

The semen microbiome of miniature pony stallions
C. Giselle Cooke A * , Zamira Gibb B , Christopher G. Grupen C , Kathrin Schemann D , Nandan Deshpande D and Joanna E. Harnett A *
A * , Zamira Gibb B , Christopher G. Grupen C , Kathrin Schemann D , Nandan Deshpande D and Joanna E. Harnett A *
A
B
C
D
Abstract
Little is known about the microbial composition of stallion semen.
To describe the microbiota detected in equine semen of healthy miniature pony stallions.
Semen specimens were collected using a Missouri artificial vagina at a single time point. PacBio (Pacific Biosciences) genomic DNA sequencing of the 16S rRNA gene was performed on these specimens, following which next-generation microbiome bioinformatics platform QIIME2 was used to process fastq files and analyse the amplicon data. The data were categorised into genus, family, class, order and phylum.
Firmicutes and Bacteroidetes phyla predominated (76%), followed by Proteobacteria (15%). Bacteroidales, Clostridiales and Cardiobacteriales predominated the microbial rank of order (86%). Class was mainly composed of Bacteroidia, Clostridia and Gammaproteobacteria (87%), while family was mainly composed of Porphyromonadaceae, Family_XI and Cardiobacteriaceae (62%). At the level of genus, 80% of the abundance was composed of seven genera, namely Porphyromonas, Suttonella, Peptoniphilus, Fastidiosipila, Ezakiella, Petrimonas and an unknown taxon.
The findings indicate that specific microbiota may be characteristic of healthy miniature pony stallions’ semen with some inter-individual variations observed.
Larger equine studies involving fertile and infertile subjects could be informed by this study and could explore the relationship of the semen microbiome to male fertility.
Keywords: equine, fertility, microbiome, microbiota, pony, reproduction, semen, stallion.
Introduction
Exploring the composition of microbial communities that inhabit anatomical regions is an important area of research that seeks to understand the role of microbiota in the prevention of disease and supporting normal health and function. More specifically, interest in the microbial communities that inhabit the reproductive tract of humans (Hou et al. 2013; Baud et al. 2019) and animals (Heil et al. 2019; Rowe et al. 2020; Ong et al. 2022), and the microbial associations with reproductive health and fertility has increased. Within the context of equine reproduction, genitourinary infections in the stallion and contamination of semen with bacteria, yeasts and fungi from passage along the distal urethra (Hughes and Loy 1975; Rota et al. 2011) potentially affect sperm quality (Al-Kass et al. 2020a) and increase the risk of endometritis for the mare (Madsen and Christensen 1995), impairing chances of conception and live foal birth outcomes. Bacterial contamination of stored equine semen poses significant challenges for breeders, requiring the addition of antibiotics to extended semen samples to prevent bacterial growth and transmission via artificial insemination, a practice which can also result in the development of antibiotic-resistant organisms and compromised sperm quality (Malaluang et al. 2021). With this concern, and in the context of equine fertility, pathogens have been studied more extensively by veterinarian researchers than the commensal bacteria that inhabit the stallion semen.
Over the past decade the equine intestinal microbiome has been characterised in both healthy and diseased horses and foals (Al Jassim and Andrews 2009; Costa et al. 2015, 2016; Kauter et al. 2019); however, it is only very recently that the semen microbiome of stallions has been described (Al-Kass et al. 2020a; Quiñones-Pérez et al. 2021). To date, impaired progressive motility of stallion sperm has been correlated with the presence of the bacterium Clostridiales Incertae Sedis XI (Quiñones-Pérez et al. 2022), while improved total sperm motility was observed in the presence of Peptoniphilaceae family (Quiñones-Pérez et al. 2022). In the case of stallion semen, a predominance of Porphyromonadaceae, Peptoniphilaceae, Corynebacteriaceae and Prevotellaceae families characterised the microbiome profile of healthy, fertile individuals (Quiñones-Pérez et al. 2021).
