
Advances in legume research in the genomics era
Ashley N. Egan A D and Mohammad Vatanparast B C
A D and Mohammad Vatanparast B C
A Department of Bioscience, Aarhus University, Ny Munkegade 116, DK-8000 Aarhus C, Denmark.
B Department of Geosciences and Natural Resource Management, University of Copenhagen, DK- 1958 Frederiksberg C, Denmark.
C Department of Botany, National Museum of Natural History, Smithsonian Institution, Washington, DC 20560, USA.
D Present address, Department of Biology, Utah Valley University, 800 W University Parkway, Orem, UT 84058, USA. Corresponding author. Email: aegan@uvu.edu
Australian Systematic Botany 32(6) 459-483 https://doi.org/10.1071/SB19019
Submitted: 4 March 2019 Accepted: 16 July 2019 Published: 30 September 2019
Journal Compilation © CSIRO 2019 Open Access CC BY-NC-ND
Abstract
Next-generation sequencing (NGS) technologies and applications have enabled numerous critical advances in legume biology, from marker discovery to whole-genome sequencing, and will provide many new avenues for legume research in the future. The past 6 years in particular have seen revolutionary advances in legume science because of the use of high-throughput sequencing, including the development of numerous types of markers and data useful for evolutionary studies above and below the species level that have enabled resolution of relationships that were previously unattainable. Such resolution, in turn, affords opportunities for hypothesis testing and inference to improve our understanding of legume biodiversity and the patterns and processes that have created one of the most diverse plant families on earth. In addition, the genomics era has seen significant advances in our understanding of the ecology of legumes, including their role as nitrogen fixers in global ecosystems. The accumulation of genetic and genomic data in the form of sequenced genomes and gene-expression profiles made possible through NGS platforms has also vastly affected plant-breeding and conservation efforts. Here, we summarise the knowledge gains enabled by NGS methods in legume biology from the perspectives of evolution, ecology, and development of genetic and genomic resources.
Additional keywords: crop genomes, Fabaceae, genome-wide research, Leguminosae, next-generation sequencing, phylogenomics, RADseq, sequence capture, target enrichment.
Introduction
In the late 20th century, Sanger sequencing (Sanger et al. 1977) transformed biology and medicine, enabling many genetic advances, the greatest being completion of the human genome project (International Human Genome Sequencing Consortium 2001). Three decades later, second- or next-generation sequencing (NGS) ushered in the genomics era, producing massive amounts of sequence data at a fraction of the cost and time. For comparison, the cost of sequencing a human genome in September of 2001 by Sanger sequencing was ~US$95 million; today sequencing the same genome will cost less than US$1500 (The Human Genome Research Institute; see https://www.genome.gov/sequencingcosts/, accessed 28 August 2019).
Numerous NGS methods have been introduced, each with strengths and weaknesses (for review, see Egan et al. 2012; Soltis et al. 2013; Reuter et al. 2015). NGS technologies provide unprecedented opportunities in fields such as crop genomics, molecular systematics, evolutionary genomics or plant breeding, prompting scientific understanding across the tree of life. In particular, plant biology has blossomed through NGS applications, including transcriptomics (e.g. Matasci et al. 2014; Wen et al. 2015), phylogenomics (Ruhfel et al. 2014; Wickett et al. 2014; Soltis et al. 2018), genome-wide single-nucleotide polymorphism (SNP) sequencing by genome-reduction techniques (Andrews et al. 2016; Jiang et al. 2016) and whole-genome sequencing (Zhang et al. 2011; Koboldt et al. 2013).
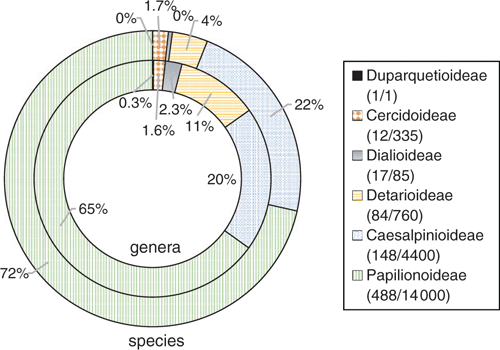
Leguminosae (Fabaceae) is the third-largest family of flowering plants after Orchidaceae and Asteraceae (Lewis et al. 2005), comprising ~770 genera and ~19 500 species (Legume Phylogeny Working Group 2013a, 2017). Our understanding of the classification and evolutionary relationships within the family has been transformed in recent years, with the impact of NGS methods as summarised by Doyle (2013). The family was recently reclassified from the classical three subfamilies (Caesalpinioideae, Mimosoideae, Papilionoideae) into six subfamilies, corresponding to the six main clades, on the basis of an international effort involving ~100 scientists, using molecular systematics (Fig. 1; Legume Phylogeny Working Group 2013b, 2017). Fabaceae is distributed in all of the world’s vegetation types (biomes) except polar ice. Legumes are second only to grasses in economic importance, with uses common to nearly all facets of life, including food, medicine, oils, timber, fibres, industry, fodder, soil stabilisation and soil enrichment (Graham and Vance 2003). Legume research has benefited from NGS innovations, with studies employing genomic sequencing techniques to address questions in evolution, ecology, conservation and plant breeding. The sequencing of legume genomes, both nuclear and plastid, along with transcriptomic and other genomic profiles, has greatly improved genetic and genomic data resources, providing foundations on which researchers can build. This review is wide in scope, both in terms of genomic methods, as well as their impacts on legume subdisciplines, from systematics to plant breeding. This review expands on that of Doyle (2013), focusing especially on work published during the intervening 6 years. We summarise recent insights across selected topics and review ongoing research regarding NGS technologies and how they are contributing to our understanding of legumes.
|
Evolutionary aspects
Given the incredible diversity and vast ecological and economic importance of legumes, understanding the patterns and processes underlying the evolution of the legume family is an important endeavour. Robust inferences of phylogenetic trees are fundamental to any subsequent analyses, above or below the species level. In this section, we summarise knowledge gains in legume evolutionary biology afforded by genomic advances.
Phylogenetics v. Phylogenomics
Investigation of evolutionary relationships among plants by using molecular data began in the late 1980s. Pioneering legume studies included size polymorphism of chloroplast DNA in Pisum L. (Palmer et al. 1985) and nuclear rDNA (rDNA) repeat-length and restriction-enzyme site locations in soybean and relatives (Doyle and Beachy 1985). Sanger sequencing revolutionised phylogenetics through ease of use and reproducibility (Sanger et al. 1977). The chloroplast gene matK is the most comprehensively sequenced phylogenetic marker in legumes. Wojciechowski et al. (2004) produced one of the earliest generic-level phylogenies of legumes by using 330 matK gene sequences, demonstrating monophyly of Papilionoideae and resolving multiple papilionoid subclades. Many subsequent studies have used matK (e.g. Bruneau et al. 2008; Simon et al. 2009; Stefanović et al. 2009; Cardoso et al. 2012; de Queiroz et al. 2015; Egan et al. 2016; Snak et al. 2016), culminating in the Legume Phylogeny Working Group (2017) phylogeny that included 3842 matK sequences representing 3696 species (~20% of the family) and 698 of the 765 genera that had been recognised. Whereas matK robustly supported each of the newly recognised six subfamilies, basal nodes key to understanding subfamilial relationships and early legume evolution remained unresolved, which was likely because of the lack of resolving power available from a single marker.
The use of nuclear markers for legume phylogenetics has lagged behind chloroplast markers, largely because of the bi-parental inheritance and the high incidence of gene duplications in the nuclear genome, which make orthology assessment and primer design difficult (Zimmer and Wen 2013). The advent of genomics has transformed and facilitated nuclear marker discovery (Zimmer and Wen 2015). For example, Scherson et al. (2005) screened several nuclear loci from the Medicago truncatula Gaertn. genome for phylogeny reconstruction in the hyper-diverse legume genus Astragalus L. Similarly, Choi et al. (2006) tested 274 putative single-copy genes garnered from comparative analysis of 15 legume genomes from six species and identified 129 single-copy loci that were tested across 95 legume species.
As helpful as NGS methods are for improving nuclear-marker discovery, nuclear loci that are putatively single-copy in one lineage may not be so in others, a phenomenon that makes finding the ‘silver bullet’ of nuclear markers difficult in plants (e.g. Manzanilla and Bruneau 2012), and which argues for having many different markers to mitigate issues with a few. This is where the true utility of NGS technologies comes in. The move from single amplicon-based Sanger sequencing to NGS-based, simultaneous sequencing of numerous markers is rapidly transforming our understanding of legume evolution (Doyle 2013), not least through development of a variety of new approaches for NGS marker data for phylogenetic and population-genetic studies. These include microsatellites, RNAseq or transcriptomics (Wang et al. 2009; Wen et al. 2015), restriction site-associated DNA tags (RADseq; Miller et al. 2007), genotyping-by-sequencing (GBS; Elshire et al. 2011), genome skimming (Straub et al. 2012), targeted enrichment (also known as sequence capture or hybrid enrichment; Gnirke et al. 2009), simultaneous amplicon sequencing (Bybee et al. 2011), and whole-genome sequencing (e.g. Stein et al. 2018; Grover et al. 2019).
Studies have capitalised on massively parallel sequencing for discovery and optimisation of microsatellite markers for population studies, for example, using transcriptome sequencing, including in legumes (e.g. Chapman 2015; Vatanparast et al. 2016; Sathyanarayana et al. 2017; Haynsen et al. 2018). Other popular NGS methods for population genetics include RADseq and GBS, which are also employed in phylogenetics below the genus level. For instance, Wong et al. (2015) used GBS across 60 accessions of seven species of Lens and produced a phylogeny in which all seven species were reciprocally monophyletic. Grillo et al. (2016) employed RADseq across 191 accessions of Medicago truncatula in Europe to investigate population structure and screen for candidate symbiosis genes. They found evidence that suggests that one gene, DMI1, is under adaptive selection. Their work detailed five distinct genetic clusters and aided in correct identification of species. That said, work by others has found that different parameter perturbations produced significant differences in phylogenetic networks of species relationships within Medicago by using RADseq (Blanco-Pastor et al. 2018), suggesting caution when using population-level markers above species level.
RNAseq is often used as the first line for phylogenetic-marker development, whether for microsatellite development, amplicon sequencing (e.g. Chapman 2015) or targeted enrichment. For example, Vatanparast et al. (2018) used 30 transcriptomes across the legume family to select over 500 nuclear markers for targeted enrichment, which were tested across 25 legume taxa. This same target set proved useful as far out as Rosales (M. Vatanparast and A.N. Egan, unpubl. data), showing that targeted enrichment can be useful for both lower- and higher-level phylogenomics, as has been found by others (Kadlec et al. 2017; Chau et al. 2018).
The first targeted enrichment-gene set in legumes selected 50 nuclear loci from the Medicago genome to resolve relationships among six Medicago species and other genera of tribe Trifolieae (de Sousa et al. 2014). Targeted enrichment may be especially useful for resolving relationships among rapidly radiated, species-rich groups by virtue of the large amount of data produced. For example, Nicholls et al. (2015) applied a targeted enrichment method using transcriptomes from three species to isolate 264 nuclear loci for sequencing Inga Mill., a genus of ~300 neotropical rainforest trees that diversified rapidly during the late Miocene (2–10 million years ago). Of these loci, 194 were used for phylogeny reconstruction across 22 Inga species, resulting in a highly resolved phylogenetic tree. Similarly, Ojeda et al. (2019) used 289 nuclear loci identified from transcriptomes across Detarioideae to construct a phylogenomic hypothesis of relationships within the Anthonotha clade, discovering an overall general trend towards petal reduction in this florally diverse group. In addition to the target-gene sets outlined here, several other, as yet unpublished, gene sets have been generated for legumes.
As legume researchers adopt NGS for phylogenomics, it could be worth considering selection and adoption of a core set of nuclear target genes for use across legumes, along lines similar to the 353 nuclear-gene set for targeted enrichment in angiosperms (Johnson et al. 2019). Doing so would also facilitate barcoding efforts (Hollingsworth et al. 2016). With the recent addition of the first genome sequences for mimosoids and cercidoids (Table 1, Fig. 2), plus a significant number of new transcriptomes, it would now be possible to design a legume-wide, legume-specific bait set more efficiently. Such a design might include genes specifically selected for phylogenetics, as well as genes that are related to particular legume functional traits such as nodulation, compound-leaf development or floral symmetry, as has been done for Caryophyllales (Moore et al. 2018). Whereas the number of target gene sets used in legumes will undoubtedly increase, including a core subset of genes in every target-gene set would enable published sequences from different studies to be combined for wider analyses. Doing so would foster wide collaboration among legume systematists, while still enabling project-specific objectives to be met. That said, issues with the conflation of orthology and paralogy need to be dealt with when using universal gene sets because of whole-genome duplications (WGDs) and differential gene birth and death events, issues that are particularly troubling if targets are used for DNA barcoding.

|

|
Species diversity
Understanding the dynamics of diversification within the third-largest species-rich plant family is complex and only a few attempts have been made to estimate species diversification and speciation–extinction rates within legumes (e.g. Sanderson and Wojciechowski 1996; Richardson et al. 2001; Scherson et al. 2008). Koenen et al. (2013) estimated species diversification rates and tested for rate shifts by using species-level, time-calibrated phylogenies of the following four legume clades: Calliandra Benth., Indigofereae, Lupinus L. and Mimosa L., finding evidence for significant among-lineage diversification-rate variation. A major challenge for diversification studies is having a well resolved phylogeny, a non-trivial task when dealing with rapid, recent radiations (Hughes et al. 2015). The most species-rich legume tribe, Galegeae, includes Astragalus, the largest genus of any biological group, with nearly 3000 species (Kazempour Osaloo et al. 2003; Podlech et al. 2014), as well as Oxytropis DC., its sister genus with between 310 and 450 species (Malyshev 2008). Recent molecular studies of these groups exemplify the difficulties of resolving phylogenies for rapidly radiating lineages (e.g. Azani et al. 2017; Bagheri et al. 2017). Where phylogenies based on data from single or a few genes lack resolution, genomic-scale data may provide a solution. Application of anchored enrichment of 527 gene regions (i.e. targeted enrichment) in Oxytropis produced a robust phylogeny compared to relying on conventional markers (Shahi Shavvon et al. 2017).
Next-generation sequencing data provide more than just phylogenetic resolution and their application to elucidating adaptive radiations and diversification is just beginning. This is exemplified in ground-breaking studies on Lupinus, a genus of ~280 species, with a series of nested and parallel rapid radiations in North and South America (Hughes and Eastwood 2006) where the genus exhibits startling diversity in growth form and habitat in the Andes, a radiation that is likely to have been spurred by Pleistocene glacial cycles (Nevado et al. 2018). Furthermore, lineage-specific diversification rates were detected across the phylogeny, but the ability to ascertain the processes underpinning rapid species radiations remained elusive. Today, investigating adaptive radiations from a genomic perspective is shedding light on how speciation and trait diversification occur. Contreras-Ortiz et al. (2018) used RADseq to investigate species diversification in Andean lupines and found evidence for both adaptive, ecological and non-adaptive, geographical drivers influencing their radiation. Using transcriptomes from slowly and rapidly diversifying lupin lineages, Nevado et al. (2016) verified, for the first time in plants, the role of adaptive evolution in rapid radiations. Rapidly diversifying lineages had two to three times more positively selected genes than did slowly diversifying lineages, suggesting a genome-wide response to adaptation. Further, the rapidly diversifying Andean lineage exhibited a higher gene-expression divergence than did the slowly diversifying lineages, suggesting that underlying genomic shifts in expression happened during adaptive radiations. Also, shifts in gene-expression level were non-randomly clustered around significant evolutionary time points, including at the base of the Andean clade when lupins moved into novel, extreme montane environments, and near a branch signifying a shift from annual to perennial life-history. NGS has gone beyond simply detecting shifts in diversification rates, by enabling the determination of the how and why behind diversification in plants.
Polyploidy
Whole-genome duplication (WGD), or polyploidy, is a major evolutionary process underlying speciation in plants (Van de Peer et al. 2017), and legumes are no exception (Doyle 2012). Polyploid crop legumes, such as peanut, alfalfa and soybean are well known and, in soybean (Glycine max [L.] Merr.), studies have confirmed genomic evidence for both ancient and recent polyploidisation events just before the radiation of the genus c. 10–12 million years ago, and between 40 and 66 million years ago (Egan and Doyle 2010). Legume WGDs were further characterised by Cannon et al. (2015), by using transcriptomic and genomic data from 20 diverse legumes and 17 outgroups to determine that WGDs coincide with the origins of major legume lineages. A follow-up study incorporating genome data for the genus Cercis L. (Cercidoideae), suggested that Cercis may represent the only extant legume lineage lacking a polyploid history, providing a plausible hypothesis of what the ancestral legume genome looked like (Stai et al. 2019). In contrast, Koenen et al. (2019) used thousands of nuclear genes and 72 protein-coding chloroplast genes to find evidence for WGDs at the stem of all legumes, as well as nested WGDs subtending radiation of subfamilies Papilionoideae and Detarioideae. Koenen et al. (2019) also described difficulties in resolving the initial divergence of the legume family tree and suggested that polyploidy may play a key role in the lack of support for deep-branching relationships in the family. Further improvements in long-read NGS platforms may solve such recalcitrant nodes and facilitate research into polyploidy and its ramifications.
Genome sequencing also enables us to detect and characterise more recent WGDs. Allopolyploidy within Glycine has been shown to be rampant and complex, but genomic data have helped unravel relationships within the genus by using transcriptomics (Bombarely et al. 2014) and GBS (Sherman‐Broyles et al. 2017). Similarly, comparison of rDNA, genomic and fluorescence in situ hybridisation data and whole plastome sequences generated by Illumina-based sequencing of Stylosanthes scabra Vogel, an important forage legume, and its hypothesised genome donors S. hamata (L.) Taub. or S. seabrana B.L.Maass & ‘t Mannetje (A genome) and S. viscosa (L.) Sw. (B genome), have provided evidence for an allopolyloid origin of S. scabra and showed the genomic impacts of subsequent homogenisation following ‘genomic shock’ (Marques et al. 2018). Capitalising on a target-gene set derived from the Medicago truncatula genome (de Sousa et al. 2014), Eriksson et al. (2017) tested the hypothesis that M. prostrata is a homoploid hybrid, while accounting for the impact and signature of introgression from M. sativa as a contributor of genetic variation therein. Eriksson et al. (2018) characterised the allopolyploid origin of two Medicago species and showed the importance of allele phasing. Using NGS methods to investigate the mechanisms of polyploidisation can resolve duplicated regions by phasing and comparative analyses.
Ecological aspects
Quantification and integration of information on species, genetic, population and ecosystem diversity from NGS-based metagenomic methods can contribute to biodiversity and conservation assessment and understanding of evolutionary history, population processes, community assembly, and ecosystem equilibrium and services (Papadopoulou et al. 2015; de la Harpe et al. 2017). As primary mediators of nitrogen fixation, legumes are an integral source of fixed nitrogen within terrestrial communities from grasslands to forests. Here, we review some of the recent advances obtained through genomic studies within ecological arenas.
