

Origins and population genetics of sambar deer (Cervus unicolor) introduced to Australia and New Zealand
Lee A. Rollins A B * , Daniel Lees B , Andrew P. Woolnough C D , Andrea J. West B , Michael Perry E and David M. Forsyth A F
A B * , Daniel Lees B , Andrew P. Woolnough C D , Andrea J. West B , Michael Perry E and David M. Forsyth A F
A Evolution & Ecology Research Centre, School of Biological, Earth and Environmental Sciences, University of New South Wales, Sydney, NSW 2052, Australia.
B Centre for Integrative Ecology, School of Life and Environmental Sciences, Deakin University, Locked Bag 20000, Geelong, Vic. 3216, Australia.
C Department of Jobs, Precincts and Regions, 121 Exhibition Street, Melbourne, Vic. 3000, Australia.
D Research, Innovation and Commercialisation, The University of Melbourne, Parkville, Vic. 3010, Australia.
E Department of Conservation, 59 Marine Parade, Napier 4110, New Zealand.
F Vertebrate Pest Research Unit, New South Wales Department of Primary Industries, 1447 Forest Road, Orange, NSW 2800, Australia.
Wildlife Research 50(9) 716-727 https://doi.org/10.1071/WR22120
Submitted: 7 July 2022 Accepted: 22 May 2023 Published: 20 July 2023
© 2023 The Author(s) (or their employer(s)). Published by CSIRO Publishing. This is an open access article distributed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License (CC BY-NC-ND)
Abstract
Context: Some populations of introduced species cause significant undesirable impacts but can also act as reservoirs for genetic diversity. Sambar deer (Cervus unicolor) are ‘Vulnerable’ in their native range and invasive in Australia and New Zealand. Genetic data can be used to determine whether these introduced populations might serve as genetic reservoirs for declining native populations and to identify spatial units for management.
Aims: We aimed to identify the provenance of sambar deer in Australia and New Zealand, and to characterise their genetic diversity and population structure.
Methods: We used mitochondrial control region sequences and 18 nuclear microsatellite loci of 24 New Zealand and 63 Australian sambar deer collected across continuous habitat in each location. We estimated genetic diversity and population differentiation by using pairwise FST, AMOVA, and Structure analyses. We compared our data with 27 previously published native and invasive range sequences to identify phylogenetic relationships.
Key results: Sambar deer in Australia and New Zealand are genetically more similar to those in the west of the native range (South and Central Highlands of India, and Sri Lanka), than to those in the east (eastern India, and throughout Southeast Asia). Nuclear genetic diversity was lower than in the native range; only one mitochondrial haplotype was found in each introduced population. Australian and New Zealand sambar deer were genetically distinct but there was no population structure within either population.
Conclusions: The genetic differences we identified between these two introduced populations at putatively neutral loci indicate that there also may be underlying diversity at functional loci. The lack of population genetic structure that we found within introduced populations suggests that individuals within these populations do not experience barriers to dispersal across the areas sampled.
Implications: Although genetic diversity is reduced in the introduced range compared with the native range, sambar deer in Australia and New Zealand harbour unique genetic variants that could be used to strengthen genetic diversity in populations under threat in the native range. The apparent high levels of gene flow across the areas we sampled suggest that localised control is unlikely to be effective in Australia and New Zealand.
Keywords: Cervidae, invasive species, management units, microsatellite, mitochondrial DNA, population genetics, Rusa unicolor, sambar deer.
Introduction
Globally, many native populations of ungulates are at risk of extinction owing to habitat loss and hunting (Ripple et al. 2015). Non-native ungulate populations are increasingly being recognised for their potential as sources for re-introduction to their native range, either to re-establish a population that has gone extinct or for ‘genetic rescue’ in declining populations (Bradshaw et al. 2006; Wallach et al. 2015; Hill et al. 2019). Genetic rescue is the addition of genetic material to a population to reduce its risk of extinction. Despite this potential benefit, translocating animals from non-native populations into their native range has several major challenges (Armstrong and Seddon 2008). First, non-native populations could be genetically very different from the extant native population, which could result in outbreeding depression (the introduction of maladaptive alleles that reduce fitness in offspring). For example, a recent study of amphibians found that interbreeding individuals from different populations lowered offspring fitness in comparison to within-population crosses (Byrne and Silla 2020). However, the circumstances that increase the probability of outbreeding depression have been characterised, and these findings can assist conservation management (Frankham et al. 2011). Second, because non-native populations often originate from a small number of individuals, they are likely to contain reduced genetic diversity compared with native populations (Dlugosch and Parker 2008). This reduction in diversity may be accentuated by subsequent genetic drift and can result in a reduction in fitness (Frankham et al. 1999). In this context, genetic studies of non-native ungulate populations are needed to help determine their suitability for translocation.
Genetic analysis is also invaluable for the management of non-native ungulates that become invasive and have significant environmental, economic and social impacts (Côté et al. 2004; Bengsen et al. 2014; Latham et al. 2017). Despite the need to effectively manage invasive populations, determining the scale at which a population should be managed can be challenging. Managers need to know whether areas that could potentially be managed separately are demographically independent populations (sometimes called management units, MUs), or whether the areas have regular exchange, in which case their control will need to be coordinated if it is to be effective (Allendorf and Luikart 2007). Identifying MUs among large mainland populations is often not straightforward (Hampton et al. 2004; Robertson and Gemmell 2004). Population genetic analyses are an increasingly affordable way to identify spatial structure in invasive populations that can help delimit MUs (e.g. feral pigs (Sus scrofa), Hampton et al. 2004; Cowled et al. 2008; dromedary camels (Camelus dromedarius), Spencer et al. 2012).
Sambar deer (Cervus unicolor: nomenclature follows Jackson et al. 2015) are the largest (~178 kg) cervid species native to Southeast Asia, India and China (Leslie 2011). The species is assessed as ‘Vulnerable’ owing to sustained declines in its native range from overhunting by humans and habitat loss (Timmins et al. 2015). There is debate about the phylogeography and taxonomy of sambar deer, with five (Mattioli 2011) or seven (Leslie 2011; Timmins et al. 2015) subspecies being recognised. However, a recent mitogenomic analysis using historical samples collected between the late 19th and mid-20th century did not support those subspecific descriptions; rather, three clades were identified that were associated with Myanmar/India, Sri Lanka, and Sunda (i.e. Sumatra, Mentawai, and Borneo; Martins et al. 2018). Genetic variation in native Indian sambar deer has been investigated using mitochondrial and nuclear markers, identifying multiple genetic groups (two and three respectively; Gupta 2014; Gupta et al. 2015).