A range of technologies are used to characterise microbial communities and the genes they contain. Advances in 16s rRNA gene sequencing technology have provided insights into the microbial genes present in stallion semen (Al-Kass et al. 2020a) and enabled identification of microbiota that have not been identified via culture-based techniques or MALDI-TOF MS (Matrix Assisted Laser Desorption Ionisation-Time Of Flight mass spectrometry) technology, (Al-Kass et al. 2020b), which rapidly identifies microorganisms using an accurate high-throughput methodology. The adoption of polymerase chain reaction (PCR) sampling and full length 16S rRNA gene sequencing is associated with strengths in identifying the presence of viable and non-viable bacteria – anaerobes and aerobes (Böttger 1989) – thus enabling the identification of lesser known microbes and revealing those which may be difficult to culture (Woo et al. 2008). Microbial technologies using 16S rRNA gene sequencing have been estimated to identify up to 99% of microbes present in a sample to genus level (Mignard and Flandrois 2006), hence increasing the scope and insight into the composition of microbial communities that inhabit different regions.
Environmental factors and nutrition have been shown to influence the intestinal microbiome composition in horses (Julliand and Grimm 2017; Garber et al. 2020; Mach et al. 2020; Paßlack et al. 2020; Bull et al. 2021; Theelen et al. 2021) and their general health. The environment and nutrition are also known to impact fertility, yet little is known about the composition of the semen microbiota of healthy stallions and of different breeds of horses. To date, no studies have reported the semen microbiome profiles of miniature pony stallions. The aim of this study was to add to the existing knowledge base regarding the composition of equine seminal microbiota, by identifying bacteria in more detail than previously reported, from phylum to genus, in four miniature pony stallions. (Fig. 1).
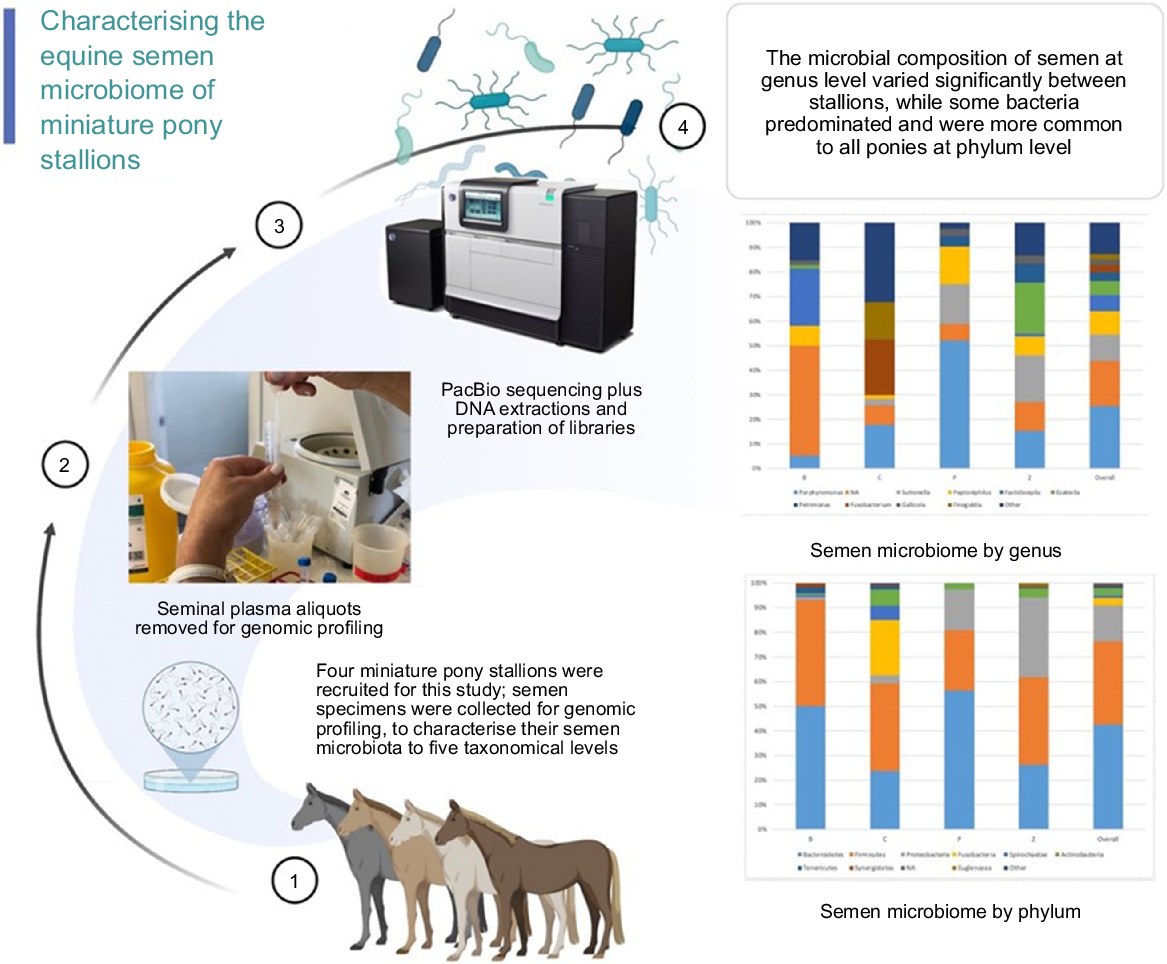
Materials and methods
Subjects
Four crossbred miniature pony stallions, aged between 2 and 15 years and stabled at an equestrian centre in New South Wales, Australia, were recruited for this study in October 2019. They comprised a cohort of horses that routinely provide semen specimens for scientific purposes and in accordance with the ethical requirements of this study.