Forestry and range management
From providing essential ecosystem and natural resources to sustaining natural and human infrastructures, legumes are fundamental and abundant components of biomes across the globe, from desert sands to towering trees of the Amazon rainforests and temperate grasslands where legume biodiversity is directly related to the overall biological diversity and community health. Metagenomic comparison of above- and below-ground plant-species richness in a grassland employed Sanger sequencing and Roche 454 pyrosequencing to illustrate that below-ground diversity was higher, demonstrating the power of NGS methods to detect dormant plant diversity (Hiiesalu et al. 2012). Assessments of the portion of biodiversity that legumes represent in grasslands, and perhaps other ecosystems, through metagenomic barcoding may help in maintaining and managing biodiversity in ecosystems, be they native or range managed, particularly as loss of legume biodiversity is directly linked to a decline in the nitrogen budget and the overall health and biodiversity of plant communities (Spehn et al. 2002).
Legumes are particularly prevalent in Neotropical forests and savannas, where plot data suggest that they make up more than 11% of species (Oliveira-Filho et al. 2013; Yahara et al. 2013). With increasing pressures on forest-ecosystem dynamics, including fragmentation, over-logging of particular species, and wholesale deforestation, understanding how to mitigate the effects of such events and their toll on ecosystem health, as well as the roles that legumes play therein, are urgent needs. Genomic methods are poised to help (Neale and Kremer 2011). Creation of genomic-marker sets is an important starting point. For example, RADseq has been used to generate 330 SNPs for population genetic analyses in Robinia pseudoacacia L., an economcially important eastern North American tree widely cultivated and invasive in Europe and elsewhere (Verdu et al. 2016); employment of such markers may help forest managers capitalise on its use and limit its dynamic spread. Similarly, several studies have generated SNP marker sets for Dipteryx Schreb., an economically important genus of Neotropical canopy trees, various species of which are threatened. These include development of microsatellite markers (Soares et al. 2012), a nuclear and plastid SNP MassArray panel (Honorio Coronado et al. 2019), and a draft genome (Jimenez-Madrigal 2018), providing useful genetic data resources for management and conservation through detailed GD assessment. Gailing et al. (2017) used RADseq to produce a framework genetic-linkage map for Gleditsia triacanthos L., a common North American hardwood forest tree, providing an important genetic resource for future quantitative trait-locus mapping.
Landscape genomic data coupled with an assessment of GD can promote sound forest and range management, quantify the impacts of deforestation, fragmentation, climate change and habitat restoration. For example, Acacia koa A.Gray, a Hawaiian endemic and one of two dominant canopy hardwood tree species in Hawaiian forests, is under increasing pressures from logging and changing climate. Gugger et al. (2018) used GBS to assess GD across 311 Acacia koa trees sampled over its geographical, elevational and climatic range, and found evidence for genetic differentiation among islands and a strong association between genetic structure and the mean annual rainfall. These results suggest that changing rainfall patterns could cause a genetic offset between adaptation and extant populations, placing future survival of this species at risk. Such knowledge can be used for future management planning. Similarly, Grando (2015) assessed GD across native and restored populations of Piptadenia gonoacantha (Mart.) J.F.Macbr., a species that is often used in reforestation across the Brazilian Atlantic Forest because of its rapid growth and regeneration. These data provided insights into which native populations should be used as seed sources for restoration efforts and determined that GD was similar across native and restored sites, which is evidence of successful capture and maintenance of biodiversity during restoration efforts. As NGS becomes more affordable for high sample numbers, landscape genomics and GD assessment using such data can ensure sound scientific foundations for natural-resource management and biodiversity conservation in the face of global change.
Conservation biology and genetics
As one of the largest plant families and key components of tropical and temperate forest ecosystems, legumes are the focus of global legume-diversity assessment (GLDA; Yahara et al. 2013) to quantify biodiversity and species loss stemming from rapid deforestation taking place across South-East Asia. Within the GLDA, biodiversity assessments of rosewoods (Dalbergia spp.; Vatanparast et al. 2013), Bauhinia L., Mucuna Adans. (Moura et al. 2016) and Desmodium Desv. have been prioritised. These ongoing studies are incorporating species-distribution modelling with biodiversity metrics. To assess conservation and biodiversity metrics correctly, a complete time-calibrated phylogenetic tree of a target group is required. However, for many lineages or communities, a complete phylogeny at the species level is not available. Combining biodiversity metrics with enhanced NGS-based phylogenies can enable greater understanding of the contribution of legumes to the overall biodiversity and aid in conservation efforts (for a review on biodiversity metrics, see Kellar et al. 2015). Similarly, Ahrendsen et al. (2016) used Illumina shotgun sequencing of rosid species in a Nebraska grassland to isolate ~80 plastid genes for 45 species, 22 of which were legumes. This enabled the reconstruction of a plastid phylogeny for a complete community for divergence dating and estimation of conservation metrics, illustrating the potential of genomics methods for conservation-biology research.
Conservation often involves efforts to characterise genetic diversity within vulnerable or endangered species by using microsatellite markers. Genomic sequencing has revolutionised discovery of such markers (Zalapa et al. 2012), whether species-specific (Abdelkrim et al. 2009) or for use across a broader taxonomic group (Hodel et al. 2016), including for several legumes (e.g. Borges et al. 2015; Morris et al. 2016). For example, microsatellite markers developed from the Lotus japonicus (Regel) K.Larsen genome (Sato et al. 2008) were used through cross-species amplification in Lotus sessilifolius DC., a species endemic to Macaronesia (Yang et al. 2018). To determine which populations should be prioritised for conservation management, eight populations across four islands were assessed for their GD across 11 microsatellite markers, highlighting a population from Tejina-Milán as strongly distinct and of low genetic diversity relative to the others, suggesting this as one population to target. Next-generation sequencing platforms coupled with amplicon sequencing can now be used to obtain microsatellite data en masse (Zhan et al. 2017).
Although microsatellites are a time-tested and effective tool for assessing GD, issues of small sample sizes and low numbers of markers limit their power for assessing population structure and dynamics. Reduced representation techniques such as GBS or RADseq, or targeted enrichment and amplicon-sequencing methods linked to RADseq and GBS (e.g. GTseq and Rapture), offer cost-effective methods for generating orders of magnitude more marker sites (thousands to millions of SNPs) than do microsatellites (Meek and Larson 2019) for assessing GD and establishing population and species relationships. For example, Harrison et al. (2019) garnered thousands of SNPs by GBS to compare genetic variation among infraspecific taxa of the Astragalus lentiginosus Hook. species complex including var. piscinensis Barneby, that inhabits just 8 km2. They showed that, in spite of rarity, significant genetic diversity and population structure exists within and among varieties. Harrison et al. (2019) exemplified the power of NGS to produce large numbers of SNPs for population-genomic and conservation studies.
Invasion biology
The numerous markers generated by reduced-representation genomic methods can also resolve recent population divergences such as those arising from anthropogenic plant invasions (Chown et al. 2015) and help understand whether pre-adaption to the novel environment or rapid adaptive changes account for invasions. For example, Helliwell et al. (2018) used 9658 SNPs genotyped across 446 accessions of Medicago polymorpha L. within its native European and introduced New World ranges. They showed that latitudinal variation in phenology that facilitated invasion resulted from rapid evolutionary adaptation across this clinal gradient following a single introduction and subsequent range expansion. Similarly, M. S. Haynsen and A. N. Egan (unpubl. data; A. N. Egan, pers. comm.) genotyped ~600 loci by using GBS over 600 individuals of kudzu (Pueraria montana (Lour.) Merr. var. lobata (Willd.) Maesen & Almeida ex Sanjappa & Pradeep), a notorious invasive vine that has now spread over half of the USA, to detail its introduction history.
Understanding the evolutionary mechanisms behind invasiveness and tracing patterns of introduction history are important for managing invasive species. Alternatively, investigating the reasons behind dieback within an invasive species inside its introduced range may also aid management. For example, Steinrucken (2017) used Illumina metagenomic sequencing of fungal and bacterial soil and plant communities within healthy and diseased populations in the native and introduced ranges of Parkinsonia aculeata L., an invasive caesalpinioid legume tree introduced from Venezuela to Australia, to determine that fungal endophytes, not bacterial ones, were likely to be responsible for dieback in the invasive range; this knowledge may prove useful for biological control of P. aculeata in Australia. Studies that integrate gene-expression profiling with population-level sampling will be able to truly determine how invasiveness arises, knowledge that we as yet lack at the genomic level.
Nitrogen fixation
Legumes provide numerous inputs to agricultural and natural ecosystems, with perhaps the most important being soil enrichment by nitrogen fixation. The majority of legume species have the ability to form symbioses with rhizobial bacteria that transform atmospheric nitrogen to ammonia, making atmospheric nitrogen bioavailable in the soil as amino acids and other cellular constituents. Metagenomic approaches are commonly used to characterise soil microbiomes (Andújar et al. 2015), advancing understanding of the interactions of legumes and the environment through nodulation and nitrogen fixation (e.g. Afkhami and Stinchcombe 2016). Birnbaum et al. (2018) investigated symbionts of Acacia rostellifera Benth. across the natural-soil fertility gradient of the Jurien Bay Dune chronosequence. Using Illumina-based nif metabarcoding, they delineated which species of Rhizobiaceae inhabited nodules and how the composition of nodule symbionts changed with soil fertility. They noted that the older soils with the lowest soil phosphorus had more unclassified operational taxonomic units, suggesting a shift to a unique set of nitrogen-fixing bacteria more adapted to limited soil fertility. Associations between legume species and their root-nodule symbionts can be both generalist and specific, but our understanding of how specificity is controlled remains fragmentary. Keller et al. (2018) attempted to delineate the hows and whys of host specificity by meta-sequencing three Lupinus nodulomes, discovering differential compatibility between lupine and Bradyrhizobium and that different hormone, secondary metabolite and plant-defence mechanisms activated depended on host compatibility. Similarly, GBS was used to show lack of local adaptation to different prevailing soil species of Ensifer between northern and southern populations of Medicago lupulina L. in North America (Harrison et al. 2017). Knowledge of which nitrogen-fixing bacteria are optimal is of key importance to maximise crops yields. High-throughput sequencing of nodules or rhizospheres has determined the symbionts of legume crops, forage legumes and invasive species, including cowpea (Chidebe et al. 2018), lucerne (alfalfa; Wigley et al. 2017), rooibos tea (Le Roux et al. 2017) and silver wattle (Kamutando et al. 2017). Furthermore, characterisation and sequencing of bacterial genomes, including those that are key to nitrogen fixation, is now routine. For example, a new species, Rhizobium hidalgonense, was recently isolated and characterised from a Phaseolus vulgaris L. nodule growing in acidic soil (Yan et al. 2017), and draft genome sequences of Bradyrhizobium (Tian et al. 2015) and another new species, Ensifer aridi (Le Quéré et al. 2017), have been completed and characterised.
The evolutionary origin of nodulation has baffled researchers for many years, and particularly whether it evolved once or multiple times, whether some sort of cryptic precursor could have predisposed lineages in the nitrogen-fixing clade of angiosperms to evolve nodulation, and whether polyploidy may have been involved in its origination (Werner et al. 2014; Doyle, 2016). Griesmann et al. (2018) used a comparative genomics approach to address this question by sequencing genomes from across the nitrogen-fixing clade including several non-nodulating species, to look for genes known to be vital to nitrogen-fixing nodulation (NFN). They discovered that all NFN genes were conserved in all but one nodulating species and found evidence for multiple independent losses of the nodule-inception (NIN) gene in 10 of 13 non-nodulating species, attesting to the key role of this gene within the nodulation pathway, and suggesting multiple evolutionary losses of nodulation (van Velzen et al. 2019). Comparative analysis of the legume Medicago truncatula and the non-legume Parasponia andersonii Planch. also supported the idea of a single origin and multiple losses of nodulation across the nitrogen-fixing angiosperm clade (van Velzen et al. 2018, 2019). However, others disagree, citing differences in transcriptome profiles as evidence for a two-step process in origination of nodulation (Battenberg et al. 2018). In addition to working out its origin, understanding the subsequent evolution of nodulation is also important. One unique example is the ability of some members of photosynthesising Bradyrhizobium strains to prompt nodulation and fix nitrogen in the absence of nodABC genes that are key to modulating nodule formation. Strains of this bacterium form symbioses with Aeschynomene evenia C.Wright, and transcriptomics has enabled the creation of a gene-map to delineate genes involved in this unique type of symbiosis (Chaintreuil et al. 2016).
Genetic and genomics resources
The genomics era and all genetic knowledge owes homage to the pea (Pisum sativum L.), the legume crop that captured Gregor Mendel’s attention and led to his monumental discovery of genetic inheritance. The importance of nitrogen-fixing legumes as the most important protein and rotation crops has driven high-throughput sequencing and genomics to establish genetic and genomic data to underpin plant-breeding research. Here, we discuss some of these resources and subsequent discoveries.
Sequenced genomes
With soybean providing nearly 70% of the world’s edible protein, the completion of the soybean genome (Schmutz et al. 2010) marked an important milestone in legume research. Even though the soybean genome was not sequenced using NGS methods, it remains a gold standard in plant-genome sequencing, providing a vital reference for other work, including the assembly and annotation of other legume genomes. Draft genome sequences of the model legumes Lotus japonicus (Sato et al. 2008) and Medicago truncatula (Young et al. 2011), and common bean, Phaseolus vulgaris (Schmutz et al. 2014), based on Sanger sequencing, have been improved to reference quality by using NGS data (e.g. Tang et al. 2014).
Plant genomes are complex relative to other eukaryotic genomes, owing to large genome sizes, higher repetitive fraction, prevalence of polyploidy and difficulty of obtaining high-molecular weight DNA caused by presence of the cell wall, polysaccharides and secondary metabolites that impair enzymes or damage DNA (Jiao and Schneeberger 2017). Nevertheless, the advent of NGS has enabled whole-genome sequencing of a rapidly growing number of species and multiple accessions within species (Fig. 2, Table 1), as costs have fallen (from the ~US$20 million cost of the soybean genome; Marris 2008) and techniques have advanced such that a single laboratory can now rapidly produce multiple genomes. For example, Griesmann et al. (2018) sequenced draft genomes of four legume genera for comparative genomic analysis of nodulation genes, and Chang et al. (2019) sequenced three African orphan legume crop species to support crop breeding. Liu et al. (2019) set out to barcode 761 vascular plants from the Riuli Botanical Garden by using NGS whole-genome sequencing, 71 of which are legumes (because identification and genome assembly remain incomplete, these genomes are not listed in Table 1).
Owing to plant genome size and complexity, NGS-based plant genomes often have poorer assembly statistics than do those of vertebrates and are commonly assembled only to draft status. However, a draft genome is still worthwhile! For example, genomic sequencing of multiple accessions of the recent domesticate, Lupinus angustifolius L., has allowed mapping of key disease resistance and domestication traits (Yang et al. 2013; Hane et al. 2017). Even without assembly to draft status, sequencing the full genomic content of an organism can yield important information. For example, Kang et al. (2015) sequenced the genomes of Vigna angularis var. nipponensis (Ohwi) Ohwi & H.Ohashi and V. nepalensis Tateishi & Maxted, wild relatives of cultivated adzuki bean, V. angularis var. angularis (Willd.) Ohwi & H.Ohashi. Even though the reads were not fully assembled, nor genes called de novo, variation in the two wild relatives called against the draft genome of the cultivated bean provided insights into the timing of domestication and variation across the genus.
Early NGS platforms provided vast amounts of data; however, limitations, such as short read length and limited read output, made genome assembly from such platforms alone challenging. As technologies have improved and new mapping methods have been devised, draft genome assemblies can now be improved on after the fact. For example, subterraneum clover (Trifolium subterraneum L.), an annual relative of the forage legumes T. repens L. (white clover) and T. pratense L. (red clover), was sequenced using Illumina and 454 pyrosequencing as a reference in the genus Trifolium L. (Hirakawa et al. 2016), producing 27 228 scaffolds representing the draft genome, TSUd_r1.1. Subsequently, Kaur et al. (2017) applied a Bionano Genomics (San Diego, CA, USA) optical map and a transcriptome atlas to improve the TSUd_r1.1 assembly by anchoring unplaced contigs, correcting mis-assemblies, and improving gene annotation, resulting in the coalecense of 264 contigs into 97 super-scaffolds representing 43% of the genome and a 1.4-fold increase in the scaffold N50 to create the Tsub_Refv2.0 assembly. Similarly, also building on TSUd_r1.1, application of Hi-C contact mapping, a chromosome conformation technology, corrected misjoins, anchored and oriented scaffolds into eight chromosome-length pseudomolecules that included 95% of the sequenced bases in the input assembly, with the resulting TrSub3 assembly improving the initial scaffold N50 from 287 kb to 56 Mb (Dudchenko et al. 2018).
Whereas Illumina sequencing produces the most data for the lowest cost, long-read sequencing is increasingly affordable and can sequence through repetitive regions that often make plant genomes difficult to assemble. For example, Lonardi et al. (2019) used PacBio (Pacific Biosciences of California, Inc., Menlo Park, CA, USA) to sequence the genome of cowpea (Vigna unguiculata [L.] Walp) by using 56.8 Gb of sequence data with a read N50 of 14 595 bp, i.e. longer than many de novo assembled contigs and scaffolds. In addition, they employed two Bionano optical maps and a novel ‘stitching’ assembly method that combined eight different assemblies created using three different programs. Long-read lengths coupled with iterative cross-checking methods to remove chimeric joins enabled a highly accurate and complete assembly that was able to push through the long repetitive regions that break other plant assemblies into numerous contigs. They also detected a 4.2-Mb chromosomal inversion that may confer resistance to a parasitic weed, Striga gesnerioides (Willd.) Vatke. Cross-platform genome-sequencing strategies that use both short- and long-read platforms coupled with physical mapping are, thus, good contemporary approaches for plant-genome sequencing, including enhancement of existing assemblies from draft to reference quality.
As the number of sequenced genomes has grown, genetic and genome-scope databases have been developed, including Phytozome (phytozome.jgi.doe.gov, accessed 30 May 2019) and the Legume Information System (legumeinfo.org, accessed 30 May 2019; Dash et al. 2016), to facilitate access to genome content and information. These databases can be integrated with programs and platforms such as CyVerse (cyverse.org, accessed 30 May 2019) to enable public access to datasets, management and integration of personal data and access to high-performance computing platforms, thus enabling collaborative and critical breakthroughs through the combined power of genetic and computational resource platforms. However, none of these databases integrates other types of information, such as morphological-trait variation or geographical-distribution data, prompting legume systematists to devise a legume portal that can integrate across platforms and data types (Bruneau et al. 2019). Development of integrated database systems across genomic, taxonomic, geographical, morphological, population and expression-profile data will enhance our ability to parse and use information from NGS data and plant genomes.