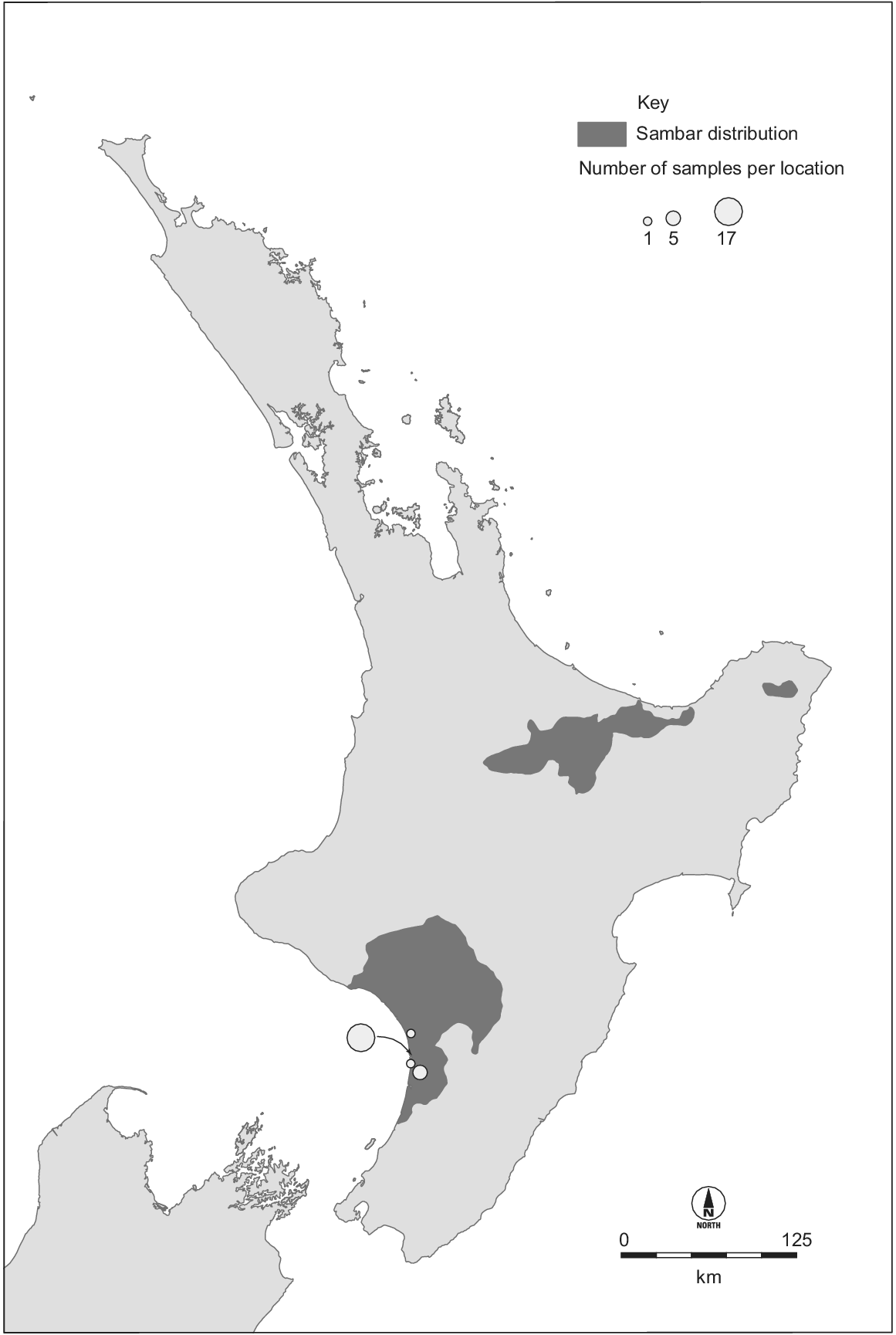
Sambar deer have been introduced outside their native range, with wild populations being established in New Zealand, Australia, USA, and South Africa (Leslie 2011; Timmins et al. 2015). The largest non-native populations are in Australia and New Zealand, both descending from animals imported in the late 1800s (Donne 1924; Bentley 1998). There is uncertainty about the origins of the individuals introduced, owing to poor historical records and the unresolved phylogeography of this species. The New Zealand sambar deer population originates from perhaps just two individuals (Donne 1924). At least one pair of sambar deer was imported from Sri Lanka in 1875 and released at Manawatu, western North Island (Donne 1924; Harris 1971; Banwell 2006). A population established and subsequently increased in size and range. Illegal hunting reduced the population to ≤30 in 1893 (Harris 1971), but following formal protection it increased to ~100 individuals in 1900 (Shailer 1957). The population continued to increase such that all protections were removed in 1930, and by 1947 the population was thought to be ~500 (Wodzicki 1950). In 1957, the Manawatu population was thought to occupy 1000 km2 (Harris 1966). Two other sambar deer populations have subsequently established in New Zealand, both from sambar deer captured from the Manawatu herd (Fig. 1) (Harris 1971; Nugent et al. 2021). Today, the New Zealand population occupies ~11 000 km2, and its impacts are minor compared with more widespread and abundant invasive deer species (Nugent et al. 2021).
Distribution of Cervus unicolor in New Zealand (Fraser and Nugent 2005) and our sampling locations. The two eastern populations originate from the western population, which we sampled. The three populations occupy ~11 000 km2 and number in the thousands.
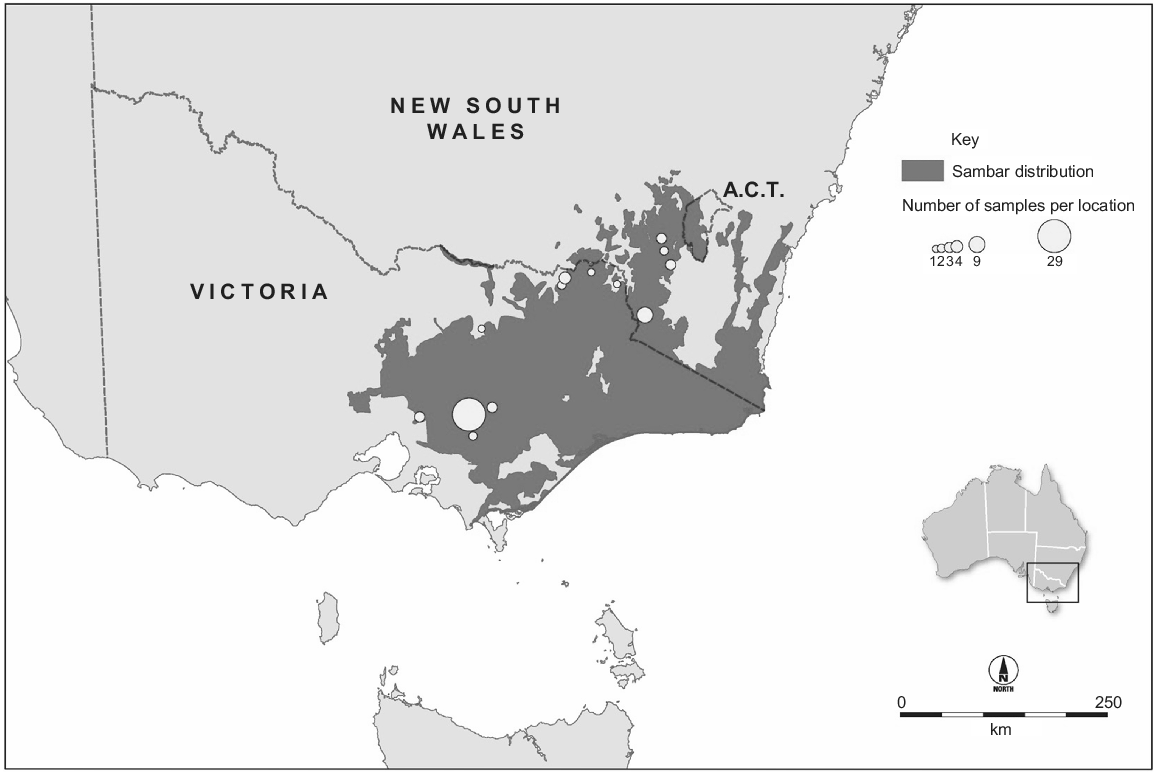
The Australian population is much larger than the New Zealand population. In Australia, sambar deer were imported to Royal Park, Victoria, from Sri Lanka in 1861, and introductions from India (1845) and the Philippines (1859) have also been reported (Downes 1983; Bentley 1998). Although the ‘Sri Lankan’ animals were kept in an enclosure, all deliberate or accidental introductions of this species in south-eastern Australia originate from these animals. They were released at multiple locations in Victoria during 1863–1873 (Bentley 1998). The main ‘Eastern Victoria’ sambar deer population established in the Koo Wee Rup swamp, and this population numbered ‘some hundreds’ in the 1880s (Bentley 1998). This population subsequently colonised to the north and north-east into the Great Dividing Range, and to the south-east, and by 1980 had colonised Kosciuszko National Park in New South Wales (Dunn 1985). In 2015, this ‘Eastern Victoria’ population occupied ~67 000 km2 of Victoria, New South Wales and the Australian Capital Territory (Fig. 2). On the basis of harvest estimates (Moloney et al. 2022), the population must number in the hundreds of thousands of animals. This population has caused significant social, economic and environmental impacts (Lindeman and Forsyth 2008; Forsyth and Davis 2011; Davis et al. 2016). Shooting and exclusion fencing are being used to manage these impacts at small spatial scales (Bennett et al. 2015; Fahey 2017; Comte et al. 2023, this issue), but larger-scale management of this population could be improved by knowledge about its spatial structure. In particular, it is unclear how many MUs exist across its Australian range. Although the ecology and impacts of this species have been studied widely, less is known about the population genetics of Australian sambar deer. Within Victoria, three genetic groups were identified using microsatellite data, including the geographically isolated Mount Cole and French Island sambar deer, and the more contiguously distributed population in the south-east of the state (Davies et al. 2022). That study also investigated genetic diversity using sequence data from a portion of the mitochondrial control region, and identified two haplotypes within Victoria, including a private haplotype in Mount Cole. Davies et al. (2022) conducted phylogenetic analyses including sequences from the native range, which indicated that Australian sambar deer were likely to have been sourced from Sri Lanka, although branch support for this conclusion was weak. A second genetic study in Australia used reduced representation sequencing to produce a panel of single-nucleotide polymorphisms (SNPs) and identified four genetically distinct groups across south-eastern Australia as well as the presence of sambar and rusa deer (Cervus timorensis) hybrids in three geographically separated regions (Hill et al. 2023, this issue).
