83a6c02733b1b1aeaab41CAS |
Maher B (2008) The case of the missing heritability. Nature 456, 18–21.
| The case of the missing heritability.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtlCjtrnM&md5=9c6f92e3d640f5fa9af932623fdbecc1CAS |
Mao W, Hunt HD, Cheng HH (2010) Cloning and functional characterization of chicken stem cell antigen 2. Developmental and Comparative Immunology 34, 360–368.
| Cloning and functional characterization of chicken stem cell antigen 2.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXjvVWntA%3D%3D&md5=715e5b2c4f529ecd62b59fbcc1dcf853CAS |
Maynard Smith J, Haigh J (1974) The hitch-hiking effect of a favourable gene. Genetical Research 23, 23–35.
| The hitch-hiking effect of a favourable gene.Crossref | GoogleScholarGoogle Scholar |
Muir WM, Wong GK, Zhang Y, Wang J, Groenen MAM, Crooijmans RPMA, Megens H-J, Zhang H, Okimoto R, Vereijken A, Jungerius A, Albers GAA, Taylor Lawley C, Delany ME, MacEachern S, Cheng HH (2008) Genome-wide assessment of world-wide chicken SNP genetic diversity indicates significant absence of rare alleles in commercial breeds. Proceedings of the National Academy of Sciences, USA 105, 17 312–17 317.
| Genome-wide assessment of world-wide chicken SNP genetic diversity indicates significant absence of rare alleles in commercial breeds.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhsVWlurjJ&md5=f98a605cfa310788d9a045eb95fd2443CAS |
Niikura M, Kim T, Hunt HD, Burnside J, Morgan RW, Dodgson JB, Cheng HH (2007) Marek’s disease virus up-regulates major histocompatibility complex class II cell surface expression in infected cells. Virology 359, 212–219.
| Marek’s disease virus up-regulates major histocompatibility complex class II cell surface expression in infected cells.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhs1Snsr4%3D&md5=b15aca797b51b83f31a2b643c199e4ddCAS |
O’Neill MJ, Ingram RS, Vrana PB, Tilghman SM (2000) Allelic expression of IGF2 in marsupials and birds. Development Genes and Evolution 210, 18–20.
| Allelic expression of IGF2 in marsupials and birds.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXntlalsL4%3D&md5=640dded73993d100faa0e582580b3974CAS |
Okazaki W, Purchase HG, Burmester BR (1970) Protection against Marek’s disease by vaccination with a herpesvirus of turkeys. Avian Diseases 14, 413–429.
| Protection against Marek’s disease by vaccination with a herpesvirus of turkeys.Crossref | GoogleScholarGoogle Scholar | 1:STN:280:DyaE3c3jtVSktA%3D%3D&md5=d36f7179052b74c7edf738e8f63f51afCAS |
Pastinen T (2010) Genome-wide allele-specific analysis: insights into regulatory variation. Nature Reviews. Genetics 11, 533–538.
| Genome-wide allele-specific analysis: insights into regulatory variation.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXovFOhtr8%3D&md5=a69a1e1644d806df8954f38bd6693a96CAS |
Pickrell JK, Marioni JC, Pai AA, Degner JF, Engelhardt BE, Nkadori E, Veyrieras J-B, Stephens M, Gilad Y, Pritchard JK (2010) Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature 464, 768–772.
| Understanding mechanisms underlying human gene expression variation with RNA sequencing.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXivFKns7Y%3D&md5=6f8c3beff48eb29b334480a1df1c1256CAS |
Purchase HG (1985) Clinical disease and its economic impact. In ‘Marek’s disease, scientific basis and methods of control’. (Ed LN Payne) pp. 17–42. (Martinus Nkjhoff Publishing: Boston, MA)
Serre D, Gurd S, Ge B, Sladek R, Sinnett D, Harmsen E, Bibikova M, Chudin E, Barker DL, Dickinson T, Fan JB, Hudson TJ (2008) Differential allelic expression in the human genome: a robust approach to identify genetic and epigenetic cis-acting mechanisms regulating gene expression. PLOS Genetics 4, e1000006
| Differential allelic expression in the human genome: a robust approach to identify genetic and epigenetic cis-acting mechanisms regulating gene expression.Crossref | GoogleScholarGoogle Scholar |
Stamatoyannopoulos JA (2004) The genomics of gene expression. Genomics 84, 449–457.
| The genomics of gene expression.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXmtlOksbk%3D&md5=61794b09b9d4ce73b99cfea214b9a1b5CAS |
Valouev A, Johnson DS, Sundquist A, Medina C, Anton E, Batzoglou S, Myers RM, Sidow A (2008) Genome-wide analysis of transcription factor binding sites based on ChIP-seq data. Nature Methods 5, 829–834.
| Genome-wide analysis of transcription factor binding sites based on ChIP-seq data.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtVGgsb3F&md5=f30758dc8be9c84ea3faacb39dd3bf57CAS |
Witter RL (1997) Increased virulence of Marek’s disease virus field isolates. Avian Diseases 41, 149–163.
| Increased virulence of Marek’s disease virus field isolates.Crossref | GoogleScholarGoogle Scholar | 1:STN:280:DyaK2s3kt1ehuw%3D%3D&md5=0752a7011008def9c31d0aad00b9a4c2CAS |
Witter RL (2001) Protective efficacy of Marek’s disease vaccine. In ‘Marek’s disease’. (Ed. K Hirai) pp. 57–90. (Springer-Verlag: New York)
Witter RL, Nazerian K, Purchase HG, Burgoyne GH (1970) Isolation from turkeys of a cell-associated herpesvirus antigenically related to Marek’s disease virus. American Journal of Veterinary Research 31, 525–538.
Wray GA (2007) The evolutionary significance of cis-regulatory mutations. Nature Reviews. Genetics 8, 206–216.
| The evolutionary significance of cis-regulatory mutations.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhslOgt7w%3D&md5=c9ad56e38c77649a7d63c50e95666d06CAS |
Yan H, Yuan W, Velculescu VE, Vogelstein B, Kinzler KW (2002) Allelic variation in human gene expression. Science 297, 1143
| Allelic variation in human gene expression.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XmsVOiu7s%3D&md5=b72f83d12b64d6932111c3981743004cCAS |
Yokomine T, Hata K, Tsudzuki M, Sasaki H (2006) Evolution of the vertebrate DNMT3 gene family: a possible link between existence of DNMT3L and genomic imprinting. Cytogenetic and Genome Research 113, 75–80.
| Evolution of the vertebrate DNMT3 gene family: a possible link between existence of DNMT3L and genomic imprinting.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XjtVSqtL4%3D&md5=fe7677efb2ba7f56bf7559c2e4e35e28CAS |