The ponies grazed on native grasses in adjacent paddocks, and lucerne hay was added to their diet throughout the year. No exercise regime was undertaken other than free paddock roaming, and the ponies were stabled overnight. The body condition score of the ponies was 6–7 on the US body condition score chart (Henneke et al. 1983).
Study design
Semen samples were collected on the same morning from each pony with a minimum of two days rest between each semen collection. Seminal specimens were prepared and subjected to genomic profiling analysis of bacterial microbial composition, and classified by phylum, order, class, family, genus, and alpha and beta diversity for a descriptive analysis.
Collection of semen samples
After careful washing of each stallion’s prepuce and penis with warm water to remove smegma and other residues, semen collections were undertaken. Semen was collected from the miniature pony stallions using a Missouri artificial vagina (Minitube Australia, Ballarat, Vic.) with an in-line gel filter, and a phantom mount. Following semen collection, 250 μL was immediately removed and diluted with 20 μL of DNA Microshield, after which it was stored at −30°C until analyses.
Microbial DNA profiling of semen microbiota
ZymoBiomics DNA Miniprep kit (Zymo Research) was used to extract DNA from 150 μL specimens of seminal fluid, having been firstly digested with Proteinase K at 55°C for 30 min, following the manufacturer’s instructions. Full-length 16S rRNA gene sequencing (Hypervariable regions V1-9) was performed in preparation of the PacBio libraries, with SMRTBell® Library Preparation and Sequencing Procedure and Checklist with 15 ng extracted DNA input for the semen samples library preparation. Initially, PCR was conducted with KAPA HiFi HotStart ReadyMix (Roche), with 20 cycles in 25 μL reactions following protocol cycling conditions. Amplified DNA was indexed using the barcoded F/R Universal Primers Plate 96 v2 – part number 100-466-100 (Pacific Biosciences), and PCR products were purified by using AMPure PB (Agencourt Bioscience), in accordance with PacBio’s protocol. Two primers were employed in this study. Primer *27F, 5’ block /5AmMC6/ with universal sequence gcagtcgaacatgtagctgactcaggtcac 16S rRNA gene Primer AGRGTTYGATYMTGGCTCAG. The second primer used was **1492R 5’Block /5AmMC6/ with universal sequence tggatcacttgtgcaagcatcacatcgtag, 16S Primer RGYTACCTTGTTACGACTT.
Verification of amplicon sizes was carried out with a Fragment Analyser 5200 (Agilent Technologies) and Qubit 4.0 Fluorometer (ThermoFisher Scientific) using the High Sensitivity dsDNA assay. Pooling of barcoded amplicons was performed before the library construction process with the SMRTBell® Express Template Prep Kit 2.0 (Pacific Biosciences). Semen specimens were pooled in groups of 20, then sequenced on separate chips. A PacBio sequel (Pacific Biosciences) sequencing process was used with v3 sequencing chemistry in CCS collection mode for 20 h on a 1M v3 LR SMRT Cell. 16S rRNA gene sequencing used in this protocol is 1600 base pairs. Following this, CCS analysis was run prior to demultiplexing and fastq conversion. The CCS (Circular Consensus Sequence) analysis algorithm is applied to processes which require distinguishing of closely related molecules of DNA.