Organelle genomes
With the abundance of chloroplast DNA, compared to nuclear, within a cell, 10–20% of NGS reads are chloroplast, such that entire chloroplast genomes can be assembled from even low-coverage genome skimming. Furthermore, the shorter length and less-repetitive nature of the chloroplast makes assembly easier, even for degraded herbarium material (Bakker et al. 2016). Thus, the use and sequencing of chloroplast genomes has rocketed forward with the advent of NGS methods. Some have even suggested the use of whole chloroplast genomes as DNA barcodes for plants (Li et al. 2015).
Comparative phylogenetic analyses have shown that structural changes in the chloroplast genome are often synapomorphies for large legume clades (Wang et al. 2018). For example, the loss of one copy of the inverted repeat marks the inverted repeat lacking clade (IRLC), a large papilionoid clade including tribes Cicereae, Hedysareae, Trifolieae and Fabeae (Vicieae), among others (Wojciechowski et al. 2000). Recent sequencing of chloroplasts from eight Cercidoideae genera showed structural diversification characteristic of that subfamily (Wang et al. 2018). In contrast, comparative analyses of chloroplast genomes determined that Papilionoideae have reduced genome sizes and are more divergent from the ancestral angiosperm chloroplast-genome organisation than are other subfamilies (Schwarz et al. 2015). Structural changes have also been identified at a generic level. Trifolium subterraneum L. was shown to have a highly unusual chloroplast genome greatly expanded by repetitive regions (Cai et al. 2008). To determine the evolutionary origin of this unique plastome type, Sveinsson and Cronk (2014) sequenced eight other Trifolium chloroplast genomes, showing that the expanded T. subterraneum-type plastome is shared by members of ‘core Trifolium’, providing a synapomorphy for what they called the ‘refractory clade’. The expansion of the inverted repeat region across the large Ingeae + Acacias.s. clade of mimosoid legumes (Wang et al. 2017) has provided another synapomorphic structural plastid mutation apparently characterising that clade, and is associated with shifts in evolutionary rate or selection pressures on proximate chloroplast gene regions (Mensous et al. 2017).
Numerous chloroplast genomes have been sequenced in legumes; the NCBI archive of full-length chloroplast genomes includes 284 accessions (97.2% circularised, 2.8% linear; 66.4% complete, 35.6% partial), representing 72 genera and 202 species, ranging from 120 289 bp in Lathyrus odoratus L. to 178 887 bp in Ebenopsis ebano (Berlandier) Barneby & Grimes, including 118 NCBI designated reference sequences (Table S1, available as Supplementary material to this paper). This list is by no means exhaustive and some accessions may represent duplicate chloroplast assemblies at various stages; however, every effort to remove duplicates was taken. Even so, this list exemplifies the power of the chloroplast for evolutionary studies in legumes, including for phylogeny estimation of recalcitrant clades. For example, whole chloroplast alignments showed that 13 of 15 Guibourtia Benn. species were reciprocally monophyletic, with evidence of a single dispersal event from the Old to the New World c. 12 million years ago (Tosso et al. 2018). Of the nearly 300 legume accessions with sequenced chloroplasts, 95 are from Acacia, a lineage of >1000 species. Williams et al. (2016) used whole chloroplast sequences of 65 Acacia species to build a robust backbone constraint tree, adding amplicon sequences of four chloroplast and two nuclear ribosomal loci for 508 other Acacia species, to produce a more robust phylogeny than from amplicon sequences alone. This suggests that combining legacy Sanger sequencing datasets with large, phylogenomic datasets will likely yield positive results.
Another byproduct of NGS is sequencing of the mitochondrion, which has significantly fewer genomes sequenced than does the chloroplast (Table 2). The first legume mitochondrion sequenced was that of Vigna radiata (L.) Wilczek (Alverson et al. 2011) and the Sanger-based sequence of the Vicia faba L. mitochondrion was not far behind (Negruk 2013). The first legume mitochondria sequenced by NGS methods were Lotus japonicus and Millettia pinnata (L.) Panigrahi, on the Illumina platform (Kazakoff et al. 2012). Soon after, the mitochondrial genomes of soybean (Chang et al. 2013) and Medicago truncatula (Bi et al. 2016) were sequenced on the 454 platform, and Vigna angularis was compiled using both 454 and Illumina sequence reads (Naito et al. 2013). The paucity of sequenced plant mitochondrial genomes is largely due to the highly recombinant nature of mitochondria, which makes assembly difficult. Shi et al. (2018) combined PacBio with Illumina reads to investigate the repetitive complement of the Styphnolobium japonicum (L.) Schott mitochondrial genome, and discovered that small repeats (<100 bp) had a disproportionate impact on the evolution of Styphnolobium mitochondria through mediation of recombination along intronic regions. The first non-papilionoid legume mitochondrion to be sequenced was Leucaena trichandra (Zucc.) Urb., completed using PacBio (Kovar et al. 2018) to obtain long-reads, enabling assessment of variable assemblies and investigation of mitochondrial genome-size variation in legumes. Further, overlaying transcriptomic data enabled comparative study of RNA editing among Leucaena species, providing knowledge that can yield useful information regarding close species relationships and hybrid origins (Kovar et al. 2018).

|
Despite these advances, the repetitive nature of the mitochondrion and its evolutionary implications are only beginning to be understood, particularly with regard to cross-species interactions. That said, Sanchez-Puerta et al. (2019) presented an elegant study of host–parasite horizontal gene transfer between the holoparasite Lophophytum mirabile Schott & Endl. and its host Acacia ligulata A.Cunn., showing that ~60% of the L. mirabile mitochondrion is derived from its host, including 34 of 43 protein-coding genes (also see Sanchez-Puerta et al. 2017; Kovar et al. 2018). In addition, ~26 of its native genes were replaced by host genes through homologous recombination, and with a large portion of intergenic regions also host-derived. This work provided incontrovertible support for mitochondrial-to-mitochondrial horizontal gene transfer, a phenomenon well documented in Amborella trichopoda Baill. (Rice et al. 2013). Furthermore, Kovar et al. (2018) also showed that non-coding mitochondrial DNA was horizontally transferred, suggesting capture of an entire mimosoid mitochondrial genome during the evolutionary history of the Lophophytum parasite. Knowledge such as this may shed light on the incredible length diversity of plant mitochondrial genomes, which range from 271 618 bp in Medicago truncatula to 698 138 bp in Acacia ligulata. At the time of writing, 15 legume mitochondria had been sequenced (Table 2). As this number expands, broader comparative analyses become possible; sequencing of the wild soybean, Glycine soja (Asaf et al. 2018), provided insights into a likely progenitor to the soybean mitochondrion, whereas that of Ammopiptanthus mongolicus (Kom.) S.H.Cheng provided a glimpse into a Tertiary relic (Yu et al. 2018). Comparative analysis of the most recently sequenced legume mitochondrion, namely that of Castanospermum australe A.Cunn & C.Fraser, enabled phylogenetic analysis of 33 mitochondrial genes across legumes, producing a fully supported phylogeny (Zhang et al. 2019), suggesting that despite the fewer phylogenetically informative sites than in chloroplast genomes (Palmer and Herbon 1988), the mitochondrion has potential utility for molecular systematics of legumes.
Plant breeding
Simply put, the genomics era has revolutionised plant breeding in legumes. We cannot possibly summarise it all here; however, some studies are essential for discussion. Major goals of plant breeding are improvement of crop traits useful for humans and adaptation to environment such as increasing yield, freeze and drought tolerance, disease resistance, nutritional quality and seed size. A genome sequence provides the basis for genome-wide association studies (GWAS), functional genomics, quantitative trait-loci (QTL) analysis and linkage studies, SNP variant detection, and genetic modification by CRISPR, among many others. For example, sequencing of the licorice genome (Glycyrrhiza uralensis Fisch.) enabled assessment of genes involved in flavonoid and saponin synthesis, producing candidate genes for improving yield of glycyrrhizin, an active chemical component used in traditional Chinese medicine (Mochida et al. 2017). Of wider importance are several crop genomes, in addition to those already discussed. Chickpea (Cicer arietinum L.) is surpassed only by soybean, peanut and Phaseolus ssp. as a widely grown legume crop (Fig. 3, Table S2, available as Supplementary material to this paper). Its sequenced genomes (Jain et al. 2013; Varshney et al. 2013), coupled with resequencing of numerous cultivars and wild accessions from 10 countries, enabled discovery of gene regions associated with key domestication, agronomic and disease-resistance traits (Varshney et al. 2013). Likewise, sequencing of Cajanus cajan [L.] Millsp., the pigeon pea (Singh et al. 2012; Varshney et al. 2012; Mahato et al. 2018), one of the most widely cultivated and consumed orphan legume crops in India, highlighted drought-tolerance genes important during its domestication (Varshney et al. 2012). These genomic resources have aided successful translational improvements of both chickpea and pigeon pea (for review, see Varshney 2016).

|
To illustrate the types of NGS applications and advances made in legume-crop breeding, we focus on Vigna Savi, a pantropical genus of over 100 species of which 10 species have been domesticated and are cultivated in both warm, humid and dry, seasonal climates (Table 3; Harouna et al. 2018). As such, Vigna species provide food for nearly half of the world’s population. Ten species or varieties of Vigna have had their genomes sequenced (Tables 1, 3), including six crop species (Kang et al. 2014, 2015; Chang et al. 2019; Lonardi et al. 2019) and four wild relatives (Kang et al. 2014, 2015; Lestari et al. 2014). The impacts of these genome sequences are just beginning to be realised. For example, comparative searches of the V. angularis and V. radiata genomes for homologues of ONSEN-like sequences, a heat-activated retrotransposon isolated from Arabidopsis thaliana (L.) Heynh., produced several key hits in V. angularis, sequences that proved to be polymorphic in different accessions (Masuta et al. 2018). These retrotransposons were associated with accumulation of extrachromosomal DNA and were employed to successfully induce retrotransposition of V. angularis ONSEN-like elements in regenerated V. angularis callus tissue, providing a new tool for molecular breeding in Vigna.

|
Many plant-breeding efforts require a genetic map. RADseq was used to create a high-density genetic-linkage map by using 170 individuals to enable QTL detection of loci related to yield in cowpea (Pan et al. 2017), a species that includes two broad cultivar types, namely, bushy, short-podded grain grown predominantly in Africa, and long-podded climbing vegetables grown mostly in Asia. Genomic scans confirmed that pod length was selected for during domestication of the vegetable variety (Xu et al. 2017). Furthermore, GWAS for genomic regions controlling pod length between cultivars of these two types discovered 72 SNPs whose pod-length association was verified across 299 cowpea accessions. This knowledge, coupled with transcriptomic analysis, suggested the involvement of sugar, gibberellin and nutrition as key factors in pod-length regulation and that cell proliferation rather than cell elongation was key to pod length.
Applications of transcriptomics are often used to study stress responses such as low temperature-stress resistance in V. subterranea, providing gene modules for plant-breeding improvement (Bonthala et al. 2016), among many others. These studies often identify candidate genes useful for functional genomic characterisation. For instance, acidic soils are often characterised by accumulation of aluminium, with aluminium toxicity causing reduced yields in crop plants. Rice bean (V. umbellata (Thunb.) Ohwi & H.Ohashi) is tolerant of soils with high aluminium accumulation. Studies of such resistance in other plants have suggested a role for the abscisic acid (ABA) pathway in dealing with aluminium toxicity. Transcriptomic analysis and functional genomic studies in rice bean support this hypothesis (Fan et al. 2019), but its involvement was shown to be dependent on AB15, a transcription factor that mediated changes in cell-wall modification and osmoregulation.
Another focus of plant-breeding efforts involve resistance to pests and pathogens. Micro-RNAs (miRNAs) are short (20–24 nucleotides) non-coding RNAs that act to regulate gene expression, particularly during response to stress, such as that inflicted on a plant by viral infection. The mungbean yellow mosaic India virus (MYMIV) significantly decreases yield across many South-East Asian countries where Vigna species are staple crops. Understanding the mechanisms of infection and host response are key to crop survival. Kundu et al. (2017) identified miRNAs involved in the stress response of V. mungo (L.) Hepper to MYMIV infection, by comparing gene-expression patterns and miRNAs across resistant and non-resistant cultigens, with putative target genes known to be involved in pathogen-stress response, such as NB-LRR, ARF, SOD, SPB, and Basic blue copper protein, linked to and validated as being regulated by miRNAs in stressed and non-stressed plants. Another proverbial pest problem plaguing legume crops is bruchid beetles (Callosobruchus Pic. spp.), which infest seeds in the field, then multiply and destroy seed during storage. Bruchid resistance has been found in wild mungbean, V. radiata var. sublobata (Roxb.) Verdc., and in one mungbean cultivar. Schafleitner et al. (2016) used GBS to map inbred recombinant lines for each of these resistant populations and discovered one QTL associated with bruchid resistance shared between both resistant entities. The markers associated with this QTL were validated as 100% predictive of bruchid resistance, providing an excellent screening tool for developing resistant cultivars. Similar genome-based research across the full spectrum of important legume crops is underway. Rapidly expanding knowledge of gene functional pathways from comparative genomics and the development of genome-based selection, is accelerating and revolutionising legume-crop breeding; these efforts will aid in future crop and food security.
This short summary would be incomplete without some discussion of the genomics of crop wild relatives, which can contribute key genes and diversity to crops to increase pest and disease resistance and extend environmental tolerances of important crop legumes. One of the first crop wild relatives to have its genome sequenced was Glycine soja Sieb. and Zucc. (Kim et al. 2010), one of the first genome re-sequencing projects in plants. In the study of Kim et al. (2010), G. soja was sequenced and its genome assembled against that of soybean, with a comparison between the two suggesting that G. max diverged from G. soja c. 0.27 million years ago, i.e. hundreds of thousands of years before domestication of soybean. The draft genome of Glycine latifolia (Benth.) C.A.Newell & Hymowitz, a perennial wild relative of soybean, was recently sequenced using only linked-reads from a single 10X Genomics (Pleasanton, CA, USA) Chromium library (Liu et al. 2018), presenting a valuable resource of alleles and genes for soybean improvement. Like soybean, the cultivated peanut (or groundnut; Arachis hypogaea L.) is of polyploid origin. The large allotetraploid peanut genome sequence (http://peanutbase.org/peanut_genome, accessed 30 May 2019; Chen et al. 2019; ~2.7 Gb) comprises the two recently diverged subgenomes of its diploid ancestors, Arachis duranensis Krapov. & W.C.Greg. and A. ipaensis Krapov. & W.C.Greg, which were used to assist assembly of the domesticated-peanut genome and detect genetic recombination among peanut subgenomes, providing key information regarding the origin of the cultivated peanut (Bertioli et al. 2016). As climate changes and demands for better yield increase, wild relatives offer plant breeders sources of diverse and adapted traits to incorporate into cultivars.
Securing the future of legume germplasm diversity
Given the central roles that legumes play in agriculture, ecosystems and the global nitrogen cycle, securing the future of legume genetic resources is both urgent and of paramount importance. Application of genomics methods, from capturing the genome of a fading species to guiding restoration and management efforts and assisting and speeding up plant breeding, can enable advances in legume research that were previously unattainable that will help secure their future.
A good example is Ammopiptanthus S.H.Cheng, a genus of two evergreen broadleaf desert shrub species endemic to central Asia. This taxonomically isolated genus is hypothesised to be a relic from the Tertiary, having adapted to aridification from a moist and humid climate of the evergreen broadleaf forest characteristic of the Tethyan flora (Zhang et al. 2015). As a relict, Ammopiptanthus has evolved drought-, cold- and wind-resistance, among other stress-tolerant characteristics. Both species are considered threatened because of low seed set and increasing anthropogenic disturbance, with A. nanus (Popov) S.H.Cheng listed as Critically Endangered (www.iucnredlist.org, accessed 19 February 2019). Ammopiptanthus has been the focus of abiotic stress studies, using transcriptomics to understand its drought (Zhou et al. 2012) and cold (Pang et al. 2013) adaptations. These studies have enabled the cloning, characterisation and validation of candidate genes shown as beneficial in other model study systems, conferring salinity and heat tolerance to Escherichia coli by the A. nanus betaine aldehyde dehydrogenase gene (Yu et al. 2014), or cold tolerance by the A. nanus antifreeze gene AnAFP to both E. coli and tobacco (Deng et al. 2014), among many others. Recently, the A. nanus genome was sequenced on the PacBio platform (Table 1; Gao et al. 2018), providing an essential resource for functional genomics and improvement studies, with Ammopiptanthus fast becoming a model for understanding drought and cold tolerance.
Along with alfalfa (Medicago sativa) and several related species, clovers (Trifiolium spp.) are important forage crops and key components of natural grazing systems. Red clover (Trifolium pratense) is an excellent short-term hay-rotation crop and pasture or field restoration plant. Its dual use as protein-rich fodder and soil-fertility enhancer led to its early adoption in crop-rotation schemes and is one reason why some tout it as a perfect plant for conservation agriculture, a movement aimed at sustainable intensification of food and forage for increasing world needs (McKenna et al. 2018). The recent sequencing of its genome (Ištvánek et al. 2014; De Vega et al. 2015) has provided another forage genome resource for comparative analyses to assist breeding (Annicchiarico et al. 2015). Furthermore, transcriptomic studies have enabled advances in seed set (Kovi et al. 2017), drought (Yates et al. 2014) and leaf senescence (Chao et al. 2018a). For example, Chao et al. (2018b) used PacBio to sequence and analyse full-length transcripts, enabling the detection of over 30 000 novel isoforms and 5492 alternative splicing events, the majority of which involved intron retention. Using two-dimensional difference gel electrophoresis, Bertrand et al. (2016) characterised the proteome of red clover, particularly with reference to freezing tolerance; selection detailed the involvement of a small number of cold-regulated proteins, suggesting them as potential targets for breeding programs. These studies illustrated the incredible protein diversity arising from the complement of known genes and provided a third layer of information on top of genomic and expression-level knowledge.
The impacts of climate change will be particularly felt on food security, with changes in temperature and rainfall patterns affecting what, where and how crops will flourish. Genomics can help improve food security by providing fundamental molecular-data resources through genome sequencing, comparative genomics and rapid evaluation of genetic variation, to enable assessment of adaptation priority, determine import of specific traits for breeding programs, and discover adaptive genes and traits (Mousavi‐Derazmahalleh et al. 2019), as previously discussed. Many have suggested that the potential of legumes to enhance food security and reduce meat consumption has not been realised nor explored adequately (Foyer et al. 2016). Phaseolus, with a sequenced genome and well-developed breeding programs, provides a good example. Key areas of inquiry remain unexplored. For instance, phenotyping of the world’s germplasm collections is important to understand the diversity currently held and how that compares to wild populations (McClean et al. 2011), and with the advent of genomics techniques, the time in now ripe for doing so en masse. Genotyping-by-sequencing and genome–environment associations within wild populations of common bean have discovered several genomic regions associated with drought adaptation, pinpointing genomic signatures potentially useful for marker-assisted selection (Cortés and Blair 2018). Yet, much still needs to be done. As wild relatives and related species often harbour greater GD than do cultivars, efforts to assess such genetic and phenotypic diversity and unlock the adaptive potential of these entities should be a priority for future food security (Porch et al. 2013). Efforts to identify gaps in germplasm collections for Phaseolus have begun (Ramírez-Villegas et al. 2010), yet, much is still needed (Dohle et al. 2019).