Distribution of the major populations of Cervus unicolor in Australia (Forsyth et al. 2015) and our sampling locations. The population occupies ~67 000 km2 and numbers in the hundreds of thousands.
Genetic data are often used to characterise introduced populations such as those studied here. They can be used to clarify patterns of gene flow and may be useful for tracking contemporary dispersers and human-assisted translocations (Rollins et al. 2006). Further, the identification of population subdivision for a pest animal may provide targets for localised control (Rollins et al. 2009). Where population structure exists, dispersal rates and barriers to dispersal can be identified (Rollins et al. 2006). In instances where invasion history is unclear and samples from putative native sources are available, population of origin can be identified (Bonizzoni et al. 2004). Understanding the extent of genetic variation can also be useful for management. Introductions of multiple, genetically distinct native populations can result in invasive populations of greater genetic diversity (Kolbe et al. 2004), whereas single-source introductions often show a decrease in genetic diversity in the invasive range, which also declines with increasing distance from the introduction point (Rollins et al. 2009). Additionally, genetic diversity can indicate the evolutionary potential (robustness) of the population. This raises an interesting point regarding species such as sambar deer that face threatening processes in their native range but are successful invaders elsewhere. Although a genetically depauperate introduced population may be less likely to persist (Lee 2002, but see Tsutsui et al. 2000), it could still serve as a reintroduction source to bolster threatened native populations.
Here we report an investigation into the origins and population structure of sambar deer in Australia and New Zealand. Our first objective was to evaluate their potential use as a source of reintroduction by resolving the origins of these populations via the incorporation of recent genetic data from the native range of this species (Gupta et al. 2015; Martins et al. 2018) into our analyses, and by determining whether genetic outliers exist in our data that may indicate the presence of hybrids. Our second objective was to estimate genetic diversity in the Australian and New Zealand populations, including whether there was evidence of reduced genetic diversity following these introductions and whether unique alleles are present in these introductions. Even if these introduced populations have reduced diversity, the presence of unique alleles could indicate functional diversity that could assist native populations. Our third objective was to determine whether spatial structure exists within the Australian population, so as to assist the management of this species. We discuss the genetic value of the Australian and New Zealand populations in the context of the phylogeography of this species, and how the spatial structure of the Australian population helps on-ground management there.
Materials and methods
Sample collection and DNA extraction
Samples were collected from 87 sambar deer harvested by government and recreational hunters in Australia and New Zealand. We included New Zealand samples collected from an area with an approximately 12 km radius near the original release site (n = 24) (Fig. 1). We also included samples collected from localities spread across the length of the major Australian distribution (New South Wales and Victoria, n = 34) as well as a large group of samples from a single area in the south of this distribution (Yarra Valley Reservoir; n = 29) (Fig. 2). Samples were collected from ear clips, muscle tissue, or faecal material (taken directly from the gut) and either frozen, or preserved in DMSO/NaCl solution (Seutin et al. 1991), or Longmire buffer (Longmire et al. 1988). Sampling location was recorded. DNA was extracted using a Gentra Puregene Tissue Kit (QIAGEN), following manufacturer’s instructions.
Mitochondrial sequencing
A portion of the mitochondrial control region was sequenced using primers designed for cervids (Cerv.tPro and CervCRH; Balakrishnan et al. 2003). DNA was amplified in a 20 μL reaction containing 1.5 mM MgCl2, 0.5 μM of each primer, 0.2 mM dNTPs and 0.2 units of Taq DNA polymerase. Cycling conditions included a 5-min step at 94°C, 30 cycles of 94°C for 30 s, 54°C for 15 s, and 72°C for 30 s, followed by a final extension step for 10 min at 72°C. A volume of 5 μL of each polymerase chain reaction (PCR) product was mixed with 1 μL of ExoSAP-IT (Thermo Fisher Scientific) and incubated, following manufacturer’s instructions. Cleaned PCR products were sequenced using BigDye chemistry (Thermo Fisher Scientific), precipitated in ethanol and sequenced on an ABI 3730 (Applied Biosystems). A subset of samples was sequenced in both directions to confirm accuracy of sequencing. Sequences were aligned using ClustalX2 v2.0.12 (Larkin et al. 2007). DNA polymorphism was estimated using DnaSP v5 (Librado and Rozas 2009).
We also aligned our sequences with the sequence of 27 haplotypes characterised in sambar deer sampled across the native range (Randi et al. 2001; Gupta et al. 2015; haplotype nomenclature used here and original sample names are described in Supplementary Material, Table S1, and GenBank accession numbers are listed) and those previously sequenced in Victoria (Davies et al. 2020). For this analysis, we included a 143 bp segment of the mitochondrial control region. Sequences were aligned as above. Using these data, we constructed a median joining haplotype network using Network v10.2.0.0 (Bandelt et al. 1999) and post-processed it by using the maximum-parsimony calculation. The final network was drawn in Network Publisher v2.1.2.5 (Fluxus Engineering, Clare, UK).
Cross-specific microsatellite screening
Initial microsatellite screening was conducted using DNA from individuals sampled from the Yarra Valley in Victoria, Australia. We selected from the literature 48 pairs of microsatellite primers that have previously been shown to amplify in cervids (Table S2). We included universal primer sequences (M13) on forward primers following Neilan et al. (1997). We used the DNA from two individuals for initial primer screening. PCR was conducted for each pair of primers by using a Multiplex PCR Kit (QIAGEN), following manufacturer’s instructions, and a step-down PCR protocol consisting of 10 cycles each at the following annealing temperatures: 70°C, 64°C, 58°C, 54°C, 50°C. PCR products were visualised on an agarose gel. We chose 36 pairs of primers that amplified under these conditions for further screening for polymorphism, by using a panel of eight individuals and fluorescently labelled M13 universal primers. These samples were genotyped on an ABI 3730, by using GS-500 (Liz) in each capillary as a size standard. Allele sizes were estimated on GeneMapper v3.7 (Applied Biosystems). Twelve loci that were invariant in these samples and two that amplified poorly were removed from further screening (Supplementary Material, Table S2).