Taxonomical classifications of microbiota
The semen microbiota of the subjects were categorised according to taxonomical classifications of phylum, order, class, family and genus.
Bioinformatics for microbiome profiling
Prior to data analysis, the quality of the raw reads was checked using the package FastQC (Ver. 0.11.9). An average 80–95% of the raw reads were retained across the samples. This was followed by inspection of the length distribution of the reads. Most of the sequences were found to be in the expected range of approximately 1400 to 1550 bp. A filter was applied to retain the sequences between 1000 and 1600 bp length. The next-generation microbiome bioinformatics platform QIIME2 (version q2cli ver. 2020.8.0) was used for analysing the amplicon data and further generation of Amplicon Sequence Variants (ASVs) was conducted from the raw amplicon data, using the algorithm DADA2 (ver. 1.19.2). DADA2-formatted training fasta files were derived from the Silva Project’s ver. 128 release and used for taxonomy assignment. Three output files were generated using DADA2. These included the sequence feature count tables, the taxonomy identification file per ASV and a fasta file containing sequences of all ASVs. These files were exported in a phyloseq format for further analysis using QIIME2. These files along with the sample metadata files were used as input for different modules in the QIIME2 workflow.
Data produced by QIIME 2 exists as QIIME 2 artifacts, which contains data and metadata. The feature tables imported from DADA2 were filtered to retain the features, which were observed in at least two samples. A rooted phylogenetic tree was generated using the filtered feature table for downstream diversity analysis.
Data analysis