Conclusions
From evolution and ecology to classical and applied genetics, the genomics era has and will continue to contribute to our understanding of legume biology in many important ways, and this is set to expand and accelerate in coming years as sequencing costs continue to fall. Partial or whole genomes of hundreds to thousands of legume species are expected to be available in the near future, with the 10 000 genomes project already targeting over 300 legume genera (Cheng et al. 2018). While such an unprecedented accumulation of genomic data presents significant computational and analytical challenges, there will soon be unparalleled opportunities to address large-scale comparative genomic questions. Analyses of numerous legume genomes will provide massive insights into synteny, micro- and macro-level gene and genome duplication, and chromosome structure coupled with gene expression analyses and recombination perspectives. At the same time, variant detection-panel screening across population-level sampling schemes will enable exceptional advances in understanding patterns and processes of evolution at micro- and macro-scales. Such knowledge will help secure the future of legumes as vital components of ecological and economic security.
Conflicts of interest
A. N. Egan is also an Associate Editor of the ‘Advances in Legume Systematics 13’ special issue. Despite this relationship, she did not at any stage have Associate Editor-level access to this manuscript while in peer review, as is the standard practice when handling manuscripts submitted by an editor to this journal. Australian Systematic Botany encourages its editors to publish in the journal and they are kept totally separate from the decision-making process for their manuscripts. The authors have no further conflicts of interest to declare.
Declaration of Funding
This research did not receive any specific funding. However, A. N. Egan was supported in part by a subaward from the Carlsberg Foundation to Henrik Balslev (grant #CF14-0245) and by the USA National Science Foundation (DEB-1352217).
Acknowledgements
A. N. Egan thanks Dr Henrik Balslev and the Department of Biosciences at Aarhus University for providing a sabbatical academic home from which to pursue this work and Dr Jeffrey J. Doyle and Dr Colin Hughes, as well as an anonymous reviewer, for helpful comments on the manuscript.
References
Abdelkrim J, Robertson BC, Stanton JA, Gemmell NJ (2009) Fast, cost-effective development of species-specific microsatellite markers by genomic sequencing. BioTechniques 46, 185–192.| Fast, cost-effective development of species-specific microsatellite markers by genomic sequencing.Crossref | GoogleScholarGoogle Scholar | 19317661PubMed |
Afkhami ME, Stinchcombe JR (2016) Multiple mutualist effects on genome-wide expression in the tripartite association between Medicago truncatula, nitrogen‐fixing bacteria and mycorrhizal fungi. Molecular Ecology 25, 4946–4962.
| Multiple mutualist effects on genome-wide expression in the tripartite association between Medicago truncatula, nitrogen‐fixing bacteria and mycorrhizal fungi.Crossref | GoogleScholarGoogle Scholar | 27543961PubMed |
Ahrendsen DL, Aust SK, Kellar PR (2016) Biodiversity assessment using next-generation sequencing: comparison of phylogenetic and functional diversity between Nebraska grasslands. Plant Systematics and Evolution 302, 89–108.
| Biodiversity assessment using next-generation sequencing: comparison of phylogenetic and functional diversity between Nebraska grasslands.Crossref | GoogleScholarGoogle Scholar |
Alverson AJ, Zhuo S, Rice DW, Sloan DB, Palmer JD (2011) The mitochondrial genome of the legume Vigna radiata and the analysis of recombination across short mitochondrial repeats. PLoS One 6, e16404
| The mitochondrial genome of the legume Vigna radiata and the analysis of recombination across short mitochondrial repeats.Crossref | GoogleScholarGoogle Scholar | 21283772PubMed |
Andrews KR, Good JM, Miller MR, Luikart G, Hohenlohe PA (2016) Harnessing the power of RADseq for ecological and evolutionary genomics. Nature Reviews. Genetics 17, 81–92.
| Harnessing the power of RADseq for ecological and evolutionary genomics.Crossref | GoogleScholarGoogle Scholar | 26729255PubMed |
Andújar C, Arribas P, Ruzicka F, Crampton-Platt A, Timmermans MJTN, Vogler AP (2015) Phylogenetic community ecology of soil biodiversity using mitochondrial metagenomics. Molecular Ecology 24, 3603–3617.
| Phylogenetic community ecology of soil biodiversity using mitochondrial metagenomics.Crossref | GoogleScholarGoogle Scholar | 25865150PubMed |
Annicchiarico P, Barrett B, Brummer EC, Julier B, Marshall AH (2015) Achievements and challenges in improving temperate perennial forage legumes. Critical Reviews in Plant Sciences 34, 327–380.
| Achievements and challenges in improving temperate perennial forage legumes.Crossref | GoogleScholarGoogle Scholar |
Asaf S, Khan AL, Al-Harrasi A, Kim TH, Lee IJ (2018) The first complete mitochondrial genome of wild soybean (Glycine soja). Mitochondrial DNA. Part B, Resources 3, 527–528.
| The first complete mitochondrial genome of wild soybean (Glycine soja).Crossref | GoogleScholarGoogle Scholar |
Azani N, Bruneau A, Wojciechowski MF, Zarre S (2017) Molecular phylogenetics of annual Astragalus (Fabaceae) and its systematic implications. Botanical Journal of the Linnean Society 184, 347–365.
| Molecular phylogenetics of annual Astragalus (Fabaceae) and its systematic implications.Crossref | GoogleScholarGoogle Scholar |
Bagheri A, Maassoumi AA, Rahiminejad MR, Brassac J, Blattner FR (2017) Molecular phylogeny and divergence times of Astragalus section Hymenostegis: an analysis of a rapidly diversifying species group in Fabaceae. Scientific Reports 7, 14033
| Molecular phylogeny and divergence times of Astragalus section Hymenostegis: an analysis of a rapidly diversifying species group in Fabaceae.Crossref | GoogleScholarGoogle Scholar | 29070910PubMed |
Bakker FT, Lei D, Yu J, Mohammadin S, Wei Z, van de Kerke S, Gravendeel B, Nieuwenhuis M, Staats M, Alquezar-Planas DE, Holmer R (2016) Herbarium genomics: plastome sequence assembly from a range of herbarium specimens using an iterative organelle genome assembly pipeline. Biological Journal of the Linnean Society. Linnean Society of London 117, 33–43.
| Herbarium genomics: plastome sequence assembly from a range of herbarium specimens using an iterative organelle genome assembly pipeline.Crossref | GoogleScholarGoogle Scholar |
Battenberg K, Potter D, Tabuloc CA, Chiu JC, Berry AM (2018) Comparative transcriptomic analysis of two actinorhizal plants and the legume Medicago truncatula supports the homology of root nodule symbioses and is congruent with a two-step process of evolution in the nitrogen-fixing clade of angiosperms. Frontiers in Plant Science 9, 1256
| Comparative transcriptomic analysis of two actinorhizal plants and the legume Medicago truncatula supports the homology of root nodule symbioses and is congruent with a two-step process of evolution in the nitrogen-fixing clade of angiosperms.Crossref | GoogleScholarGoogle Scholar | 30349546PubMed |
Bertioli DJ, Cannon SB, Froenicke L, Huang G, Farmer AD, Cannon EK, Liu X, Gao D, Clevenger J, Dash S, Ren L, Moretzsohn MC, et al (2016) The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut. Nature Genetics 48, 438–446.
| The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut.Crossref | GoogleScholarGoogle Scholar | 26901068PubMed |
Bertioli DJ, Jenkins J, Clevenger J, Dudchenko O, Gao D, Seijo G, Leal-Bertioli SC, Ren L, Farmer AD, Pandey MK, Samoluk SS, et al (2019) The genome sequence of segmental allotetraploid peanut Arachis hypogaea. Nature Genetics 51, 877–884.
| The genome sequence of segmental allotetraploid peanut Arachis hypogaea.Crossref | GoogleScholarGoogle Scholar | 31043755PubMed |
Bertrand A, Bipfubusa M, Castonguay Y, Rocher S, Szopinska-Morawska A, Papadopoulos Y, Renaut J (2016) A proteome analysis of freezing tolerance in red clover (Trifolium pratense L.). BMC Plant Biology 16, 65
| A proteome analysis of freezing tolerance in red clover (Trifolium pratense L.).Crossref | GoogleScholarGoogle Scholar | 26965047PubMed |
Bi C, Wang X, Xu Y, Wei S, Shi Y, Dai X, Yin T, Ye N (2016) The complete mitochondrial genome of Medicago truncatula. Mitochondrial DNA – B. Resources 1, 122–123.
| The complete mitochondrial genome of Medicago truncatula.Crossref | GoogleScholarGoogle Scholar |
Birnbaum C, Bissett A, Teste FP, Laliberté E (2018) Symbiotic N2-fixer community composition, but not diversity, shifts in nodules of a single host legume across a 2-million-year dune chronosequence. Microbial Ecology 76, 1009–1020.
| Symbiotic N2-fixer community composition, but not diversity, shifts in nodules of a single host legume across a 2-million-year dune chronosequence.Crossref | GoogleScholarGoogle Scholar | 29663039PubMed |
Blanco-Pastor JL, Bertrand YJ, Liberal IM, Wei Y, Brummer EC, Pfeil BE (2018) Robustness of RADseq for evolutionary network reconstruction from gene trees. bioRxiv 2018, 414243.
Bombarely A, Coate JE, Doyle JJ (2014) Mining transcriptomic data to study the origins and evolution of a plant allopolyploid complex. PeerJ 2, e391
| Mining transcriptomic data to study the origins and evolution of a plant allopolyploid complex.Crossref | GoogleScholarGoogle Scholar | 24883252PubMed |
Bonthala VS, Mayes K, Moreton J, Blythe M, Wright V, May ST, Massawe F, Mayes S, Twycross J (2016) Identification of gene modules associated with low temperatures response in bambara groundnut by network-based analysis. PLoS One 11, e0148771
| Identification of gene modules associated with low temperatures response in bambara groundnut by network-based analysis.Crossref | GoogleScholarGoogle Scholar | 26859686PubMed |
Borges DB, Mariano-Neto E, Gaiotto FA (2015) Development of microsatellite primers for Melanoxylon brauna (Fabaceae): an endangered and endemic tree from the Brazilian Atlantic Forest. Conservation Genetics Resources 7, 65–68.
| Development of microsatellite primers for Melanoxylon brauna (Fabaceae): an endangered and endemic tree from the Brazilian Atlantic Forest.Crossref | GoogleScholarGoogle Scholar |
Bruneau A, Mercure M, Lewis GP, Herendeen PS (2008) Phylogenetic patterns and diversification in the caesalpinioid legumes. Botany 86, 697–718.
| Phylogenetic patterns and diversification in the caesalpinioid legumes.Crossref | GoogleScholarGoogle Scholar |
Bruneau A, Borges LM, Allkin R, Egan AN, de la Estrella M, Javadi F, Klitgård B, Miller JT, Murphy DJ, Sinou C, Vatanparast M, Zhang R (2019) Towards a new online species-information system for legumes. Australian Systematic Botany 32, 495–518.
| Towards a new online species-information system for legumes.Crossref | GoogleScholarGoogle Scholar |
Bybee SM, Bracken-Grissom H, Haynes BD, Hermansen RA, Byers RL, Clement MJ, Udall JA, Wilcox ER, Crandall KA (2011) Targeted amplicon sequencing (TAS): a scalable next-gen approach to multilocus, multitaxa phylogenetics. Genome Biology and Evolution 3, 1312–1323.
| Targeted amplicon sequencing (TAS): a scalable next-gen approach to multilocus, multitaxa phylogenetics.Crossref | GoogleScholarGoogle Scholar | 22002916PubMed |
Cai Z, Guisinger M, Kim HG, Ruck E, Blazier JC, McMurtry V, Kuehl JV, Boore J, Jansen RK (2008) Extensive reorganization of the plastid genome of Trifolium subterraneum (Fabaceae) is associated with numerous repeated sequences and novel DNA insertions. Journal of Molecular Evolution 67, 696–704.
| Extensive reorganization of the plastid genome of Trifolium subterraneum (Fabaceae) is associated with numerous repeated sequences and novel DNA insertions.Crossref | GoogleScholarGoogle Scholar | 19018585PubMed |
Cannon SB, McKain MR, Harkess A, Nelson MN, Dash S, Deyholos MK, Peng Y, Joyce B, Stewart CN, Rolf M, Kutchan T, et al (2015) Multiple polyploidy events in the early radiation of nodulating and nonnodulating legumes. Molecular Biology and Evolution 32, 193–210.
| Multiple polyploidy events in the early radiation of nodulating and nonnodulating legumes.Crossref | GoogleScholarGoogle Scholar | 25349287PubMed |
Cardoso D, de Queiroz LP, Pennington RT, de Lima HC, Fonty E, Wojciechowski MF, Lavin M (2012) Revisiting the phylogeny of papilionoid legumes: new insights from comprehensively sampled early-branching lineages. American Journal of Botany 99, 1991–2013.
| Revisiting the phylogeny of papilionoid legumes: new insights from comprehensively sampled early-branching lineages.Crossref | GoogleScholarGoogle Scholar | 23221500PubMed |
Chaintreuil C, Rivallan R, Bertioli DJ, Klopp C, Gouzy J, Courtois B, Leleux P, Martin G, Rami JF, Gully D, Parrinello H, Séverac D, et al (2016) A gene-based map of the Nod factor-independent Aeschynomene evenia sheds new light on the evolution of nodulation and legume genomes. DNA Research 23, 365–376.
| A gene-based map of the Nod factor-independent Aeschynomene evenia sheds new light on the evolution of nodulation and legume genomes.Crossref | GoogleScholarGoogle Scholar | 27298380PubMed |
Chang S, Wang Y, Lu J, Gai J, Li J, Chu P, Guan R, Zhao T (2013) The mitochondrial genome of soybean reveals complex genome structures and gene evolution at intercellular and phylogenetic levels. PLoS One 8, e56502
| The mitochondrial genome of soybean reveals complex genome structures and gene evolution at intercellular and phylogenetic levels.Crossref | GoogleScholarGoogle Scholar | 24367547PubMed |
Chang Y, Liu H, Liu M, Liao X, Sahu SK, Fu Y, Song B, Cheng S, Kariba R, Muthemba S, Hendre PS, Mayes S, et al (2019) The draft genomes of five agriculturally important African orphan crops. GigaScience 8, giy152
| The draft genomes of five agriculturally important African orphan crops.Crossref | GoogleScholarGoogle Scholar | 30689836PubMed |
Chao Y, Xie L, Yuan J, Guo T, Li Y, Liu F, Han L (2018a) Transcriptome analysis of leaf senescence in red clover (Trifolium pratense L.). Physiology and Molecular Biology of Plants 24, 753–765.
| Transcriptome analysis of leaf senescence in red clover (Trifolium pratense L.).Crossref | GoogleScholarGoogle Scholar | 30150852PubMed |
Chao Y, Yuan J, Li S, Jia S, Han L, Xu L (2018b) Analysis of transcripts and splice isoforms in red clover (Trifolium pratense L.) by single-molecule long-read sequencing. BMC Plant Biology 18, 300
| Analysis of transcripts and splice isoforms in red clover (Trifolium pratense L.) by single-molecule long-read sequencing.Crossref | GoogleScholarGoogle Scholar | 30477428PubMed |
Chapman MA (2015) Transcriptome sequencing and marker development for four underutilized legumes. Applications in Plant Sciences 3, 1400111
| Transcriptome sequencing and marker development for four underutilized legumes.Crossref | GoogleScholarGoogle Scholar | 25699221PubMed |
Chau JH, Rahfeldt WA, Olmstead RG (2018) Comparison of taxon‐specific versus general locus sets for targeted sequence capture in plant phylogenomics. Applications in Plant Sciences 6, e1032
| Comparison of taxon‐specific versus general locus sets for targeted sequence capture in plant phylogenomics.Crossref | GoogleScholarGoogle Scholar | 29732262PubMed |
Chen X, Li H, Pandey MK, Yang Q, Wang X, Garg V, Li H, Chi X, Doddamani D, Hong Y, Upadhyaya H, Guo H, et al (2016) Draft genome of the peanut A-genome progenitor (Arachis duranensis) provides insights into geocarpy, oil biosynthesis, and allergens. Proceedings of the National Academy of Sciences of the United States of America 113, 6785–6790.
| Draft genome of the peanut A-genome progenitor (Arachis duranensis) provides insights into geocarpy, oil biosynthesis, and allergens.Crossref | GoogleScholarGoogle Scholar | 27247390PubMed |
Chen X, Lu Q, Liu H, Zhang J, Hong Y, Lan H, Li H, Wang J, Liu H, Li S, Pandey MK, Zhang Z, et al (2019) Sequencing of cultivated peanut, Arachis hypogaea, yields insights into genome evolution and oil improvement. Molecular Plant 12, 920–934.
| Sequencing of cultivated peanut, Arachis hypogaea, yields insights into genome evolution and oil improvement.Crossref | GoogleScholarGoogle Scholar | 30902685PubMed |
Cheng S, Melkonian M, Smith SA, Brockington S, Archibald JM, Delaux PM, Li FW, Melkonian B, Mavrodiev EV, Sun W, Fu Y, Yang H, et al (2018) 10KP: a phylodiverse genome sequencing plan. GigaScience 7, giy013
| 10KP: a phylodiverse genome sequencing plan.Crossref | GoogleScholarGoogle Scholar | 29618049PubMed |
Chidebe IN, Jaiswal SK, Dakora FD (2018) Distribution and phylogeny of microsymbionts associated with cowpea (Vigna unguiculata) nodulation in three agroecological regions of Mozambique. Applied and Environmental Microbiology 84, e01712–e01717.