For the remaining 22 loci, forward primers were ordered, incorporating fluorescent labels for use in downstream genotyping of the full data set. Multiplexes were optimised with the new forward primers and a further four loci were removed during this process because of inconsistent amplification (RM095, RM188, RME25 and T108). All 18 remaining loci were included in the population genetic analyses described below.
Microsatellite screening
Three multiplex PCR assays were developed using the cross-specific primers described above (Supplementary Material, Table S3). To amplify these loci, we used a Multiplex PCR Kit following manufacturer’s instructions, except that reactions were run at one-fifth the recommended volume. The multiplex PCR protocol was the same as was used in single-locus reactions (above).
Microsatellite data were tested for departures from linkage equilibrium by using GenePop v4.0 (Rousset 2008) and Hardy–Weinberg equilibrium by using Arlequin v3.5 (Excoffier et al. 2005). Genetic diversity indices were calculated including gene diversity (calculated in Arlequin), number of alleles per locus, number of private alleles and allelic richness (calculated in FSTAT v2.9.3.2; Goudet 2002).
Population structure was assessed using several methods and, for these analyses, we partitioned data into four groups (New Zealand, New South Wales, Yarra Valley, and all other Victorian samples) to investigate whether population structure exists within the Australian range. Pairwise FST values were calculated using Arlequin. We then conducted Analysis of Molecular Variance (AMOVA) (Excoffier et al. 1992), with genetic variation partitioned among regions (New Zealand vs Australia), among localities within regions and within localities. Second, we used a Principal Coordinates Analysis (PCoA) conducted in GenAlex v. 6.5 (Peakall and Smouse 2006) to visualise genetic distances (Nei 1972). We also characterised genetic subdivision using Structure v2.3.3 (Falush et al. 2003; Pritchard et al. 2007). We selected the admixture model with correlated allele frequencies and ran 10 replicates for each value of K from K = 1 to K = 6, with a burn-in period of 100 000 Markov-chain Monte Carlo steps followed by 106 iterations for each value of K. We applied the ΔK method (Evanno et al. 2005) to our Structure results to determine the most probable number of genetic groups. To detect all levels of population structure within these data, we applied a hierarchical approach to these analyses (Coulon et al. 2008). Finally, we looked for genetic outliers in the PCoA and Structure plots that might indicate the presence of hybrids in our samples.
Results
Mitochondrial control region
We sequenced 577 base pairs of the mitochondrial control region in a total of 80 individuals from Australia (n = 59) and New Zealand (n = 21). Each introduction contained a single haplotype. There were 19 polymorphic sites between the two haplotypes (nucleotide diversity = 0.014). The New Zealand haplotype contained a single bp deletion and a 41 bp insertion.
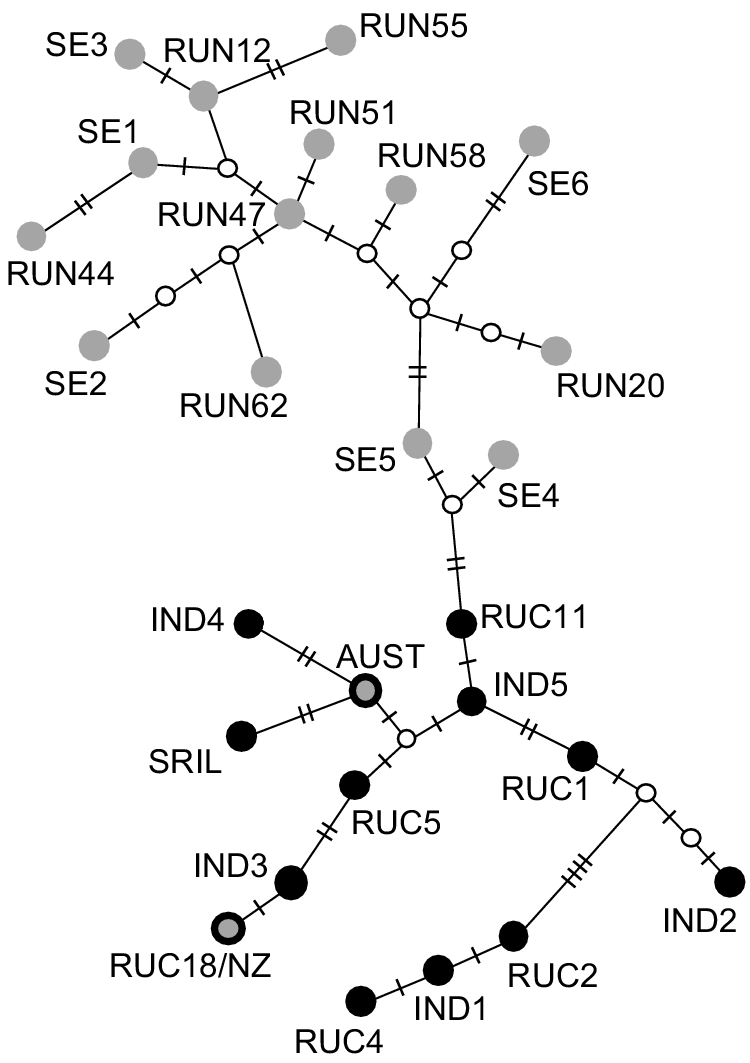
When we analysed the reduced length Australian and New Zealand haplotypes (143 bp segment; Supplementary Material, Table S4) together with previously published haplotypes (n = 25; Supplementary Material, Table S1), we found 19 polymorphic nucleotides. This segment of the Australian haplotype was identical to that of previously published Australian samples (MK473444.1 and MK473445.1, Davies et al. 2020; Fig. 3). This haplotype was most similar (2 bp difference) to a haplotype found in Sri Lanka (e.g. RUN3, Martins et al. 2018) and to a haplotype sampled in India (RUN33, Martins et al. 2018). The New Zealand haplotype from the present study was identical to a haplotype sampled in the Central Highlands of India (RUC18; Gupta et al. 2015). This short segment of DNA resolved differences between haplotypes sampled in Sri Lanka and across India, and those sampled across the rest of the native range (including the east of India; Fig. 3).
Haplotype network of 143 bp region of the mitochondrial control region of Cervus unicolor, including 27 previously sequenced haplotypes from the native range and Australia (samples described in Table S1). Intermediate nodes are denoted by small white circles. The number of nucleotide differences between haplotypes is denoted by hashmarks on network edges. The top clade includes samples collected from Bengal (India) to the eastern extent of the species native range (grey dots, no border). The bottom clade includes samples collected in India (to the west of Bengal) and in Sri Lanka (black dots). The Australian haplotype included in the current study (grey dot with heavy black border labelled ‘AUST’) groups with those from India/Sri Lanka and the New Zealand haplotype included here (grey dot with black border labelled ‘RUC18/NZ’) was identical to haplotype RUC18 sampled in the Central Highlands of India.
Microsatellites
We genotyped 87 samples at 18 microsatellite loci, none of which showed evidence of linkage nor Hardy–Weinberg disequilibrium across populations. In total, we identified 98 alleles across all loci (mean = 5.4; Supplementary Material, Table S3). Measures of genetic diversity were similar across New Zealand and Australia. Estimated gene diversity was 0.545 in New Zealand and 0.551 in Australia. We found 3.7 alleles per locus in New Zealand and 4.4 in Australia. Allelic richness estimates were similar in New Zealand (R = 3.59) and Australia (R = 3.58). However, the number of private alleles was lower in New Zealand (n = 18) than in Australia (n = 31).
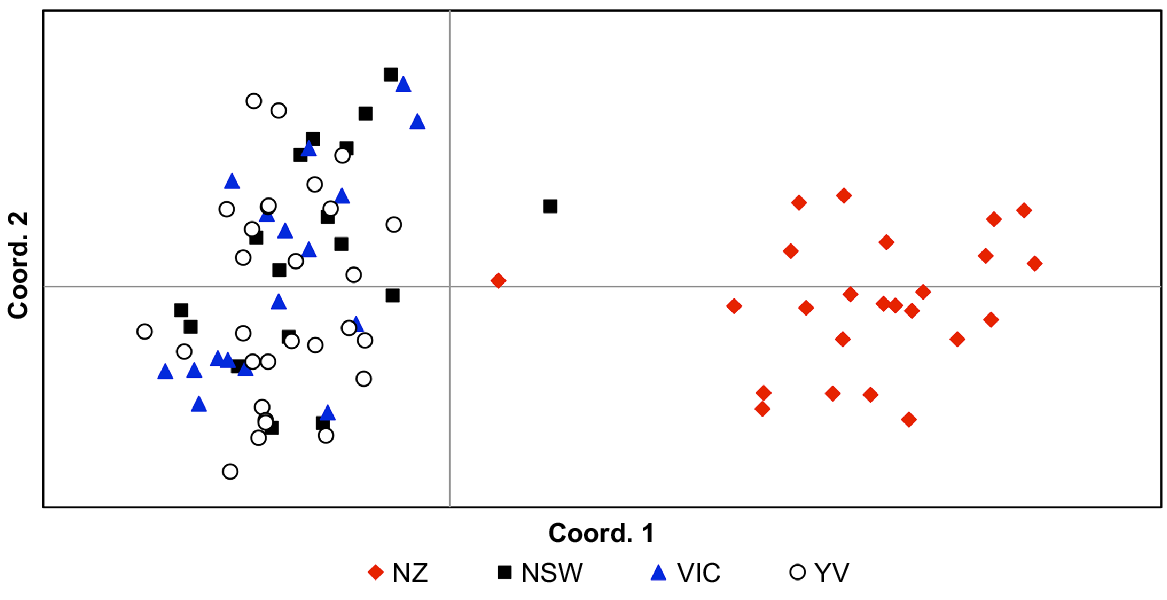
Pairwise FST values indicated significant population structure (FST = 0.23–0.24; P < 0.001) in all comparisons involving New Zealand; all other pairwise FST values comparing localities within Australia were not significant, ranging between 0.00 and 0.01. AMOVA showed that the majority of genetic variance was found within populations (67%) but high levels of variance were also identified between regions (New Zealand vs Australia; 32%). These findings are illustrated both by PCoA (Fig. 4) and Structure analyses (Fig. 5), both of which show strong genetic structure between Australia and New Zealand. Little variance was found among Australian sampling sites (1%) and hierarchical Structure analyses showed no substructuring of these data (data not shown). We found no evidence of genetic outliers in our data.
Principal Coordinates Analysis (PCoA) of genetic distances between Cervus unicolor individuals sampled from the Australian (n = 63; New South Wales, NSW; Victoria, Vic.; Yarra Valley, YV) and New Zealand (n = 24; NZ) introductions on the basis of 18 microsatellite markers (Supplementary Materials, Table S3).

Discussion
In this study, we have characterised genetic diversity of sambar deer across their introduced southern distribution in Australia and in their introduced range in New Zealand. Nuclear and mitochondrial data indicated that these two populations are genetically distinct from one another, yet both appear to have resulted from introductions from India or Sri Lanka, rather than from further east in the sambar deer native range. These results confirmed historical data that indicate that sambar deer in Australia originated in Sri Lanka (Bentley 1998). The only historical account of introductions to New Zealand suggests that a single pair of sambar deer was introduced from Sri Lanka (Harris 1971; Dunn 1985; Banwell 2006). However, our mitochondrial data suggested that the New Zealand samples included in this study had a haplotype identical to one found only in the Central Highlands of India (Haplotype RUC18; Gupta et al. 2015).
Introduced populations, such as the sambar deer studied here, may be a useful resource for the conservation of threatened species. Because sambar deer are listed as ‘Vulnerable’ in their native range (Timmins et al. 2015), introduced populations may serve as an important resource for reintroduction to those areas. Indeed, it has been recognised that introduced populations may prevent global extinction of such species (Wallach et al. 2015). The data that we presented here regarding the likely provenance of sambar deer in Australia and New Zealand could be useful for determining appropriate reintroduction sources in future, especially in the western part of the range that appears to be the source of these introductions.
One important criterion regarding the suitability of animals from a non-native population for re-introduction to their native range is whether they have hybridised with congeners (Bradshaw et al. 2006; Hill et al. 2019). Some New Zealand sambar deer were sourced from an introduced population in New Caledonia where they had reportedly hybridised with C. timorensis (Harris 1971), but this has not been confirmed with genetic data to our knowledge. Although we found no evidence of hybridisation in our microsatellite data (i.e. no strong genetic outliers), it does appear that sambar deer in Australia have hybridised with C. timorensis (Hill et al. 2023, this issue), albeit at a low frequency. In that study, hybrids were evident in population genetic structure analyses and morphologically identified hybrids were genetically intermediate on the PCA plot, including those sampled from areas overlapping with our sampling sites. Despite these concerns, hybrid animals may be advantageous for reintroduction because they carry novel genetic variants. Relevant experimental hybridisation and reintroduction projects are underway (e.g. Chan et al. 2018). The SNPs used in Hill et al. (2023, this issue) would be a useful tool for choosing suitable individuals for reintroduction because they provide suitable markers to identify hybrids.
Another important criterion is the extent to which non-native populations have experienced a reduction in genetic diversity since establishment, potentially leading to inbreeding depression and declining immunocompetence (Armstrong and Seddon 2008). Our estimates of gene diversity were lower in Australian and New Zealand sambar deer than has been estimated in native Indian populations (0.720; Gupta 2014) or across the entirety of the native range (0.786; Martins et al. 2018), by using a similar number of microsatellite markers. Although the difference could be due to different marker sets, a reduction in nuclear genetic diversity and the dearth of mitochondrial variation as seen in our data are consistent with the reported small founding sizes of these introductions. Genetic diversity levels were similar in Australia and New Zealand, suggesting that either of these populations would be an equally suitable choice for reintroduction.
Reduced genetic diversity in a non-native population does not preclude it as a source of animals for reintroduction, if the variation present is higher than that in the population being rescued, or if the introduced animals carry alleles that are rare or absent in the native population. Experimental evidence suggests that even a small number of individuals can be an effective genetic rescue of a population with low genetic diversity (Hufbauer et al. 2015). Additionally, reintroduction from multiple non-native populations with individuals that have different alleles may provide insurance for unpredictable future events experienced by the native population (‘provenancing’; Hoffmann et al. 2021); for example, sambar deer from both Australia and New Zealand could be reintroduced to the native range. Indeed, despite similar levels of genetic diversity for the two invasive sambar deer populations as identified with microsatellite markers, we found substantial genetic differentiation between them, likely resulting from differences in their population of origin. Remarkably, this estimate of differentiation is higher than that found between sambar deer and the congeneric rusa deer in their native range (FST values of ~0.23 vs 0.14; Martins et al. 2018). These differences, along with the private alleles we identified, suggest that sambar deer populations in Australia and New Zealand harbour distinct genetic variants that could contribute to diversity in the native range. However, if the threats (e.g. overhunting, habitat loss) a species experiences in their native range remain, genetic rescue and provenancing may be of little value.