Choi HK, Luckow MA, Doyle J, Cook DR (2006) Development of nuclear gene-derived molecular markers linked to legume genetic maps. Molecular Genetics and Genomics 276, 56–70.
| Development of nuclear gene-derived molecular markers linked to legume genetic maps.Crossref | GoogleScholarGoogle Scholar | 16642337PubMed |
Chown SL, Hodgins KA, Griffin PC, Oakeshott JG, Byrne M, Hoffmann AA (2015) Biological invasions, climate change and genomics. Evolutionary Applications 8, 23–46.
| Biological invasions, climate change and genomics.Crossref | GoogleScholarGoogle Scholar | 25667601PubMed |
Contreras-Ortiz N, Atchison GW, Hughes CE, Madriňán S (2018) Convergent evolution of high elevation plant growth forms and geographically structured variation in Andean Lupinus (Fabaceae). Botanical Journal of the Linnean Society 187, 118–136.
| Convergent evolution of high elevation plant growth forms and geographically structured variation in Andean Lupinus (Fabaceae).Crossref | GoogleScholarGoogle Scholar |
Cortés AJ, Blair MW (2018) Genotyping by sequencing and genome–environment associations in wild common bean predict widespread divergent adaptation to drought. Frontiers in Plant Science 9, 128
| Genotyping by sequencing and genome–environment associations in wild common bean predict widespread divergent adaptation to drought.Crossref | GoogleScholarGoogle Scholar | 29515597PubMed |
Dash S, Campbell JD, Cannon EK, Cleary AM, Huang W, Kalberer SR, Karingula V, Rice AG, Singh J, Umale PE, Weeks NT, Wilkey AP, Farmer AD, Cannon SB (2016) Legume information system (LegumeInfo. org): a key component of a set of federated data resources for the legume family. Nucleic Acids Research 44, D1181–D1188.
| Legume information system (LegumeInfo. org): a key component of a set of federated data resources for the legume family.Crossref | GoogleScholarGoogle Scholar | 26546515PubMed |
de la Harpe M, Paris M, Karger DN, Rolland J, Kessler M, Salamin N, Lexer C (2017) Molecular ecology studies of species radiations: current research gaps, opportunities and challenges. Molecular Ecology 26, 2608–2622.
| Molecular ecology studies of species radiations: current research gaps, opportunities and challenges.Crossref | GoogleScholarGoogle Scholar | 28316112PubMed |
de Queiroz LP, Pastore JF, Cardoso D, Snak C, Lima AL, Gagnon E, Vatanparast M, Holland AE, Egan AN (2015) A multilocus phylogenetic analysis reveals the monophyly of a recircumscribed papilionoid legume tribe Diocleae with well-supported generic relationships. Molecular Phylogenetics and Evolution 90, 1–9.
| A multilocus phylogenetic analysis reveals the monophyly of a recircumscribed papilionoid legume tribe Diocleae with well-supported generic relationships.Crossref | GoogleScholarGoogle Scholar | 25934529PubMed |
de Sousa F, Bertrand YJ, Nylinder S, Oxelman B, Eriksson JS, Pfeil BE (2014) Phylogenetic properties of 50 nuclear loci in Medicago (Leguminosae) generated using multiplexed sequence capture and next-generation sequencing. PLoS One 9, e109704
| Phylogenetic properties of 50 nuclear loci in Medicago (Leguminosae) generated using multiplexed sequence capture and next-generation sequencing.Crossref | GoogleScholarGoogle Scholar | 25329401PubMed |
De Vega JJ, Ayling S, Hegarty M, Kudrna D, Goicoechea JL, Ergon Å, Rognli OA, Jones C, Swain M, Geurts R, Lang C, Mayer KFX, et al (2015) Red clover (Trifolium pratense L.) draft genome provides a platform for trait improvement. Scientific Reports 5, 17394
| Red clover (Trifolium pratense L.) draft genome provides a platform for trait improvement.Crossref | GoogleScholarGoogle Scholar | 26617401PubMed |
Deng LQ, Yu HQ, Liu YP, Jiao PP, Zhou SF, Zhang SZ, Li WC, Fu FL (2014) Heterologous expression of antifreeze protein gene AnAFP from Ammopiptanthus nanus enhances cold tolerance in Escherichia coli and tobacco. Gene 539, 132–140.
| Heterologous expression of antifreeze protein gene AnAFP from Ammopiptanthus nanus enhances cold tolerance in Escherichia coli and tobacco.Crossref | GoogleScholarGoogle Scholar | 24502990PubMed |
Dohle S, Berny Mier y Teran JC, Egan AN, Kisha T, Khoury CK (2019) Wild beans (Phaseolus L.) of North America. In ‘North American Crop Wild Relatives, Vol. 2: Important Species’. (Eds SL Greene, KA Williams, CK Khoury, MB Kantar, LF Marek) pp. 99–130. (Springer: Cham, Switzerland)
Doyle JJ (2012) Polyploidy in legumes. In ‘Polyploidy and Genome Evolution’. (Eds. P Soltis, D Soltis) pp. 147–180. (Springer: New York, NY, USA)
Doyle JJ (2013) The promise of genomics for a next generation of advances in higher-level legume molecular systematics. South African Journal of Botany 89, 10–18.
| The promise of genomics for a next generation of advances in higher-level legume molecular systematics.Crossref | GoogleScholarGoogle Scholar |
Doyle JJ (2016) Chasing unicorns: nodulation origins and the paradox of novelty. American Journal of Botany 103, 1865–1868.
| Chasing unicorns: nodulation origins and the paradox of novelty.Crossref | GoogleScholarGoogle Scholar | 27756731PubMed |
Doyle JJ, Beachy RN (1985) Ribosomal gene variation in soybean (Glycine) and its relatives. Theoretical and Applied Genetics 70, 369–376.
| Ribosomal gene variation in soybean (Glycine) and its relatives.Crossref | GoogleScholarGoogle Scholar | 24253007PubMed |
Dudchenko O, Pham M, Lui C, Batra SS, Hoeger M, Nyquist SK, Durand NC, Shamim MS, Machol I, Erskine W, Aiden EL, Kaur P (2018) Hi-C yields chromosome-length scaffolds for a legume genome, Trifolium subterraneum. bioRxiv 2018, 473553.
Egan AN, Doyle J (2010) A comparison of global, gene-specific, and relaxed clock methods in a comparative genomics framework: dating the polyploid history of soybean (Glycine max). Systematic Biology 59, 534–547.
| A comparison of global, gene-specific, and relaxed clock methods in a comparative genomics framework: dating the polyploid history of soybean (Glycine max).Crossref | GoogleScholarGoogle Scholar | 20705909PubMed |
Egan AN, Schlueter J, Spooner DM (2012) Applications of next-generation sequencing in plant biology. American Journal of Botany 99, 175–185.
| Applications of next-generation sequencing in plant biology.Crossref | GoogleScholarGoogle Scholar | 22312116PubMed |
Egan AN, Vatanparast M, Cagle W (2016) Parsing polyphyletic Pueraria: delimiting distinct evolutionary lineages through phylogeny. Molecular Phylogenetics and Evolution 104, 44–59.
| Parsing polyphyletic Pueraria: delimiting distinct evolutionary lineages through phylogeny.Crossref | GoogleScholarGoogle Scholar | 27495827PubMed |
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 6, e19379
| A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species.Crossref | GoogleScholarGoogle Scholar | 21573248PubMed |
Eriksson JS, Blanco-Pastor JL, Sousa F, Bertrand YJ, Pfeil BE (2017) A cryptic species produced by autopolyploidy and subsequent introgression involving Medicago prostrata (Fabaceae). Molecular Phylogenetics and Evolution 107, 367–381.
| A cryptic species produced by autopolyploidy and subsequent introgression involving Medicago prostrata (Fabaceae).Crossref | GoogleScholarGoogle Scholar | 27919807PubMed |
Eriksson JS, de Sousa F, Bertrand YJ, Antonelli A, Oxelman B, Pfeil BE (2018) Allele phasing is critical to revealing a shared allopolyploid origin of Medicago arborea and M. strasseri (Fabaceae). BMC Evolutionary Biology 18, 9
| Allele phasing is critical to revealing a shared allopolyploid origin of Medicago arborea and M. strasseri (Fabaceae).Crossref | GoogleScholarGoogle Scholar | 29374461PubMed |
Fan W, Xu JM, Wu P, Yang ZX, Lou HQ, Chen WW, Jin JF, Zheng SJ, Yang JL (2019) Alleviation by abscisic acid of Al toxicity in rice bean is not associated with citrate efflux but depends on ABI5‐mediated signal transduction pathways. Journal of Integrative Plant Biology 61, 140–154.
| Alleviation by abscisic acid of Al toxicity in rice bean is not associated with citrate efflux but depends on ABI5‐mediated signal transduction pathways.Crossref | GoogleScholarGoogle Scholar | 29975451PubMed |
Foyer CH, Lam HM, Nguyen HT, Siddique KH, Varshney RK, Colmer TD, Cowling W, Bramley H, Mori TA, Hodgson JM, Cooper JW, Miller AJ, et al (2016) Neglecting legumes has compromised human health and sustainable food production. Nature Plants 2, 16112
| Neglecting legumes has compromised human health and sustainable food production.Crossref | GoogleScholarGoogle Scholar | 28221372PubMed |
Gailing O, Staton ME, Lane T, Schlarbaum SE, Nipper R, Owusu SA, Carlson JE (2017) Construction of a framework genetic linkage map in Gleditsia triacanthos L. Plant Molecular Biology Reporter 35, 177–187.
| Construction of a framework genetic linkage map in Gleditsia triacanthos L.Crossref | GoogleScholarGoogle Scholar |
Gao F, Wang X, Li X, Xu M, Li H, Abla M, Sun H, Wei S, Feng J, Zhou Y (2018) Long-read sequencing and de novo genome assembly of Ammopiptanthus nanus, a desert shrub. GigaScience 7, 1–5.
| Long-read sequencing and de novo genome assembly of Ammopiptanthus nanus, a desert shrub.Crossref | GoogleScholarGoogle Scholar |
Gnirke A, Melnikov A, Maguire J, Rogov P, LeProust EM, Brockman W, Fennell T, Giannoukos G, Fisher S, Russ C, Gabriel S (2009) Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nature Biotechnology 27, 182–189.
| Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing.Crossref | GoogleScholarGoogle Scholar | 19182786PubMed |
Graham PH, Vance CP (2003) Legumes: importance and constraints to greater use. Plant Physiology 131, 872–877.
| Legumes: importance and constraints to greater use.Crossref | GoogleScholarGoogle Scholar | 12644639PubMed |
Grando C (2015) Mating system of Piptadenia gonoacantha (Mart) Macbr (Fabaceae) and genetic diversity at forest restoration sites. PhD dissertation, UNICAMP, Campinas, Sao Paulo, Brazil.
Griesmann M, Chang Y, Liu X, Song Y, Haberer G, Crook MB, Billault-Penneteau B, Lauressergues D, Keller J, Imanishi L, Roswanjaya YP, Kohlen W, et al (2018) Phylogenomics reveals multiple losses of nitrogen-fixing root nodule symbiosis. Science 361, eaat1743
| Phylogenomics reveals multiple losses of nitrogen-fixing root nodule symbiosis.Crossref | GoogleScholarGoogle Scholar | 29794220PubMed |
Grillo MA, De Mita S, Burke PV, Solórzano‐Lowell KL, Heath KD (2016) Intrapopulation genomics in a model mutualist: population structure and candidate symbiosis genes under selection in Medicago truncatula. Evolution 70, 2704–2717.
| Intrapopulation genomics in a model mutualist: population structure and candidate symbiosis genes under selection in Medicago truncatula.Crossref | GoogleScholarGoogle Scholar | 27757965PubMed |
Grover CE, Arick MA, Thrash A, Conover JL, Sanders WS, Peterson DG, Frelichowski JE, Scheffler JA, Scheffler BE, Wendel JF (2019) Insights into the evolution of the New World diploid cottons (Gossypium, subgenus Houzingenia) based on genome sequencing. Genome Biology and Evolution 11, 53–71.
| Insights into the evolution of the New World diploid cottons (Gossypium, subgenus Houzingenia) based on genome sequencing.Crossref | GoogleScholarGoogle Scholar | 30476109PubMed |
Gugger PF, Liang CT, Sork VL, Hodgskiss P, Wright JW (2018) Applying landscape genomic tools to forest management and restoration of Hawaiian koa (Acacia koa) in a changing environment. Evolutionary Applications 11, 231–242.
| Applying landscape genomic tools to forest management and restoration of Hawaiian koa (Acacia koa) in a changing environment.Crossref | GoogleScholarGoogle Scholar | 29387158PubMed |
Hane JK, Ming Y, Kamphuis LG, Nelson MN, Garg G, Atkins CA, Bayer PE, Bravo A, Bringans S, Cannon S, Edwards D, Foley R, et al (2017) A comprehensive draft genome sequence for lupin (Lupinus angustifolius), an emerging health food: insights into plant–microbe interactions and legume evolution. Plant Biotechnology Journal 15, 318–330.
| A comprehensive draft genome sequence for lupin (Lupinus angustifolius), an emerging health food: insights into plant–microbe interactions and legume evolution.Crossref | GoogleScholarGoogle Scholar | 27557478PubMed |
Harouna DV, Venkataramana PB, Ndakidemi PA, Matemu AO (2018) Under-exploited wild Vigna species potentials in human and animal nutrition: a review. Global Food Security 18, 1–11.
| Under-exploited wild Vigna species potentials in human and animal nutrition: a review.Crossref | GoogleScholarGoogle Scholar |
Harrison TL, Wood CW, Borges IL, Stinchcombe JR (2017) No evidence for adaptation to local rhizobial mutualists in the legume Medicago lupulina. Ecology and Evolution 7, 4367–4376.
| No evidence for adaptation to local rhizobial mutualists in the legume Medicago lupulina.Crossref | GoogleScholarGoogle Scholar | 28649348PubMed |
Harrison JG, Forister ML, Mcknight SR, Nordin E, Parchman TL (2019) Rarity does not limit genetic variation or preclude subpopulation structure in the geographically restricted desert forb Astragalus lentiginosus var. piscinensis. American Journal of Botany 106, 260–269.
| Rarity does not limit genetic variation or preclude subpopulation structure in the geographically restricted desert forb Astragalus lentiginosus var. piscinensis.Crossref | GoogleScholarGoogle Scholar | 30763451PubMed |
Haynsen MS, Vatanparast M, Mahadwar G, Zhu D, Moger-Reischer RZ, Doyle JJ, Crandall KA, Egan AN (2018) De novo transcriptome assembly of Pueraria montana var. lobata and Neustanthus phaseoloides kudzu transcriptomes for the development of eSSR and SNP markers: narrowing the US origin(s) of the invasive kudzu. BMC Genomics 19, 439
| De novo transcriptome assembly of Pueraria montana var. lobata and Neustanthus phaseoloides kudzu transcriptomes for the development of eSSR and SNP markers: narrowing the US origin(s) of the invasive kudzu.Crossref | GoogleScholarGoogle Scholar | 29871589PubMed |
Helliwell EE, Faber‐Hammond J, Lopez ZC, Garoutte A, von Wettberg E, Friesen ML, Porter SS (2018) Rapid establishment of a flowering cline in Medicago polymorpha after invasion of North America. Molecular Ecology 27, 4758–4774.
| Rapid establishment of a flowering cline in Medicago polymorpha after invasion of North America.Crossref | GoogleScholarGoogle Scholar | 30325569PubMed |
Hiiesalu I, Öpik MA, Metsis M, Lilje L, Davison J, Vasar M, Moora M, Zobel M, Wilson SD, Päertel M (2012) Plant species richness belowground: higher richness and new patterns revealed by next‐generation sequencing. Molecular Ecology 21, 2004–2016.
| Plant species richness belowground: higher richness and new patterns revealed by next‐generation sequencing.Crossref | GoogleScholarGoogle Scholar | 22168247PubMed |
Hirakawa H, Kaur P, Shirasawa K, Nichols P, Nagano S, Appels R, Erskine W, Isobe SN (2016) Draft genome sequence of subterranean clover, a reference for genus Trifolium. Scientific Reports 6, 30358
| Draft genome sequence of subterranean clover, a reference for genus Trifolium.Crossref | GoogleScholarGoogle Scholar | 27545089PubMed |
Hodel RG, Gitzendanner MA, Germain‐Aubrey CC, Liu X, Crowl AA, Sun M, Landis JB, Segovia‐Salcedo MC, Douglas NA, Chen S, Soltis DE (2016) A new resource for the development of SSR markers: millions of loci from a thousand plant transcriptomes. Applications in Plant Sciences 4, 1600024
| A new resource for the development of SSR markers: millions of loci from a thousand plant transcriptomes.Crossref | GoogleScholarGoogle Scholar | 27347456PubMed |
Hollingsworth PM, Li DZ, van der Bank M, Twyford AD (2016) Telling plant species apart with DNA: from barcodes to genomes. Philosophical Transactions of the Royal Society of London – B. Biological Sciences 371, 20150338
| Telling plant species apart with DNA: from barcodes to genomes.Crossref | GoogleScholarGoogle Scholar | 27481790PubMed |
Honorio Coronado EN, Blanc-Jolivet C, Mader M, García-Dávila CR, Sebbenn AM, Meyer-Sand BR, Paredes-Villanueva K, Tysklind N, Troispoux V, Massot M, Degen B (2019) Development of nuclear and plastid SNP markers for genetic studies of Dipteryx tree species in Amazonia. Conservation Genetics Resources 11, 333
| Development of nuclear and plastid SNP markers for genetic studies of Dipteryx tree species in Amazonia.Crossref | GoogleScholarGoogle Scholar |
Hughes C, Eastwood R (2006) Island radiation on a continental scale: exceptional rates of plant diversification after uplift of the Andes. Proceedings of the National Academy of Sciences of the United States of America 103, 10334–10339.
| Island radiation on a continental scale: exceptional rates of plant diversification after uplift of the Andes.Crossref | GoogleScholarGoogle Scholar | 16801546PubMed |
Hughes CE, Nyffeler R, Linder HP (2015) Evolutionary plant radiations: where, when, why and how? New Phytologist 207, 249–253.
| Evolutionary plant radiations: where, when, why and how?Crossref | GoogleScholarGoogle Scholar | 26096199PubMed |
International Human Genome Sequencing Consortium (2001) Initial sequencing and analysis of the human genome. Nature 409, 860–921.
| Initial sequencing and analysis of the human genome.Crossref | GoogleScholarGoogle Scholar | 11237011PubMed |
Ištvánek J, Jaroš M, Křenek A, Řepková J (2014) Genome assembly and annotation for red clover (Trifolium pratense; Fabaceae). American Journal of Botany 101, 327–337.
| Genome assembly and annotation for red clover (Trifolium pratense; Fabaceae).Crossref | GoogleScholarGoogle Scholar | 24500806PubMed |
Jain M, Misra G, Patel RK, Priya P, Jhanwar S, Khan AW, Shah N, Singh VK, Garg R, Jeena G, Yadav M, Kant C, et al (2013) A draft genome sequence of the pulse crop chickpea (Cicer arietinum L.). The Plant Journal 74, 715–729.
| A draft genome sequence of the pulse crop chickpea (Cicer arietinum L.).Crossref | GoogleScholarGoogle Scholar | 23489434PubMed |
Jiang Z, Wang H, Michal JJ, Zhou X, Liu B, Woods LC, Fuchs RA (2016) Genome wide sampling sequencing for SNP genotyping: methods, challenges and future development. International Journal of Biological Sciences 12, 100–108.
| Genome wide sampling sequencing for SNP genotyping: methods, challenges and future development.Crossref | GoogleScholarGoogle Scholar | 26722221PubMed |
Jiao WB, Schneeberger K (2017) The impact of third generation genomic technologies on plant genome assembly. Current Opinion in Plant Biology 36, 64–70.
| The impact of third generation genomic technologies on plant genome assembly.Crossref | GoogleScholarGoogle Scholar | 28231512PubMed |
Jimenez-Madrigal JP (2018) Next-generation sequencing technologies in tree improvement and conservation genetics of Dipteryx oleifera Benth. PhD dissertation, North Carolina State University, Raleigh, NC, USA.