The Australian and New Zealand sambar deer populations that we studied did not exhibit within-country spatial genetic structuring. These findings are consistent with historical knowledge of these populations. By contrast, other studies of sambar deer in Australia have found evidence of population structure (Davies et al. 2022; Hill et al. 2023, this issue). Davies et al. (2022) sampled several small and isolated populations that were not included in our study and whose genetic characteristics differed from the large population we sampled, and Hill et al. (2023, this issue) sampled deer from a wider geographic distribution than in our study. It is difficult to tease apart what drives differences between our results and those of Hill et al. (2023, this issue), because the samples included in our study have little geographic overlap with the boundaries of the four genetic groups identified by Hill et al. (2023, this issue). Also, microsatellite data are likely to be less sensitive to subtle population structure than are the SNP data analysed by Hill et al. (2023, this issue). Further, it is important to note that our samples were sourced primarily from the alpine regions of Victoria and New South Wales, an area that is largely contiguous, whereas samples in Hill et al. (2023, this issue) were taken from areas that are geographically separated or that include peri-urban areas.
The Australian and New Zealand populations were established at a similar time and have expanded their ranges through natural dispersal in continuous habitat (Harris 1971; Bentley 1998; Gormley et al. 2011; Nugent et al. 2021). Home ranges and dispersal behaviours of sambar deer have been little studied in either the native or introduced populations, but male deer typically have larger home ranges and disperse more frequently and further than do female deer (e.g. Catchpole et al. 2004; Kjellander et al. 2004; Loe et al. 2009). For example, in 1971, an adult male sambar deer was photographed near Berrigan, New South Wales, at least 130 km through contiguous habitat from the nearest known females (Bentley 1998). It is therefore likely that long-distance movements of individual male sambar deer contribute to the absence of population structure in this species across large expanses of contiguous habitat. Further, the generation time for sambar deer is 8–10 years (Timmins et al. 2015). The Australian and New Zealand populations both established in about the 1870s (see Introduction), and, hence, ~15–18 generations had elapsed between their introduction and our sampling. Therefore, there has been limited time available for genetic drift and selection to occur (and thus for genetic differentiation to develop; cf. Davies et al. 2022; Hill et al. 2023, this issue), even if there are barriers to dispersal within Australia. It is possible that the population differentiation reported in those studies may be driven by hybridisation with other deer species.
Our results have several implications for managing the increasingly undesirable impacts (see Introduction) of the main Australian sambar deer population located in alpine regions of southern New South Wales and Victoria, and eastern Victoria. First, the high levels of gene flow in this population indicate that localised control is unlikely to eradicate a subpopulation unless immigration can be prevented (Parkes 1990; Bomford and O’Brien 1995; Spencer et al. 2012). One way to prevent immigration into an area in which control is conducted is to erect a barrier fence, as was done during the eradication of feral pigs on Santa Cruz Island (Parkes et al. 2010). However, building and maintaining a sambar deer-proof fence in rugged Australian alpine regions and/or the densely forested terrain of eastern Victoria would likely be logistically and financially prohibitive. Hence, eradicating sambar deer from even a small part of the current main Australian range is not feasible. Second, the absence of genetic population structure with the markers we used means that they are unlikely to be informative with respect to detecting the source(s) of future extra-limital range expansions (reviewed in Signorile et al. 2016). Such range expansions could occur from natural dispersal, deliberate introduction (to establish a hunting resource; ‘translocation’) or escapes from captive herds (Fraser et al. 2000; Moriarty 2004).
To address this inability to distinguish populations by using microsatellite data, approaches such as that used by Hill et al. (2023, this issue) or other next-generation sequencing technologies (e.g. whole-genome sequencing) may increase the power of genetic data to detect subtle differences in population structure. However, it is interesting to note that in Hill et al. (2023, this issue), within the same geographic area we sampled, no sambar deer population structure was identified using SNPs (Hill et al. 2023, this issue, area denoted in green in Fig. 5). Although many emerging studies have reported superior performance of SNPs versus microsatellites in detecting population structure (Sunde et al. 2020), microsatellite datasets such as the one we have presented here remain a useful proxy for SNP datasets (Alvarez et al. 2021). Given this, and considering the higher cost and greater need for computing resources required by SNP data sets, microsatellites remain a powerful tool for non-model systems.
This study has provided useful information about the origin, genetic variation, and population structure of Australian and New Zealand sambar deer. We have discussed the potential for populations such as these to be used for reintroduction in cases where native range populations are threatened. Despite these potential benefits, we are unaware of non-native ungulate populations currently being used as the source of individuals for reintroduction to their native range. We have also outlined how the knowledge we have produced here can be used for the management of this species in its introduced range. Because introductions continue to occur worldwide and species meet the challenges of climate change and shifts in habitat availability, considering how these non-native populations can be used to assist native populations will become more important. Genetic information will continue to be a key tool in conserving native populations and managing the negative impacts of non-native populations.
Data availability
DNA sequences are available in Supplementary Material (Table S4). Microsatellite and associated metadata are available on Dryad (https://doi.org/10.5061/dryad.5mkkwh79v).
Conflicts of interest
David Forsyth was a guest Associate Editor for this special issue. Despite this relationship, he did not at any stage have editor-level access to this manuscript while in peer review, as is the standard practice when handling manuscripts submitted by an editor to this journal. Wildlife Research encourages its editors to publish in the journal and they are kept totally separate from the decision-making process for their manuscripts. The authors have no further conflicts of interest to declare.
Declaration of funding
Funding for this project was supplied by the Department of Economic Development, Jobs, Transport and Resources (State of Victoria; Invasive Animals Research Projects). LAR was supported by a Deakin University Fellowship and a University of New South Wales Scientia Fellowship. AJW was supported by a Deakin University Faculty of Science Engineering and Built Environment Summer Project Prize.
Acknowledgements
We thank Professor William Sherwin for discussions about study design. We thank members of the Deerstalkers Club (Vic.), Drs Jordan Hampton and Mark Richardson, and staff from New South Wales National Parks and Wildlife Service, Parks Victoria, and Melbourne Water for collecting samples from sambar deer in Australia. Comments from the Editor, Associate Editor and two anonymous reviewers improved the paper.
References
Alvarez I, Fernandez I, Traore A, Menendez-Arias NA, Goyache F (2021) Population structure assessed using microsatellite and SNP data: an empirical comparison in West African cattle. Animals (Basel) 11, 151.
| Crossref | Google Scholar |
Armstrong DP, Seddon PJ (2008) Directions in reintroduction biology. Trends in Ecology & Evolution 23, 20-25.
| Crossref | Google Scholar |
Balakrishnan CN, Monfort SL, Gaur A, Singh L, Sorenson MD (2003) Phylogeography and conservation genetics of Eld’s deer (Cervus eldi). Molecular Ecology 12, 1-10.
| Crossref | Google Scholar |
Bandelt HJ, Forster P, Rohl A (1999) Median-joining networks for inferring intraspecific phylogenies. Molecular Biology and Evolution 16, 37-48.
| Crossref | Google Scholar |
Bengsen AJ, Gentle MN, Mitchell JL, Pearson HE, Saunders GR (2014) Impacts and management of wild pigs Sus scrofa in Australia. Mammal Review 44, 135-147.
| Crossref | Google Scholar |
Bennett A, Haydon S, Stevens M, Coulson G (2015) Culling reduces fecal pellet deposition by introduced sambar (Rusa unicolor) in a protected water catchment. Wildlife Society Bulletin 39, 268-275.
| Crossref | Google Scholar |
Bomford M, O’Brien P (1995) Eradication or control for vertebrate pests? Wildlife Society Bulletin 23, 249-255.
| Google Scholar |
Bonizzoni M, Guglielmino CR, Smallridge CJ, Gomulski M, Malacrida AR, Gasperi G (2004) On the origins of medfly invasion and expansion in Australia. Molecular Ecology 13, 3845-3855.
| Crossref | Google Scholar |
Bradshaw CJA, Isagi Y, Kaneko S, Bowman DMJS, Brook BW (2006) Conservation value of non-native banteng in northern Australia. Conservation Biology 20, 1306-1311.
| Crossref | Google Scholar |
Byrne PG, Silla AJ (2020) An experimental test of the genetic consequences of population augmentation in an amphibian. Conservation Science and Practice 2, e194.
| Crossref | Google Scholar |
Catchpole EA, Fan Y, Morgan BJT, Clutton-Brock TH, Coulson T (2004) Sexual dimorphism, survival and dispersal in red deer. Journal of Agricultural, Biological, and Environmental Statistics 9, 1-26.
| Crossref | Google Scholar |
Chan WY, Peplow LM, Menéndez P, Hoffmann AA, Van Oppen MJH (2018) Interspecific hybridization may provide novel opportunities for coral reef restoration. Frontiers in Marine Science 5, 160.
| Crossref | Google Scholar |
Comte S, Thomas E, Bengsen AJ, Bennett A, Davis NE, Brown D, Forsyth DM (2023) Cost-effectiveness of volunteer and contract ground-based shooting of sambar deer in Australia. Wildlife Research
| Crossref | Google Scholar |
Côté SD, Rooney TP, Tremblay J-P, Dussault C, Waller DM (2004) Ecological impacts of deer overabundance. Annual Review of Ecology, Evolution, and Systematics 35, 113-147.
| Crossref | Google Scholar |
Coulon A, Fitzpatrick JW, Bowman R, Stith BM, Makarewich CA, Stenzler LM, Lovette IJ (2008) Congruent population structure inferred from dispersal behaviour and intensive genetic surveys of the threatened Florida scrub-jay (Aphelocoma coerulescens). Molecular Ecology 17, 1685-1701.
| Crossref | Google Scholar |
Cowled BD, Elsworth P, Lapidge SJ (2008) Additional toxins for feral pig (Sus scrofa) control: identifying and testing Achilles’ heels. Wildlife Research 35, 651-662.
| Crossref | Google Scholar |
Davies C, Wright W, Wedrowicz F, Hogan FE (2020) A DNA toolbox for non-invasive genetic studies of sambar deer (Rusa unicolor). Australian Mammalogy 42, 58-66.
| Crossref | Google Scholar |
Davies C, Wright W, Wedrowicz F, Pacioni C, Hogan FE (2022) Delineating genetic management units of sambar deer (Rusa unicolor) in south-eastern Australia, using opportunistic tissue sampling and targeted scat collection. Wildlife Research 49, 147-157.
| Crossref | Google Scholar |
Davis NE, Bennett A, Forsyth DM, Bowman DMJS, Lefroy EC, Wood SW, Woolnough AP, West P, Hampton JO, Johnson CN (2016) A systematic review of the impacts and management of introduced deer (family Cervidae) in Australia. Wildlife Research 43, 515-532.
| Crossref | Google Scholar |
Dlugosch KM, Parker IM (2008) Founding events in species invasions: genetic variation, adaptive evolution, and the role of multiple introductions. Molecular Ecology 17, 431-449.
| Crossref | Google Scholar |
Dunn J (1985) Sambar deer in the Kosciusko National Park. Australian Deer 10, 3-5.
| Google Scholar |
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Molecular Ecology 14, 2611-2620.
| Crossref | Google Scholar |
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131, 479-491.
| Crossref | Google Scholar |
Excoffier L, Laval G, Schneider S (2005) Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evolutionary Bioinformatics Online 1, 47-50.
| Google Scholar |
Fahey B (2017) Tools for managing wild deer: fencing. In ‘2016 National Wild Deer Management Workshop Proceedings’, Adelaide, 17−18 November 2016. (Eds D Forsyth, T Pople, B Page, A Moriarty, D Ramsey, J Parkes, A Wiebkin, C Lane). (Invasive Animals Cooperative Research Centre: Canberra, ACT, Australia)
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164, 1567-1587.
| Crossref | Google Scholar |
Forsyth DM, Davis NE (2011) Diets of non-native deer in Australia estimated by macroscopic versus microhistological rumen analysis. The Journal of Wildlife Management 75, 1488-1497.
| Crossref | Google Scholar |
Forsyth DM, Stamation KA, Woodford L (2015) Distributions of Sambar Deer, Rusa Deer and Sika Deer in Victoria. Arthur Rylah Institute for Environmental Research Unpublished Client Report for the Biosecurity Branch, Department of Economic Development, Jobs, Transport and Resources. Arthur Rylah Institute for Environmental Research, Department of Environment, Land, Water and Planning, Melbourne, Vic., Australia.
Frankham R, Lees K, Montgomery ME, England PR, Lowe EH, Briscoe DA (1999) Do population size bottlenecks reduce evolutionary potential? Animal Conservation 2, 255-260.
| Crossref | Google Scholar |
Frankham R, Ballou JD, Eldridge MDB, Lacy RC, Ralls K, Dudash MR, Fenster CB (2011) Predicting the probability of outbreeding depression. Conservation Biology 25, 465-475.
| Crossref | Google Scholar |
Fraser KW, Cone JM, Whitford EJ (2000) A revision of the established ranges and new populations of 11 introduced ungulate species in New Zealand. Journal of the Royal Society of New Zealand 30, 419-437.
| Crossref | Google Scholar |
Gormley AM, Forsyth DM, Griffioen P, Lindeman M, Ramsey DSL, Scroggie MP, Woodford L (2011) Using presence-only and presence–absence data to estimate the current and potential distributions of established invasive species. Journal of Applied Ecology 48, 25-34.
| Crossref | Google Scholar |
Goudet J (2002) FSTAT, a program to estimate and test gene diversities and fixation indices version 2.9.3.2. Available from http://www2.unil.ch/popgen/softwares/fstat.htm [updated from Goudet (1995)])
Gupta SK, Kumar A, Gaur A, Hussain SA (2015) Detection of 40 bp insertion-deletion (INDEL) in mitochondrial control region among sambar (Rusa unicolor) populations in India. BMC Research Notes 8, 581.
| Crossref | Google Scholar |
Hampton JO, Spencer PBS, Alpers DL, Twigg LE, Woolnough AP, Doust J, Higgs T, Pluske J (2004) Molecular techniques, wildlife management and the importance of genetic population structure and dispersal: a case study with feral pigs. Journal of Applied Ecology 41, 735-743.
| Crossref | Google Scholar |
Harris LH (1971) Notes on the introduction and history of sambar deer in New Zealand. New Zealand Wildlife 35, 33-42.
| Google Scholar |
Hill E, Linacre A, Toop S, Murphy N, Strugnell J (2019) Widespread hybridization in the introduced hog deer population of Victoria, Australia, and its implications for conservation. Ecology and Evolution 9, 10828-10842.
| Crossref | Google Scholar |
Hill E, Murphy N, Li-Williams S, Davies C, Forsyth DM, Comte S, Rollins LA, Hogan F, Wedrowicz F, Crittle T, Thomas E, Woodford L, Pacioni C (2023) Hybridisation rates, population structure, and dispersal of sambar deer (Cervus unicolor) and rusa deer (Cervus timorensis) in south-eastern Australia. Wildlife Research In press.