Johnson MG, Pokorny L, Dodsworth S, Botigué LR, Cowan RS, Devault A, Eiserhardt WL, Epitawalage N, Forest F, Kim JT, Leebens-Mack JH, Leitch IJ, et al (2019) A universal probe set for targeted sequencing of 353 nuclear genes from any flowering plant designed using k-medoids clustering. Systematic Biology 68, 594–606.
| A universal probe set for targeted sequencing of 353 nuclear genes from any flowering plant designed using k-medoids clustering.Crossref | GoogleScholarGoogle Scholar | 30535394PubMed |
Kadlec M, Bellstedt DU, Le Maitre NC, Pirie MD (2017) Targeted NGS for species level phylogenomics: ‘made to measure’ or ‘one size fits all’? PeerJ 5, e3569
| Targeted NGS for species level phylogenomics: ‘made to measure’ or ‘one size fits all’?Crossref | GoogleScholarGoogle Scholar | 28761782PubMed |
Kamutando CN, Vikram S, Kamgan-Nkuekam G, Makhalanyane TP, Greve M, Le Roux JJ, Richardson DM, Cowan D, Valverde A (2017) Soil nutritional status and biogeography influence rhizosphere microbial communities associated with the invasive tree Acacia dealbata. Scientific Reports 7, 6472
| Soil nutritional status and biogeography influence rhizosphere microbial communities associated with the invasive tree Acacia dealbata.Crossref | GoogleScholarGoogle Scholar | 28747705PubMed |
Kang YJ, Kim SK, Kim MY, Lestari P, Kim KH, Ha BK, Jun TH, Hwang WJ, Lee T, Lee J, Shim S, Yoon MY, et al (2014) Genome sequence of mungbean and insights into evolution within Vigna species. Nature Communications 5, 5443
| Genome sequence of mungbean and insights into evolution within Vigna species.Crossref | GoogleScholarGoogle Scholar | 25384727PubMed |
Kang YJ, Satyawan D, Shim S, Lee T, Lee J, Hwang WJ, Kim SK, Lestari P, Laosatit K, Kim KH, Ha TJ, Chitikineni A, et al (2015) Draft genome sequence of adzuki bean, Vigna angularis. Scientific Reports 5, 8069
| Draft genome sequence of adzuki bean, Vigna angularis.Crossref | GoogleScholarGoogle Scholar | 25626881PubMed |
Kang SH, Lee HO, Ahn BO, Kim CK (2019a) The complete mitochondrial genome sequence of Senna occidentalis (Fabales: Fabaceae). Mitochondrial DNA – B. Resources 4, 85–86.
| The complete mitochondrial genome sequence of Senna occidentalis (Fabales: Fabaceae).Crossref | GoogleScholarGoogle Scholar |
Kang SH, Won SY, Kim CK (2019b) The complete mitochondrial genome sequences of Senna tora (Fabales: Fabaceae). Mitochondrial DNA – B. Resources 4, 1283–1284.
| The complete mitochondrial genome sequences of Senna tora (Fabales: Fabaceae).Crossref | GoogleScholarGoogle Scholar |
Kaur P, Bayer PE, Milec Z, Vrána J, Yuan Y, Appels R, Edwards D, Batley J, Nichols P, Erskine W, Doležel J (2017) An advanced reference genome of Trifolium subterraneum L. reveals genes related to agronomic performance. Plant Biotechnology Journal 15, 1034–1046.
| An advanced reference genome of Trifolium subterraneum L. reveals genes related to agronomic performance.Crossref | GoogleScholarGoogle Scholar | 28111887PubMed |
Kazakoff SH, Imelfort M, Edwards D, Koehorst J, Biswas B, Batley J, Scott PT, Gresshoff PM (2012) Capturing the biofuel wellhead and powerhouse: the chloroplast and mitochondrial genomes of the leguminous feedstock tree Pongamia pinnata. PLoS One 7, e51687
| Capturing the biofuel wellhead and powerhouse: the chloroplast and mitochondrial genomes of the leguminous feedstock tree Pongamia pinnata.Crossref | GoogleScholarGoogle Scholar | 23272141PubMed |
Kazempour Osaloo S, Maassoumi AA, Murakami N (2003) Molecular systematics of the genus Astragalus L. (Fabaceae): phylogenetic analyses of nuclear ribosomal DNA internal transcribed spacers and chloroplast gene ndhF sequences. Plant Systematics and Evolution 242, 1–32.
| Molecular systematics of the genus Astragalus L. (Fabaceae): phylogenetic analyses of nuclear ribosomal DNA internal transcribed spacers and chloroplast gene ndhF sequences.Crossref | GoogleScholarGoogle Scholar |
Kellar PR, Ahrendsen DL, Aust SK, Jones AR, Pires JC (2015) Biodiversity comparison among phylogenetic diversity metrics and between three North American prairies. Applications in Plant Sciences 3, 1400108
| Biodiversity comparison among phylogenetic diversity metrics and between three North American prairies.Crossref | GoogleScholarGoogle Scholar | 26191461PubMed |
Keller J, Imperial J, Ruiz-Argüeso T, Privet K, Lima O, Michon-Coudouel S, Biget M, Salmon A, Aïnouche A, Cabello-Hurtado F (2018) RNA sequencing and analysis of three Lupinus nodulomes provide new insights into specific host-symbiont relationships with compatible and incompatible Bradyrhizobium strains. Plant Science 266, 102–116.
| RNA sequencing and analysis of three Lupinus nodulomes provide new insights into specific host-symbiont relationships with compatible and incompatible Bradyrhizobium strains.Crossref | GoogleScholarGoogle Scholar | 29241560PubMed |
Kim MY, Lee S, Van K, Kim TH, Jeong SC, Choi IY, Kim DS, Lee YS, Park D, Ma J, Kim WY, Kim BC, et al (2010) Whole-genome sequencing and intensive analysis of the undomesticated soybean (Glycine soja Sieb. and Zucc.) genome. Proceedings of the National Academy of Sciences of the United States of America 107, 22032–22037.
| Whole-genome sequencing and intensive analysis of the undomesticated soybean (Glycine soja Sieb. and Zucc.) genome.Crossref | GoogleScholarGoogle Scholar | 21131573PubMed |
Koboldt DC, Steinberg KM, Larson DE, Wilson RK, Mardis ER (2013) The next-generation sequencing revolution and its impact on genomics. Cell 155, 27–38.
| The next-generation sequencing revolution and its impact on genomics.Crossref | GoogleScholarGoogle Scholar | 24074859PubMed |
Koenen EJM, de Vos JM, Atchison GW, Simon MF, Schrire BD, de Souza ER, de Queiroz LP, Hughes CE (2013) Exploring the tempo of species diversification in legumes. South African Journal of Botany 89, 19–30.
| Exploring the tempo of species diversification in legumes.Crossref | GoogleScholarGoogle Scholar |
Koenen EJ, Ojeda DI, Steeves R, Migliore J, Bakker FT, Wieringa JJ, Kidner C, Hardy O, Pennington RT, Herendeen PS, Bruneau A (2019) The origin and early evolution of the legumes are a complex paleopolyploid phylogenomic tangle closely associated with the Cretaceous–Paleogene (K-Pg) boundary. bioRxiv 2019, 577957.
Kovar L, Nageswara-Rao M, Ortega-Rodriguez S, Dugas DV, Straub S, Cronn R, Strickler SR, Hughes CE, Hanley KA, Rodriguez DN, Langhorst BW, Dimalanta ET, Bailey CD (2018) PacBiol.-based mitochondrial genome assembly of Leucaena trichandra (Leguminosae) and an intrageneric assessment of mitochondrial RNA editing. Genome Biology and Evolution 10, 2501–2517.
| PacBiol.-based mitochondrial genome assembly of Leucaena trichandra (Leguminosae) and an intrageneric assessment of mitochondrial RNA editing.Crossref | GoogleScholarGoogle Scholar | 30137422PubMed |
Kovi MR, Amdahl H, Alsheikh M, Rognli OA (2017) De novo and reference transcriptome assembly of transcripts expressed during flowering provide insight into seed setting in tetraploid red clover. Scientific Reports 7, 44383
| De novo and reference transcriptome assembly of transcripts expressed during flowering provide insight into seed setting in tetraploid red clover.Crossref | GoogleScholarGoogle Scholar | 28287176PubMed |
Kundu A, Paul S, Dey A, Pal A (2017) High throughput sequencing reveals modulation of microRNAs in Vigna mungo upon mungbean yellow mosaic India virus inoculation highlighting stress regulation. Plant Science 257, 96–105.
| High throughput sequencing reveals modulation of microRNAs in Vigna mungo upon mungbean yellow mosaic India virus inoculation highlighting stress regulation.Crossref | GoogleScholarGoogle Scholar | 28224923PubMed |
Le Quéré A, Tak N, Gehlot HS, Lavire C, Meyer T, Chapulliot D, Rathi S, Sakrouhi I, Rocha G, Rohmer M, Severac D, Filali-Maltouf A, Munive JA (2017) Genomic characterization of Ensifer aridi, a proposed new species of nitrogen-fixing rhizobium recovered from Asian, African and American deserts. BMC Genomics 18, 85
| Genomic characterization of Ensifer aridi, a proposed new species of nitrogen-fixing rhizobium recovered from Asian, African and American deserts.Crossref | GoogleScholarGoogle Scholar | 28088165PubMed |
Le Roux JJ, Keet JH, Mutiti B, Ellis AG (2017) Cultivation may not dramatically alter rhizobial community diversity or structure associated with rooibos tea (Aspalathus linearis Burm. f.) in South Africa. South African Journal of Botany 110, 87–96.
| Cultivation may not dramatically alter rhizobial community diversity or structure associated with rooibos tea (Aspalathus linearis Burm. f.) in South Africa.Crossref | GoogleScholarGoogle Scholar |
Legume Phylogeny Working Group (2013a) Legume phylogeny and classification in the 21st century: progress, prospects and lessons for other species-rich clades. Taxon 62, 217–248.
| Legume phylogeny and classification in the 21st century: progress, prospects and lessons for other species-rich clades.Crossref | GoogleScholarGoogle Scholar |
Legume Phylogeny Working Group (2013b) Towards a new classification system for legumes: progress report from the 6th international legume conference. South African Journal of Botany 89, 3–9.
| Towards a new classification system for legumes: progress report from the 6th international legume conference.Crossref | GoogleScholarGoogle Scholar |
Legume Phylogeny Working Group (2017) A new subfamily classification of the Leguminosae based on a taxonomically comprehensive phylogeny. Taxon 66, 44–77.
| A new subfamily classification of the Leguminosae based on a taxonomically comprehensive phylogeny.Crossref | GoogleScholarGoogle Scholar |
Lestari P, Kang YJ, Han KS, Gwag JG, Moon JK, Kim YH, Lee YH, Lee SH (2014) Genome-wide single nucleotide polymorphism discovery and validation in adzuki bean. Molecular Breeding 33, 497–501.
| Genome-wide single nucleotide polymorphism discovery and validation in adzuki bean.Crossref | GoogleScholarGoogle Scholar |
Lewis G, Schrire B, Mackinder B, Lock M (2005) ‘Legumes of the World.’ (Royal Botanic Gardens, Kew: London, UK)
Li XW, Yang Y, Henry RJ, Rossetto M, Wang YT, Chen SL (2015) Plant DNA barcoding: from gene to genome. Biological Reviews of the Cambridge Philosophical Society 90, 157–166.
| Plant DNA barcoding: from gene to genome.Crossref | GoogleScholarGoogle Scholar |
Liu Q, Chang S, Hartman GL, Domier LL (2018) Assembly and annotation of a draft genome sequence for Glycine latifolia, a perennial wild relative of soybean. The Plant Journal 95, 71–85.
| Assembly and annotation of a draft genome sequence for Glycine latifolia, a perennial wild relative of soybean.Crossref | GoogleScholarGoogle Scholar | 29671916PubMed |
Liu H, Wei J, Yang T, Mu W, Song B, Yang T, Fu Y, Wang X, Hu G, Li W, Zhou H, Chang Y, Chen X, Liang X (2019) Molecular digitization of a botanical garden: high-depth whole genome sequencing of 689 vascular plant species from the Ruili Botanical Garden. GigaScience 8, giz007
| Molecular digitization of a botanical garden: high-depth whole genome sequencing of 689 vascular plant species from the Ruili Botanical Garden.Crossref | GoogleScholarGoogle Scholar | 30689848PubMed |
Lonardi S, Muñoz-Amatriaín M, Liang Q, Shu S, Wanamaker SI, Lo S, Tanskanen J, Schulman AH, Zhu T, Luo MC, Alhakami H (2019) The genome of cowpea (Vigna unguiculata [L.] Walp.). The Plant Journal 98, 767–782.
| The genome of cowpea (Vigna unguiculata [L.] Walp.).Crossref | GoogleScholarGoogle Scholar | 31017340PubMed |
Lu Q, Li H, Hong Y, Zhang G, Wen S, Li X, Zhou G, Li S, Liu H, Liu H, Liu Z, Varshney RK, Chen X, Liang X (2018) Genome sequencing and analysis of the peanut B-genome progenitor (Arachis ipaensis). Frontiers in Plant Science 9, 604
| Genome sequencing and analysis of the peanut B-genome progenitor (Arachis ipaensis).Crossref | GoogleScholarGoogle Scholar | 29774047PubMed |
Mahato AK, Sharma AK, Sharma TR, Singh NK (2018) An improved draft of the pigeonpea (Cajanus cajan (L.) Millsp.) genome. Data in Brief 16, 376–380.
| An improved draft of the pigeonpea (Cajanus cajan (L.) Millsp.) genome.Crossref | GoogleScholarGoogle Scholar | 29234695PubMed |
Malyshev LI (2008) Phenetics of subgenera and sections in the genus Oxytropis DC. (Fabaceae) bearing on ecology and phylogeny. Contemporary Problems of Ecology 1, 440–444.
| Phenetics of subgenera and sections in the genus Oxytropis DC. (Fabaceae) bearing on ecology and phylogeny.Crossref | GoogleScholarGoogle Scholar |
Manzanilla V, Bruneau A (2012) Phylogeny reconstruction in the Caesalpinieae grade (Leguminosae) based on duplicated copies of the sucrose synthase gene and plastid markers. Molecular Phylogenetics and Evolution 65, 149–162.
| Phylogeny reconstruction in the Caesalpinieae grade (Leguminosae) based on duplicated copies of the sucrose synthase gene and plastid markers.Crossref | GoogleScholarGoogle Scholar | 22699157PubMed |
Marques A, Moraes L, Aparecida dos Santos M, Costa I, Costa L, Nunes T, Melo N, Simon MF, Leitch AR, Almeida C, Souza G (2018) Origin and parental genome characterization of the allotetraploid Stylosanthes scabra Vogel (Papilionoideae, Leguminosae), an important legume pasture crop. Annals of Botany 122, 1143–1159.
| Origin and parental genome characterization of the allotetraploid Stylosanthes scabra Vogel (Papilionoideae, Leguminosae), an important legume pasture crop.Crossref | GoogleScholarGoogle Scholar | 29982475PubMed |
Marris E (2008) Soya genome sequenced. NATNews 2008, 1294
| Soya genome sequenced.Crossref | GoogleScholarGoogle Scholar |
Masuta Y, Kawabe A, Nozawa K, Naito K, Kato A, Ito H (2018) Characterization of a heat-activated retrotransposon in Vigna angularis. Breeding Science 68, 168–176.
| Characterization of a heat-activated retrotransposon in Vigna angularis.Crossref | GoogleScholarGoogle Scholar | 29875600PubMed |
Matasci N, Hung LH, Yan Z, Carpenter EJ, Wickett NJ, Mirarab S, Nguyen N, Warnow T, Ayyampalayam S, Barker M, Burleigh JG (2014) Data access for the 1000 Plants (1KP) project. GigaScience 3, 17
| Data access for the 1000 Plants (1KP) project.Crossref | GoogleScholarGoogle Scholar | 25625010PubMed |
McClean PE, Burridge J, Beebe S, Rao IM, Porch TG (2011) Crop improvement in the era of climate change: an integrated, multi-disciplinary approach for common bean (Phaseolus vulgaris). Functional Plant Biology 38, 927–933.
| Crop improvement in the era of climate change: an integrated, multi-disciplinary approach for common bean (Phaseolus vulgaris).Crossref | GoogleScholarGoogle Scholar |
McKenna P, Cannon N, Conway J, Dooley J, Davies WP (2018) Red clover (Trifolium pratense) in conservation agriculture: a compelling case for increased adoption. International Journal of Agricultural Sustainability 16, 342–366.
| Red clover (Trifolium pratense) in conservation agriculture: a compelling case for increased adoption.Crossref | GoogleScholarGoogle Scholar |
Meek MH, Larson WA (2019) The future is now: amplicon sequencing and sequence capture usher in the conservation genomics era. Molecular Ecology Resources 19, 795–803.
| The future is now: amplicon sequencing and sequence capture usher in the conservation genomics era.Crossref | GoogleScholarGoogle Scholar | 30681776PubMed |
Mensous M, Van de Paer C, Manzi S, Bouchez O, Baâli-Cherif D, Besnard G (2017) Diversity and evolution of plastomes in Saharan mimosoids: potential use for phylogenetic and population genetic studies. Tree Genetics & Genomes 13, 48
| Diversity and evolution of plastomes in Saharan mimosoids: potential use for phylogenetic and population genetic studies.Crossref | GoogleScholarGoogle Scholar |
Miller MR, Dunham JP, Amores A, Cresko WA, Johnson EA (2007) Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Research 17, 240–248.
| Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers.Crossref | GoogleScholarGoogle Scholar | 17189378PubMed |
Mochida K, Sakurai T, Seki H, Yoshida T, Takahagi K, Sawai S, Uchiyama H, Muranaka T, Saito K (2017) Draft genome assembly and annotation of Glycyrrhiza uralensis, a medicinal legume. The Plant Journal 89, 181–194.
| Draft genome assembly and annotation of Glycyrrhiza uralensis, a medicinal legume.Crossref | GoogleScholarGoogle Scholar | 27775193PubMed |
Moore AJ, Vos JMD, Hancock LP, Goolsby E, Edwards EJ (2018) Targeted enrichment of large gene families for phylogenetic inference: phylogeny and molecular evolution of photosynthesis genes in the portullugo clade (Caryophyllales). Systematic Biology 67, 367–383.
| Targeted enrichment of large gene families for phylogenetic inference: phylogeny and molecular evolution of photosynthesis genes in the portullugo clade (Caryophyllales).Crossref | GoogleScholarGoogle Scholar | 29029339PubMed |
Morris AB, Scalf C, Burleyson A, Johnson LT, Trostel K (2016) Development and characterization of microsatellite primers in the federally endangered Astragalus bibullatus (Fabaceae). Applications in Plant Sciences 4, 1500126
| Development and characterization of microsatellite primers in the federally endangered Astragalus bibullatus (Fabaceae).Crossref | GoogleScholarGoogle Scholar | 27144107PubMed |
Moura TM, Vatanparast M, Tozzi AMGA, Forest F, Wilmot-Dear MC, Simon MF, Mansano VF, Kajita T, Lewis GP (2016) A molecular phylogeny and new infrageneric classification of Mucuna Adans. (Leguminosae–Papilionoideae) including insights from morphology and hypotheses about biogeography. International Journal of Plant Sciences 177, 76–89.