| Google Scholar |
Hoffmann AA, Miller AD, Weeks AR (2021) Genetic mixing for population management: From genetic rescue to provenancing. Evolutionary Applications 14, 634-652.
| Crossref | Google Scholar |
Hufbauer RA, Szűcs M, Kasyon E, Youngberg C, Koontz MJ, Richards C, Tuff T, Melbourne BA (2015) Three types of rescue can avert extinction in a changing environment. Proceedings of the National Academy of Sciences 112, 10557-10562.
| Google Scholar |
Kjellander P, Hewison AJM, Liberg O, Angibault J-M, Bideau E, Cargnelutti B (2004) Experimental evidence for density-dependence of home-range size in roe deer (Capreolus capreolus L.): a comparison of two long-term studies. Oecologia 139, 478-485.
| Crossref | Google Scholar |
Kolbe JJ, Glor RE, Rodriguez Schettino L, Lara AC, Larson A, Losos JB (2004) Genetic variation increases during biological invasion by a Cuban lizard. Nature 431, 177-181.
| Crossref | Google Scholar |
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947-2948.
| Crossref | Google Scholar |
Latham ADM, Warburton B, Byrom AE, Pech RP (2017) The ecology and management of mammal invasions in forests. Biological Invasions 19, 3121-3139.
| Crossref | Google Scholar |
Lee CE (2002) Evolutionary genetics of invasive species. Trends in Ecology & Evolution 17, 386-391.
| Crossref | Google Scholar |
Leslie DM, Jr (2011) Rusa unicolor (Artiodactyla: Cervidae). Mammalian Species 43, 1-30.
| Crossref | Google Scholar |
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451-1452.
| Crossref | Google Scholar |
Loe LE, Mysterud A, Veiberg V, Langvatn R (2009) Negative density-dependent emigration of males in an increasing red deer population. Proceedings of the Royal Society of London B: Biological Sciences 276, 2581-2597.
| Crossref | Google Scholar |
Longmire JL, Lewis AK, Brown NC, Buckingham JM, Clark LM, Jones MD, Meincke LJ, Meyne J, Ratliff RL, Ray FA, Wagner RP, Moyzis RK (1988) Isolation and molecular characterization of a highly polymorphic centromeric tandem repeat in the family falconidae. Genomics 2, 14-24.
| Crossref | Google Scholar |
Martins RF, Schmidt A, Lenz D, Wilting A, Fickel J (2018) Human-mediated introduction of introgressed deer across Wallace’s line: historical biogeography of Rusa unicolor and R. timorensis. Ecology and Evolution 8, 1465-1479.
| Crossref | Google Scholar |
Moloney PD, Gormley AM, Toop SD, Flesch JS, Forsyth DM, Ramsey DSL, Hampton JO (2022) Bayesian modelling reveals differences in long-term trends in the harvest of native and introduced species by recreational hunters in Australia. Wildlife Research 49, 673-685.
| Crossref | Google Scholar |
Moriarty A (2004) The liberation, distribution, abundance and management of wild deer in Australia. Wildlife Research 31, 291-299.
| Crossref | Google Scholar |
Nei M (1972) Genetic distance between populations. The American Naturalist 106, 283-292.
| Crossref | Google Scholar |
Neilan BA, Wilton AN, Jacobs D (1997) A universal procedure for primer labelling of amplicons. Nucleic Acids Research 25, 2938-2939.
| Crossref | Google Scholar |
Parkes JP (1990) Eradication of feral goats on islands and habitat islands. Journal of the Royal Society of New Zealand 20, 297-304.
| Crossref | Google Scholar |
Parkes JP, Ramsey DSL, Macdonald N, Walker K, McKnight S, Cohen BS, Morrison SA (2010) Rapid eradication of feral pigs (Sus scrofa) from Santa Cruz Island, California. Biological Conservation 143, 634-641.
| Crossref | Google Scholar |
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Molecular Ecology Notes 6, 288-295.
| Crossref | Google Scholar |
Pritchard JK, Wen X, Falush D (2007) Documentation for structure software: version 2.2. Available at https://web.stanford.edu/group/pritchardlab/structure.html
Randi E, Mucci N, Claro-Hergueta F, Bonnet A, Douzery EJP (2001) A mitochondrial DNA control region phylogeny of the Cervinae: speciation in Cervus and implications for conservation. Animal Conservation 4, 1-11.
| Crossref | Google Scholar |
Ripple WJ, Newsome TM, Wolf C, Dirzo R, Everatt KT, Galetti M, Hayward MW, Kerley GIH, Levi T, Lindsey PA, Macdonald DW, Malhi Y, Painter LE, Sandom CJ, Terborgh J, Van Valkenburgh B (2015) Collapse of the world’s largest herbivores. Science Advances 1, e1400103.
| Crossref | Google Scholar |
Robertson BC, Gemmell NJ (2004) Defining eradication units to control invasive pests. Journal of Applied Ecology 41, 1042-1048.
| Crossref | Google Scholar |
Rollins LA, Woolnough AP, Sherwin WB (2006) Population genetic tools for pest management: a review. Wildlife Research 33, 251-261.
| Crossref | Google Scholar |
Rollins LA, Woolnough AP, Wilton AN, Sinclair R, Sherwin WB (2009) Invasive species can’t cover their tracks: using microsatellites to assist management of starling (Sturnus vulgaris) populations in Western Australia. Molecular Ecology 18, 1560-1573.
| Crossref | Google Scholar |
Rousset F (2008) GENEPOP’007: a complete re-implementation of the GENEPOP software for Windows and Linux. Molecular Ecology Resources 8, 103-106.
| Crossref | Google Scholar |
Seutin G, White BN, Boag PT (1991) Preservation of avian blood and tissue samples for DNA analyses. Canadian Journal of Zoology 69, 82-90.
| Crossref | Google Scholar |
Signorile AL, Reuman DC, Lurz PWW, Bertolino S, Carbone C, Wang J (2016) Using DNA profiling to investigate human-mediated translocations of an invasive species. Biological Conservation 195, 97-105.
| Crossref | Google Scholar |
Spencer PBS, Giustiniano D, Hampton JO, Gee P, Burrows N, Rose K, Martin GR, Woolnough AP (2012) Identification and management of a single large population of wild dromedary camels. The Journal of Wildlife Management 76, 1254-1263.
| Crossref | Google Scholar |
Sunde J, Yilderim Y, Tibblin P, Forsman A (2020) Comparing the performance of microsatellites and RADseq in population genetic studies: analysis of data for pike (Esox lucius) and a synthesis of previous studies. Frontiers in Genetics 11, 218.
| Crossref | Google Scholar |
Timmins R, Kawanishi K, Giman B, Lynam A, Chan B, Steinmetz R, Sagar Baral H, Samba Kumar N (2015) Rusa unicolor (errata version published in 2015). The IUCN Red List of Threatened Species 2015: e.T41790A85628124. Available at http://dx.doi.org/10.2305/IUCN.UK.2015-2.RLTS.T41790A22156247.en
Tsutsui ND, Suarez AV, Holway DA, Case TJ (2000) Reduced genetic variation and the success of an invasive species. Proceedings of the National Academy of Sciences 97, 5948-5953.
| Crossref | Google Scholar |
Wallach AD, Ripple WJ, Carroll SP (2015) Novel trophic cascades: apex predators enable coexistence. Trends in Ecology & Evolution 30, 146-153.
| Crossref | Google Scholar |