| A molecular phylogeny and new infrageneric classification of Mucuna Adans. (Leguminosae–Papilionoideae) including insights from morphology and hypotheses about biogeography.Crossref | GoogleScholarGoogle Scholar |
Mousavi‐Derazmahalleh M, Bayer PE, Hane JK, Valliyodan B, Nguyen HT, Nelson MN, Erskine W, Varshney RK, Papa R, Edwards D (2019) Adapting legume crops to climate change using genomic approaches. Plant, Cell & Environment 42, 6–19.
| Adapting legume crops to climate change using genomic approaches.Crossref | GoogleScholarGoogle Scholar |
Naito K, Kaga A, Tomooka N, Kawase M (2013) De novo assembly of the complete organelle genome sequences of azuki bean (Vigna angularis) using next-generation sequencers. Breeding Science 63, 176–182.
| De novo assembly of the complete organelle genome sequences of azuki bean (Vigna angularis) using next-generation sequencers.Crossref | GoogleScholarGoogle Scholar | 23853512PubMed |
Neale DB, Kremer A (2011) Forest tree genomics: growing resources and applications. Nature Reviews. Genetics 12, 111
| Forest tree genomics: growing resources and applications.Crossref | GoogleScholarGoogle Scholar | 21245829PubMed |
Negruk V (2013) Mitochondrial genome sequence of the legume Vicia faba. Frontiers in Plant Science 4, 128
| Mitochondrial genome sequence of the legume Vicia faba.Crossref | GoogleScholarGoogle Scholar | 23675376PubMed |
Nevado B, Atchison GW, Hughes CE, Filatov DA (2016) Widespread adaptive evolution during repeated evolutionary radiations in New World lupins. Nature Communications 7, 12384
| Widespread adaptive evolution during repeated evolutionary radiations in New World lupins.Crossref | GoogleScholarGoogle Scholar | 27498896PubMed |
Nevado B, Contreras‐Ortiz N, Hughes C, Filatov DA (2018) Pleistocene glacial cycles drive isolation, gene flow and speciation in the high‐elevation Andes. New Phytologist 219, 779–793.
| Pleistocene glacial cycles drive isolation, gene flow and speciation in the high‐elevation Andes.Crossref | GoogleScholarGoogle Scholar | 29862512PubMed |
Nicholls JA, Pennington RT, Koenen EJ, Hughes CE, Hearn J, Bunnefeld L, Dexter KG, Stone GN, Kidner CA (2015) Using targeted enrichment of nuclear genes to increase phylogenetic resolution in the neotropical rain forest genus Inga (Leguminosae: Mimosoideae). Frontiers in Plant Science 6, 710
| Using targeted enrichment of nuclear genes to increase phylogenetic resolution in the neotropical rain forest genus Inga (Leguminosae: Mimosoideae).Crossref | GoogleScholarGoogle Scholar | 26442024PubMed |
Ojeda DI, Koenen E, Cervantes S, de la Estrella M, Banguera-Hinestroza E, Janssens SB, Migliore J, Demenou B, Bruneau A, Forest F, Hardy OJ (2019) Phylogenomic analyses reveal an exceptionally high number of evolutionary shifts in a florally diverse clade of African legumes. Molecular Phylogenetics and Evolution 137, 156–167.
| Phylogenomic analyses reveal an exceptionally high number of evolutionary shifts in a florally diverse clade of African legumes.Crossref | GoogleScholarGoogle Scholar | 31075505PubMed |
Oliveira-Filho AT, Cardoso D, Schrire BD, Lewis GP, Pennington RT, Brummer TJ, Rotella J, Lavin M (2013) Stability structures tropical woody plant diversity more than seasonality: insights into the ecology of high legume–succulent-plant biodiversity. South African Journal of Botany 89, 42–57.
| Stability structures tropical woody plant diversity more than seasonality: insights into the ecology of high legume–succulent-plant biodiversity.Crossref | GoogleScholarGoogle Scholar |
Palmer JD, Herbon LA (1988) Plant mitochondrial DNA evolved rapidly in structure but slowly in sequence. Journal of Molecular Evolution 28, 87–97.
| Plant mitochondrial DNA evolved rapidly in structure but slowly in sequence.Crossref | GoogleScholarGoogle Scholar | 3148746PubMed |
Palmer JD, Jorgensen RA, Thompson WF (1985) Chloroplast DNA variation and evolution in Pisum: patterns of change and phylogenetic analysis. Genetics 109, 195–213.
Pan L, Wang N, Wu Z, Guo R, Yu X, Zheng Y, Xia Q, Gui S, Chen C (2017) A high density genetic map derived from RAD sequencing and its application in QTL analysis of yield-related traits in Vigna unguiculata. Frontiers in Plant Science 8, 1544
| A high density genetic map derived from RAD sequencing and its application in QTL analysis of yield-related traits in Vigna unguiculata.Crossref | GoogleScholarGoogle Scholar | 28936219PubMed |
Pang T, Ye CY, Xia X, Yin W (2013) De novo sequencing and transcriptome analysis of the desert shrub, Ammopiptanthus mongolicus, during cold acclimation using Illumina/Solexa. BMC Genomics 14, 488
| De novo sequencing and transcriptome analysis of the desert shrub, Ammopiptanthus mongolicus, during cold acclimation using Illumina/Solexa.Crossref | GoogleScholarGoogle Scholar | 23865740PubMed |
Papadopoulou A, Taberlet P, Zinger L (2015) Metagenome skimming for phylogenetic community ecology: a new era in biodiversity research. Molecular Ecology 24, 3515–3517.
| Metagenome skimming for phylogenetic community ecology: a new era in biodiversity research.Crossref | GoogleScholarGoogle Scholar | 26174127PubMed |
Podlech D, Zarre S, Ekici M, Maassoumi AA, Sytin A (2014) A taxonomic revision of the genus Astragalus L. (Leguminosae) in the Old World. Annalen des Naturhistorischen Museums in Wien – B. Botanik und Zoologie 1, 106
Porch T, Beaver J, Debouck D, Jackson S, Kelly J, Dempewolf H (2013) Use of wild relatives and closely related species to adapt common bean to climate change. Agronomy (Basel) 3, 433–461.
| Use of wild relatives and closely related species to adapt common bean to climate change.Crossref | GoogleScholarGoogle Scholar |
Ramírez-Villegas J, Khoury C, Jarvis A, Debouck DG, Guarino L (2010) A gap analysis methodology for collecting crop genepools: a case study with Phaseolus beans. PLoS One 5, e13497
| A gap analysis methodology for collecting crop genepools: a case study with Phaseolus beans.Crossref | GoogleScholarGoogle Scholar | 20976009PubMed |
Reuter JA, Spacek DV, Snyder MP (2015) High-throughput sequencing technologies. Molecular Cell 58, 586–597.
| High-throughput sequencing technologies.Crossref | GoogleScholarGoogle Scholar | 26000844PubMed |
Rice DW, Alverson AJ, Richardson AO, Young GJ, Sanchez-Puerta MV, Munzinger J, Barry K, Boore JL, Zhang Y, Knox EB, Palmer JD (2013) Horizontal transfer of entire genomes via mitochondrial fusion in the angiosperm Amborella. Science 342, 1468–1473.
| Horizontal transfer of entire genomes via mitochondrial fusion in the angiosperm Amborella.Crossref | GoogleScholarGoogle Scholar | 24357311PubMed |
Richardson JE, Pennington TR, Pennington TD, Hollingsworth PM (2001) Rapid diversification of a species-rich genus of neotropical rain forest trees. Science 293, 2242–2245.
| Rapid diversification of a species-rich genus of neotropical rain forest trees.Crossref | GoogleScholarGoogle Scholar | 11567135PubMed |
Ruhfel BR, Gitzendanner MA, Soltis PS, Soltis DE, Burleigh JG (2014) From algae to angiosperms: inferring the phylogeny of green plants (Viridiplantae) from 360 plastid genomes. BMC Evolutionary Biology 14, 23
| From algae to angiosperms: inferring the phylogeny of green plants (Viridiplantae) from 360 plastid genomes.Crossref | GoogleScholarGoogle Scholar | 24533922PubMed |
Sanchez-Puerta M, García LE, Wohlfeiler J, Ceriotti LF (2017) Unparalleled replacement of native mitochondrial genes by foreign homologs in a holoparasitic plant. New Phytologist 214, 376–387.
| Unparalleled replacement of native mitochondrial genes by foreign homologs in a holoparasitic plant.Crossref | GoogleScholarGoogle Scholar | 27905116PubMed |
Sanchez-Puerta MV, Edera A, Gandini CL, Williams AV, Howell KA, Nevill PG, Small I (2019) Genome-scale transfer of mitochondrial DNA from legume hosts to the holoparasite Lophophytum mirabile (Balanophoraceae). Molecular Phylogenetics and Evolution 132, 243–250.
| Genome-scale transfer of mitochondrial DNA from legume hosts to the holoparasite Lophophytum mirabile (Balanophoraceae).Crossref | GoogleScholarGoogle Scholar | 30528080PubMed |
Sanderson MJ, Wojciechowski MF (1996) Diversification rates in a temperate legume clade: are there ‘so many species’ of Astragalus (Fabaceae)? American Journal of Botany 83, 1488–1502.
| Diversification rates in a temperate legume clade: are there ‘so many species’ of Astragalus (Fabaceae)?Crossref | GoogleScholarGoogle Scholar |
Sanger F, Nicklen S, Coulson AR (1977) DNA sequencing with chain-terminating inhibitors. Proceedings of the National Academy of Sciences of the United States of America 74, 5463–5467.
| DNA sequencing with chain-terminating inhibitors.Crossref | GoogleScholarGoogle Scholar | 271968PubMed |
Sathyanarayana N, Pittala RK, Tripathi PK, Chopra R, Singh HR, Belamkar V, Bhardwaj PK, Doyle JJ, Egan AN (2017) Transcriptomic resources for the medicinal legume Mucuna pruriens: de novo transcriptome assembly, annotation, identification and validation of EST–SSR markers. BMC Genomics 18, 409
| Transcriptomic resources for the medicinal legume Mucuna pruriens: de novo transcriptome assembly, annotation, identification and validation of EST–SSR markers.Crossref | GoogleScholarGoogle Scholar | 28545396PubMed |
Sato S, Nakamura Y, Kaneko T, Asamizu E, Kato T, Nakao M, Sasamoto S, Watanabe A, Ono A, Kawashima K, Fujishiro T, Katoh M, et al (2008) Genome structure of the legume Lotus japonicus. DNA Research 15, 227–239.
| Genome structure of the legume Lotus japonicus.Crossref | GoogleScholarGoogle Scholar | 18511435PubMed |
Schafleitner R, Huang SM, Chu SH, Yen JY, Lin CY, Yan MR, Krishnan B, Liu MS, Lo HF, Chen CY, Chen L-FO, Wu DC, Thi Bui TG, Ramasamy S, Tung CW, Nair R (2016) Identification of single nucleotide polymorphism markers associated with resistance to bruchids (Callosobruchus spp.) in wild mungbean (Vigna radiata var. sublobata) and cultivated V. radiata through genotyping by sequencing and quantitative trait locus analysis. BMC Plant Biology 16, 159
| Identification of single nucleotide polymorphism markers associated with resistance to bruchids (Callosobruchus spp.) in wild mungbean (Vigna radiata var. sublobata) and cultivated V. radiata through genotyping by sequencing and quantitative trait locus analysis.Crossref | GoogleScholarGoogle Scholar | 27422285PubMed |
Scherson RA, Choi HK, Cook DR, Sanderson MJ (2005) Phylogenetics of New World Astragalus: screening of novel nuclear loci for the reconstruction of phylogenies at low taxonomic levels. Brittonia 57, 354–366.
| Phylogenetics of New World Astragalus: screening of novel nuclear loci for the reconstruction of phylogenies at low taxonomic levels.Crossref | GoogleScholarGoogle Scholar |
Scherson RA, Vidal R, Sanderson MJ (2008) Phylogeny, biogeography and rates of diversification of New World Astragalus (Leguminosae) with an emphasis on South American radiations. American Journal of Botany 95, 1030–1039.
| Phylogeny, biogeography and rates of diversification of New World Astragalus (Leguminosae) with an emphasis on South American radiations.Crossref | GoogleScholarGoogle Scholar | 21632423PubMed |
Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W, Hyten DL, Song Q, Thelen JJ, Cheng J, Xu D, Hellsten U, et al (2010) Genome sequence of the palaeopolyploid soybean. Nature 463, 178–183.
| Genome sequence of the palaeopolyploid soybean.Crossref | GoogleScholarGoogle Scholar | 20075913PubMed |
Schmutz J, McClean PE, Mamidi S, Wu GA, Cannon SB, Grimwood J, Jenkins J, Shu S, Song Q, Chavarro C, Torres-Torres M, Geffroy V, et al (2014) A reference genome for common bean and genome-wide analysis of dual domestications. Nature Genetics 46, 707–713.
| A reference genome for common bean and genome-wide analysis of dual domestications.Crossref | GoogleScholarGoogle Scholar | 24908249PubMed |
Schwarz EN, Ruhlman TA, Sabir JSM, Hajrah NH, Alharbi NS, Al-Malki AL, Bailey CD, Jansen RK (2015) Plastid genome sequences of legumes reveal parallel inversions and multiple losses of rps16 in papilionoids. Journal of Systematics and Evolution 53, 458–468.
| Plastid genome sequences of legumes reveal parallel inversions and multiple losses of rps16 in papilionoids.Crossref | GoogleScholarGoogle Scholar |
Shahi Shavvon R, Kazempour Osaloo S, Maassoumii AA, Moharrek F, Karaman Erkul S, Lemmon AR, Lemmon EM, Michalak I, Muellner‐Riehl AN, Favre A (2017) Increasing phylogenetic support for explosively radiating taxa: the promise of high‐throughput sequencing for Oxytropis (Fabaceae). Journal of Systematics and Evolution 55, 385–404.
| Increasing phylogenetic support for explosively radiating taxa: the promise of high‐throughput sequencing for Oxytropis (Fabaceae).Crossref | GoogleScholarGoogle Scholar |
Shen Y, Liu J, Geng H, Zhang J, Liu Y, Zhang H, Xing S, Du J, Ma S, Tian Z (2018) De novo assembly of a Chinese soybean genome. Science China. Life Sciences 61, 871–884.
| De novo assembly of a Chinese soybean genome.Crossref | GoogleScholarGoogle Scholar | 30062469PubMed |
Sherman‐Broyles S, Bombarely A, Doyle J (2017) Characterizing the allopolyploid species among the wild relatives of soybean: utility of reduced representation genotyping methodologies. Journal of Systematics and Evolution 55, 365–376.
| Characterizing the allopolyploid species among the wild relatives of soybean: utility of reduced representation genotyping methodologies.Crossref | GoogleScholarGoogle Scholar |
Shi Y, Liu Y, Zhang S, Zou R, Tang J, Mu W, Peng Y, Dong S (2018) Assembly and comparative analysis of the complete mitochondrial genome sequence of Sophora japonica ‘JinhuaiJ2’. PLoS One 13, e0202485
| Assembly and comparative analysis of the complete mitochondrial genome sequence of Sophora japonica ‘JinhuaiJ2’.Crossref | GoogleScholarGoogle Scholar | 30485326PubMed |
Simon MF, Grether R, de Queiroz LP, Skema C, Pennington RT, Hughes CE (2009) Recent assembly of the Cerrado, a neotropical plant diversity hotspot, by in situ evolution of adaptations to fire. Proceedings of the National Academy of Sciences of the United States of America 106, 20359–20364.
| Recent assembly of the Cerrado, a neotropical plant diversity hotspot, by in situ evolution of adaptations to fire.Crossref | GoogleScholarGoogle Scholar | 19918050PubMed |
Singh NK, Gupta DK, Jayaswal PK, Mahato AK, Dutta S, Singh S, Bhutani S, Dogra V, Singh BP, Kumawat G, Pal JK, Pandit A, et al (2012) The first draft of the pigeonpea genome sequence. Journal of Plant Biochemistry and Biotechnology 21, 98–112.
| The first draft of the pigeonpea genome sequence.Crossref | GoogleScholarGoogle Scholar | 24431589PubMed |
Snak C, Vatanparast M, Silva C, Lewis GP, Lavin M, Kajita T, de Queiroz LP (2016) A dated phylogeny of the papilionoid legume genus Canavalia reveals recent diversification by a pantropical liana lineage. Molecular Phylogenetics and Evolution 98, 133–146.
| A dated phylogeny of the papilionoid legume genus Canavalia reveals recent diversification by a pantropical liana lineage.Crossref | GoogleScholarGoogle Scholar | 26860339PubMed |
Soares TN, Melo DB, Resende LV, Vianello RP, Chaves LJ, Collevatti RG, Telles MP (2012) Development of microsatellite markers for the neotropical tree species Dipteryx alata (Fabaceae). American Journal of Botany 99, e72–e73.
| Development of microsatellite markers for the neotropical tree species Dipteryx alata (Fabaceae).Crossref | GoogleScholarGoogle Scholar | 22282111PubMed |
Soltis DE, Gitzendanner MA, Stull G, Chester M, Chanderbali A, Chamala S, Jordon-Thaden I, Soltis PS, Schnable PS, Barbazuk BW (2013) The potential of genomics in plant systematics. Taxon 62, 886–898.
| The potential of genomics in plant systematics.Crossref | GoogleScholarGoogle Scholar |
Soltis DE, Moore MJ, Sessa EB, Smith SA, Soltis PS (2018) Using and navigating the plant tree of life. American Journal of Botany 105, 287–290.
| Using and navigating the plant tree of life.Crossref | GoogleScholarGoogle Scholar | 29702724PubMed |
Spehn EM, Scherer-Lorenzen M, Schmid B, Hector A, Caldeira MC, Dimitrakopoulos PG, Finn JA, Jumpponen A, O’Donnovan G, Pereira JS, Schulze ED, Troumbis AY, Korner C (2002) The role of legumes as a component of biodiversity in a cross-European study of grassland biomass nitrogen. Oikos 98, 205–218.
| The role of legumes as a component of biodiversity in a cross-European study of grassland biomass nitrogen.Crossref | GoogleScholarGoogle Scholar |
Stai JS, Yadav A, Sinou C, Bruneau A, Doyle JJ, Fernández-Baca D, Cannon SB (2019) Cercis: a non-polyploid genomic relic within the generally polyploid legume family. Frontiers in Plant Science 10, 345
| Cercis: a non-polyploid genomic relic within the generally polyploid legume family.Crossref | GoogleScholarGoogle Scholar | 31105714PubMed |
Stefanović S, Pfeil BE, Palmer JD, Doyle JJ (2009) Relationships among phaseoloid legumes based on sequences from eight chloroplast regions. Systematic Botany 34, 115–128.
| Relationships among phaseoloid legumes based on sequences from eight chloroplast regions.Crossref | GoogleScholarGoogle Scholar |
Stein JC, Yu Y, Copetti D, Zwickl DJ, Zhang L, Zhang C, Chougule K, Gao D, Iwata A, Goicoechea JL, Wei S (2018) Genomes of 13 domesticated and wild rice relatives highlight genetic conservation, turnover and innovation across the genus Oryza. Nature Genetics 50, 285–296.
| Genomes of 13 domesticated and wild rice relatives highlight genetic conservation, turnover and innovation across the genus Oryza.Crossref | GoogleScholarGoogle Scholar | 29358651PubMed |
Steinrucken TV (2017) Investigating the cause of dieback in the invasive plant, Parkinsonia aculeata. PhD thesis, Western Sydney University, Sydney, NSW, Australia.
Straub SC, Parks M, Weitemier K, Fishbein M, Cronn RC, Liston A (2012) Navigating the tip of the genomic iceberg: next‐generation sequencing for plant systematics. American Journal of Botany 99, 349–364.
| Navigating the tip of the genomic iceberg: next‐generation sequencing for plant systematics.Crossref | GoogleScholarGoogle Scholar | 22174336PubMed |
Sveinsson S, Cronk Q (2014) Evolutionary origin of highly repetitive plastid genomes within the clover genus (Trifolium). BMC Evolutionary Biology 14, 228
| Evolutionary origin of highly repetitive plastid genomes within the clover genus (Trifolium).Crossref | GoogleScholarGoogle Scholar | 25403617PubMed |
Tang H, Krishnakumar V, Bidwell S, Rosen B, Chan A, Zhou S, Gentzbittel L, Childs KL, Yandell M, Gundlach H, Mayer KF, Schwartz DC, Town CD (2014) An improved genome release (version Mt4. 0) for the model legume Medicago truncatula. BMC Genomics 15, 312
| An improved genome release (version Mt4. 0) for the model legume Medicago truncatula.Crossref | GoogleScholarGoogle Scholar | 24767513PubMed |
Tian R, Parker M, Seshadri R, Reddy TB, Markowitz V, Ivanova N, Pati A, Woyke T, Baeshen MN, Baeshen NA, Kyrpides N, Reeve W (2015) High-quality permanent draft genome sequence of Bradyrhizobium sp. Th. b2, a microsymbiont of Amphicarpaea bracteata collected in Johnson City, New York. Standards in Genomic Sciences 10, 24
| High-quality permanent draft genome sequence of Bradyrhizobium sp. Th. b2, a microsymbiont of Amphicarpaea bracteata collected in Johnson City, New York.Crossref | GoogleScholarGoogle Scholar | 26203336PubMed |
Tosso F, Hardy OJ, Doucet JL, Daïnou K, Kaymak E, Migliore J (2018) Evolution in the amphi-Atlantic tropical genus Guibourtia (Fabaceae, Detarioideae), combining NGS phylogeny and morphology. Molecular Phylogenetics and Evolution 120, 83–93.
| Evolution in the amphi-Atlantic tropical genus Guibourtia (Fabaceae, Detarioideae), combining NGS phylogeny and morphology.Crossref | GoogleScholarGoogle Scholar | 29222064PubMed |
Van de Peer Y, Mizrachi E, Marchal K (2017) The evolutionary significance of polyploidy. Nature Reviews. Genetics 18, 411–424.
| The evolutionary significance of polyploidy.Crossref | GoogleScholarGoogle Scholar | 28502977PubMed |
van Velzen R, Holmer R, Bu F, Rutten L, van Zeijl A, Liu W, Santuari L, Cao Q, Sharma T, Shen D, Roswanjaya Y, Wardhani TAK, et al (2018) Comparative genomics of the nonlegume Parasponia reveals insights into evolution of nitrogen-fixing rhizobium symbioses. Proceedings of the National Academy of Sciences of the United States of America 115, E4700–E4709.
| Comparative genomics of the nonlegume Parasponia reveals insights into evolution of nitrogen-fixing rhizobium symbioses.Crossref | GoogleScholarGoogle Scholar | 29717040PubMed |
van Velzen R, Doyle JJ, Geurts R (2019) A resurrected scenario: single gain and massive loss of nitrogen-fixing nodulation. Trends in Plant Science 24, 49–57.
| A resurrected scenario: single gain and massive loss of nitrogen-fixing nodulation.Crossref | GoogleScholarGoogle Scholar | 30409687PubMed |
Varshney RK (2016) Exciting journey of 10 years from genomes to fields and markets: some success stories of genomics-assisted breeding in chickpea, pigeonpea and groundnut. Plant Science 242, 98–107.
| Exciting journey of 10 years from genomes to fields and markets: some success stories of genomics-assisted breeding in chickpea, pigeonpea and groundnut.Crossref | GoogleScholarGoogle Scholar | 26566828PubMed |
Varshney RK, Chen W, Li Y, Bharti AK, Saxena RK, Schlueter JA, Donoghue MT, Azam S, Fan G, Whaley AM, Farmer AD, Sheridan J, et al (2012) Draft genome sequence of pigeonpea (Cajanus cajan) an orphan legume crop of resource-poor farmers. Nature Biotechnology 30, 83–89.
| Draft genome sequence of pigeonpea (Cajanus cajan) an orphan legume crop of resource-poor farmers.Crossref | GoogleScholarGoogle Scholar |
Varshney RK, Song C, Saxena RK, Azam S, Yu S, Sharpe AG, Cannon S, Baek J, Rosen BD, Tar’an B, Millan T, Zhang X, et al (2013) Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nature Biotechnology 31, 240–246.
| Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement.Crossref | GoogleScholarGoogle Scholar | 23354103PubMed |
Vatanparast M, Klitgård BB, Adema FA, Pennington RT, Yahara T, Kajita T (2013) First molecular phylogeny of the pantropical genus Dalbergia: implications for infrageneric circumscription and biogeography. South African Journal of Botany 89, 143–149.
| First molecular phylogeny of the pantropical genus Dalbergia: implications for infrageneric circumscription and biogeography.Crossref | GoogleScholarGoogle Scholar |
Vatanparast M, Shetty P, Chopra R, Doyle JJ, Sathyanarayana N, Egan AN (2016) Transcriptome sequencing and marker development in winged bean (Psophocarpus tetragonolobus; Leguminosae). Scientific Reports 6, 29070
| Transcriptome sequencing and marker development in winged bean (Psophocarpus tetragonolobus; Leguminosae).Crossref | GoogleScholarGoogle Scholar | 27356763PubMed |
Vatanparast M, Powell A, Doyle JJ, Egan AN (2018) Targeting legume loci: a comparison of three methods for targeted enrichment baits design in Leguminosae phylogenomics. Applications in Plant Sciences 6, e1036
| Targeting legume loci: a comparison of three methods for targeted enrichment baits design in Leguminosae phylogenomics.Crossref | GoogleScholarGoogle Scholar | 29732266PubMed |
Verdu CF, Guichoux E, Quevauvillers S, De Thier O, Laizet YH, Delcamp A, Gévaudant F, Monty A, Porté AJ, Lejeune P, Lassois L, Mariette S (2016) Dealing with paralogy in RAD seq data: in silico detection and single nucleotide polymorphism validation in Robinia pseudoacacia L. Ecology and Evolution 6, 7323–7333.
| Dealing with paralogy in RAD seq data: in silico detection and single nucleotide polymorphism validation in Robinia pseudoacacia L.Crossref | GoogleScholarGoogle Scholar | 28725400PubMed |
Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nature Reviews. Genetics 10, 57–63.
| RNA-Seq: a revolutionary tool for transcriptomics.Crossref | GoogleScholarGoogle Scholar | 19015660PubMed |
Wang YH, Qu XJ, Chen SY, Li DZ, Yi TS (2017) Plastomes of Mimosoideae: structural and size variation, sequence divergence, and phylogenetic implication. Tree Genetics & Genomes 13, 41
| Plastomes of Mimosoideae: structural and size variation, sequence divergence, and phylogenetic implication.Crossref | GoogleScholarGoogle Scholar |
Wang YH, Wicke S, Wang H, Jin JJ, Chen SY, Zhang SD, Li DZ, Yi TS (2018) Plastid genome evolution in the early-diverging legume subfamily Cercidoideae (Fabaceae). Frontiers in Plant Science 9, 138
| Plastid genome evolution in the early-diverging legume subfamily Cercidoideae (Fabaceae).Crossref | GoogleScholarGoogle Scholar | 29479365PubMed |
Wen J, Egan AN, Dikow R, Zimmer EA (2015) Utility of transcriptome sequencing for phylogenetic inference and character evolution. In ‘Next-Generation Sequencing in Plant Systematics’. (Eds. E Hörandl, MS Appelhans) pp. 51–91. (Koeltz Scientific Books: Königstein, Germany)
Werner GD, Cornwell WK, Sprent JI, Kattge J, Kiers ET (2014) A single evolutionary innovation drives the deep evolution of symbiotic N2-fixation in angiosperms. Nature Communications 5, 4087
| A single evolutionary innovation drives the deep evolution of symbiotic N2-fixation in angiosperms.Crossref | GoogleScholarGoogle Scholar | 24912610PubMed |
Wickett NJ, Mirarab S, Nguyen N, Warnow T, Carpenter E, Matasci N, Ayyampalayam S, Barker MS, Burleigh JG, Gitzendanner MA, Ruhfel BR, Wafula E, et al (2014) Phylotranscriptomic analysis of the origin and early diversification of land plants. Proceedings of the National Academy of Sciences of the United States of America 111, E4859–E4868.
| Phylotranscriptomic analysis of the origin and early diversification of land plants.Crossref | GoogleScholarGoogle Scholar | 25355905PubMed |
Wigley K, Moot D, Wakelin SA, Laugraud A, Blond C, Seth K, Ridgway H (2017) Diverse bacterial taxa inhabit root nodules of lucerne (Medicago sativa L.) in New Zealand pastoral soils. Plant and Soil 420, 253–262.
| Diverse bacterial taxa inhabit root nodules of lucerne (Medicago sativa L.) in New Zealand pastoral soils.Crossref | GoogleScholarGoogle Scholar |
Williams AV, Miller JT, Small I, Nevill PG, Boykin LM (2016) Integration of complete chloroplast genome sequences with small amplicon datasets improves phylogenetic resolution in Acacia. Molecular Phylogenetics and Evolution 96, 1–8.
| Integration of complete chloroplast genome sequences with small amplicon datasets improves phylogenetic resolution in Acacia.Crossref | GoogleScholarGoogle Scholar | 26702955PubMed |
Wojciechowski MF, Sanderson MJ, Steele KP, Liston A (2000) Molecular phylogeny of the ‘temperate herbaceous tribes’ of papilionoid legumes: a supertree approach. In ‘Advances in Legume Systematics, Part 9’. (Eds PS Herendeen, A Bruneau) pp. 277–298. (Royal Botanic Gardens, Kew: London, UK)
Wojciechowski M, Lavin M, Sanderson M (2004) A phylogeny of legumes (Leguminosae) based on analysis of the plastid matK gene resolves many well-supported subclades within the family. American Journal of Botany 91, 1846–1862.
| A phylogeny of legumes (Leguminosae) based on analysis of the plastid matK gene resolves many well-supported subclades within the family.Crossref | GoogleScholarGoogle Scholar | 21652332PubMed |
Wong MM, Gujaria-Verma N, Ramsay L, Yuan HY, Caron C, Diapari M, Vandenberg A, Bett KE (2015) Classification and characterization of species within the genus Lens using genotyping-by-sequencing (GBS). PLoS One 10, e0122025
| Classification and characterization of species within the genus Lens using genotyping-by-sequencing (GBS).Crossref | GoogleScholarGoogle Scholar | 25815480PubMed |
Xie M, Chung CY, Li MW, Wong FL, Wang X, Liu A, Wang Z, Leung AK, Wong TH, Tong SW, Xiao Z, Fan K, et al (2019) A reference-grade wild soybean genome. Nature Communications 10, 1216
| A reference-grade wild soybean genome.Crossref | GoogleScholarGoogle Scholar | 30872580PubMed |
Xu P, Wu X, Muñoz‐Amatriaín M, Wang B, Wu X, Hu Y, Huynh BL, Close TJ, Roberts PA, Zhou W, Lu Z, Li G (2017) Genomic regions, cellular components and gene regulatory basis underlying pod length variations in cowpea (V. unguiculata L.Walp). Plant Biotechnology Journal 15, 547–557.
| Genomic regions, cellular components and gene regulatory basis underlying pod length variations in cowpea (V. unguiculata L.Walp).Crossref | GoogleScholarGoogle Scholar | 27658053PubMed |
Yahara T, Javadi F, Onoda Y, de Queiroz LP, Faith DP, Prado DE, Akasaka M, Kadoya T, Ishihama F, Davies S, Slik JW, Yi T, et al (2013) Global legume diversity assessment: concepts key indicators and strategies. Taxon 62, 249–266.
| Global legume diversity assessment: concepts key indicators and strategies.Crossref | GoogleScholarGoogle Scholar |
Yan J, Yan H, Liu LX, Chen WF, Zhang XX, Verástegui-Valdés MM, Wang ET, Han XZ (2017) Rhizobium hidalgonense sp. nov., a nodule endophytic bacterium of Phaseolus vulgaris in acid soil. Archives of Microbiology 199, 97–104.
| Rhizobium hidalgonense sp. nov., a nodule endophytic bacterium of Phaseolus vulgaris in acid soil.Crossref | GoogleScholarGoogle Scholar | 27557842PubMed |
Yang H, Tao Y, Zheng Z, Zhang Q, Zhou G, Sweetingham MW, Howieson JG, Li C (2013) Draft genome sequence, and a sequence-defined genetic linkage map of the legume crop species Lupinus angustifolius L. PLoS One 8, e64799
| Draft genome sequence, and a sequence-defined genetic linkage map of the legume crop species Lupinus angustifolius L.Crossref | GoogleScholarGoogle Scholar | 24386387PubMed |
Yang JY, Ojeda DI, Santos-Guerra A, Molina RJ, Caujapé-Castells J, Cronk Q (2018) Population differentiation in relation to conservation: nuclear microsatellite variation in the Canary Island endemic Lotus sessilifolius (Fabaceae). Conservation Genetics Resources 10, 219–227.
| Population differentiation in relation to conservation: nuclear microsatellite variation in the Canary Island endemic Lotus sessilifolius (Fabaceae).Crossref | GoogleScholarGoogle Scholar |
Yates SA, Swain MT, Hegarty MJ, Chernukin I, Lowe M, Allison GG, Ruttink T, Abberton MT, Jenkins G, Skøt L (2014) De novo assembly of red clover transcriptome based on RNA-seq data provides insight into drought response, gene discovery and marker identification. BMC Genomics 15, 453
| De novo assembly of red clover transcriptome based on RNA-seq data provides insight into drought response, gene discovery and marker identification.Crossref | GoogleScholarGoogle Scholar | 24912738PubMed |
Young ND, Debellé F, Oldroyd GE, Geurts R, Cannon SB, Udvardi MK, Benedito VA, Mayer KF, Gouzy J, Schoof H, Van de Peer Y, Proost S, et al (2011) The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature 480, 520–524.
| The Medicago genome provides insight into the evolution of rhizobial symbioses.Crossref | GoogleScholarGoogle Scholar | 22089132PubMed |
Yu HQ, Wang YG, Yong TM, She YH, Fu FL, Li WC (2014) Heterologous expression of betaine aldehyde dehydrogenase gene from Ammopiptanthus nanus confers high salt and heat tolerance to Escherichia coli. Gene 549, 77–84.
| Heterologous expression of betaine aldehyde dehydrogenase gene from Ammopiptanthus nanus confers high salt and heat tolerance to Escherichia coli.Crossref | GoogleScholarGoogle Scholar | 25046139PubMed |
Yu T, Sun L, Cui H, Liu S, Men J, Chen S, Chen Y, Lu C (2018) The complete mitochondrial genome of a tertiary relict evergreen woody plant Ammopiptanthus mongolicus. Mitochondrial DNA – B. Resources 3, 9–11.
| The complete mitochondrial genome of a tertiary relict evergreen woody plant Ammopiptanthus mongolicus.Crossref | GoogleScholarGoogle Scholar |
Zalapa JE, Cuevas H, Zhu H, Steffan S, Senalik D, Zeldin E, McCown B, Harbut R, Simon P (2012) Using next‐generation sequencing approaches to isolate simple sequence repeat (SSR) loci in the plant sciences. American Journal of Botany 99, 193–208.
| Using next‐generation sequencing approaches to isolate simple sequence repeat (SSR) loci in the plant sciences.Crossref | GoogleScholarGoogle Scholar | 22186186PubMed |
Zhan L, Paterson IG, Fraser BA, Fraser BA, Watson B, Bradbury IR, Nadukkalam Ravindran P, Reznick D, Beiko RG, Bentzen P (2017) Megasat: automated inference of microsatellite genotypes from sequence data. Molecular Ecology Resources 17, 247–256.
| Megasat: automated inference of microsatellite genotypes from sequence data.Crossref | GoogleScholarGoogle Scholar | 27333119PubMed |
Zhang J, Chiodini R, Badr A, Zhang G (2011) The impact of next-generation sequencing on genomics. Journal of Genetics and Genomics 38, 95–109.
| The impact of next-generation sequencing on genomics.Crossref | GoogleScholarGoogle Scholar | 21477781PubMed |
Zhang ML, Huang JF, Sanderson SC, Yan P, Wu YH, Pan BR (2015) Molecular biogeography of tribe Thermopsideae (Leguminosae): a Madrean–Tethyan disjunction pattern with an African origin of core genistoides. BioMed Research International 2015, 864804
| Molecular biogeography of tribe Thermopsideae (Leguminosae): a Madrean–Tethyan disjunction pattern with an African origin of core genistoides.Crossref | GoogleScholarGoogle Scholar | 26114116PubMed |
Zhang B, Jin J, Moore MJ, Yi T-S (2019) Assembly and comparative analyses of the mitochondrial genome of Castanospermum australe (Papilionoideae, Leguminosea). Australian Systematic Botany 32, 484–494.
| Assembly and comparative analyses of the mitochondrial genome of Castanospermum australe (Papilionoideae, Leguminosea).Crossref | GoogleScholarGoogle Scholar |
Zhou Y, Gao F, Liu R, Feng J, Li H (2012) De novo sequencing and analysis of root transcriptome using 454 pyrosequencing to discover putative genes associated with drought tolerance in Ammopiptanthus mongolicus. BMC Genomics 13, 266
| De novo sequencing and analysis of root transcriptome using 454 pyrosequencing to discover putative genes associated with drought tolerance in Ammopiptanthus mongolicus.Crossref | GoogleScholarGoogle Scholar | 22721448PubMed |
Zimmer EA, Wen J (2013) Using nuclear gene data for plant phylogenetics: progress and prospects. Molecular Phylogenetics and Evolution 66, 539–550.
| Using nuclear gene data for plant phylogenetics: progress and prospects.Crossref | GoogleScholarGoogle Scholar | 23375140PubMed |
Zimmer EA, Wen J (2015) Using nuclear gene data for plant phylogenetics: progress and prospects II: next‐gen approaches. Journal of Systematics and Evolution 53, 371–379.
| Using nuclear gene data for plant phylogenetics: progress and prospects II: next‐gen approaches.Crossref | GoogleScholarGoogle Scholar |