This cross-sectional data sample was prepared in Microsoft Excel v16.67 (2022). ASV count data was aggregated first at the classification of phylum and sorted from highest to lowest frequency. This method was applied again for each of the remaining taxonomical ranks, namely order, class, family and genus. The top 10 organisms identified in each taxon for each pony, with the addition of an overall microbial profile were reported. Principal coordinates analysis (PCoA) was applied to analyse and visualise biodiversity between the semen microbiota of the four subjects using Bray–Curtis distance matrix. The alpha diversity measures evenness (abundance), richness and Shannon’s diversity index summarise the cohort’s semen microbiota. Summary statistics report the distribution of those three alpha diversity measures.
Ethic statement
All procedures undertaken in this study were approved by the University of Newcastle Animal Ethics Committee and adhere to the Australian code for the care and use of animals for scientific purposes (approval number A-2011-122). The Australian code for the care and use of animals for scientific purposes adheres to the EU standards for the protection of animals used for scientific purposes.
Results
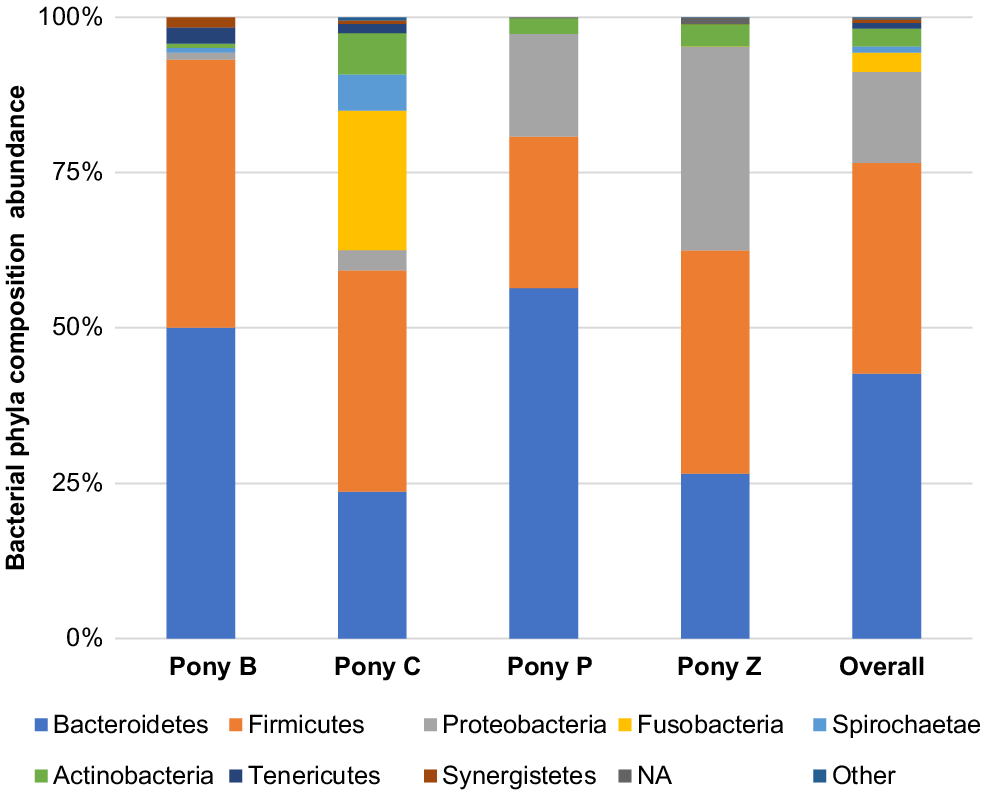
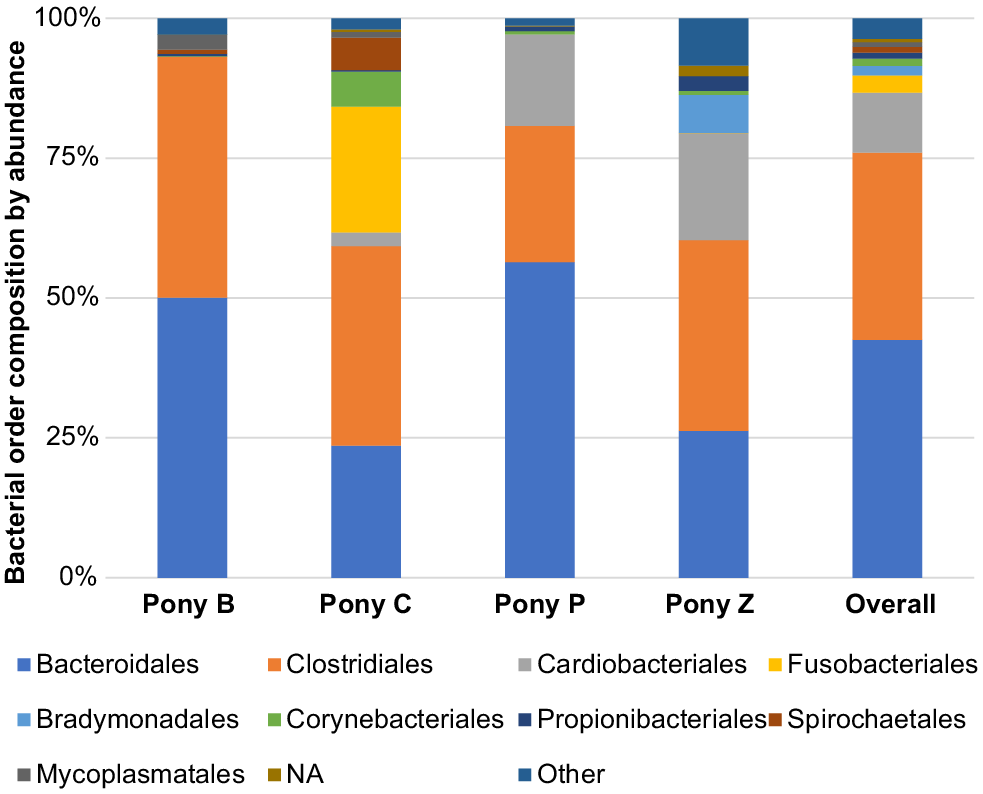
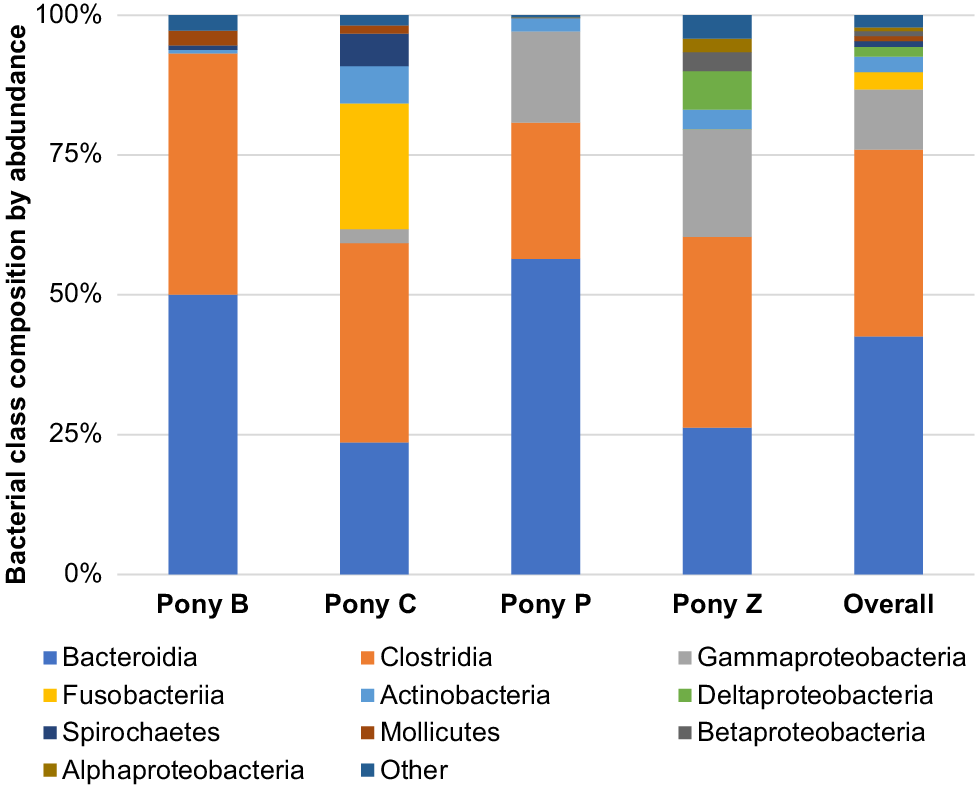
The principal results of this study indicate specific microbiota are potentially characteristic of the semen of healthy miniature pony stallions within each taxonomical ranking, with some inter-individual variations. The microbiota identified in the semen of the four equine subjects (Ponies B, C, P, Z) are presented in Figs 2–6 by percentage abundance and taxonomical classifications of phylum, order, class, family and genus. Overall, Firmicutes and Bacteroidetes phyla predominated (76%), followed by Proteobacteria (15%). Bacteroidales, Clostridiales and Cardiobacteriales predominated the microbial rank of order (86%). For class, Bacteroidia, Clostridia and Gammaproteobacteria (87%) predominated, and for family Porphyromonadaceae, Family_XI and Cardiobacteriaceae (62%) predominated. At the level of genus, 80% of the abundance was composed of seven genera – Porphyromonas, Suttonella, Peptoniphilus, Fastidiosipila, Ezakiella, Petrimonas and undescribed taxa (NA) that are yet to named.
Phylum
The eight phyla predominating the semen samples of four miniature stallions were Bacteroidetes, Firmicutes, Proteobacteria, Fusobacteria, Spirochaete, Actinobacteria, Tenericutes and Synergistetes; those less frequently represented were categorised as other (Fig. 2). Overall, the phyla Bacteroidetes and Firmicutes were most represented in this small cohort, comprising 76%, and Proteobacteria was the third most abundant phylum, comprising another 15% of the phyla identified. Less than 1% of those detected were classified as taxon undescribed (NA).
Order
Within the taxonomical classification of order, Bacteroidales, Clostridiales, Cardiobacteriales, Fusobacteriales, Bradymonadales, Corynebacteriales, Propionibacteriales, Spirochaetales and Mycoplasmatales were most abundant (Fig. 3). The three predominating orders were Bacteroidales, Clostridiales and Cardiobacteriales, accounting for 86% of the abundance distribution.
Class
Within the taxonomic classification of microbial class, Bacteroidia, Clostridia, Gammaproteobacteria, Fusobacteriia, Actinobacteria, Deltaproteobacteria, Spirochaetes, Mollicutes, Betaproteobacteria and Alphaproteobacteria were the most abundant, with 87% of the microbiota composed of Bacteroidia, Clostridia and Gammaproteobacteria (Fig. 4).
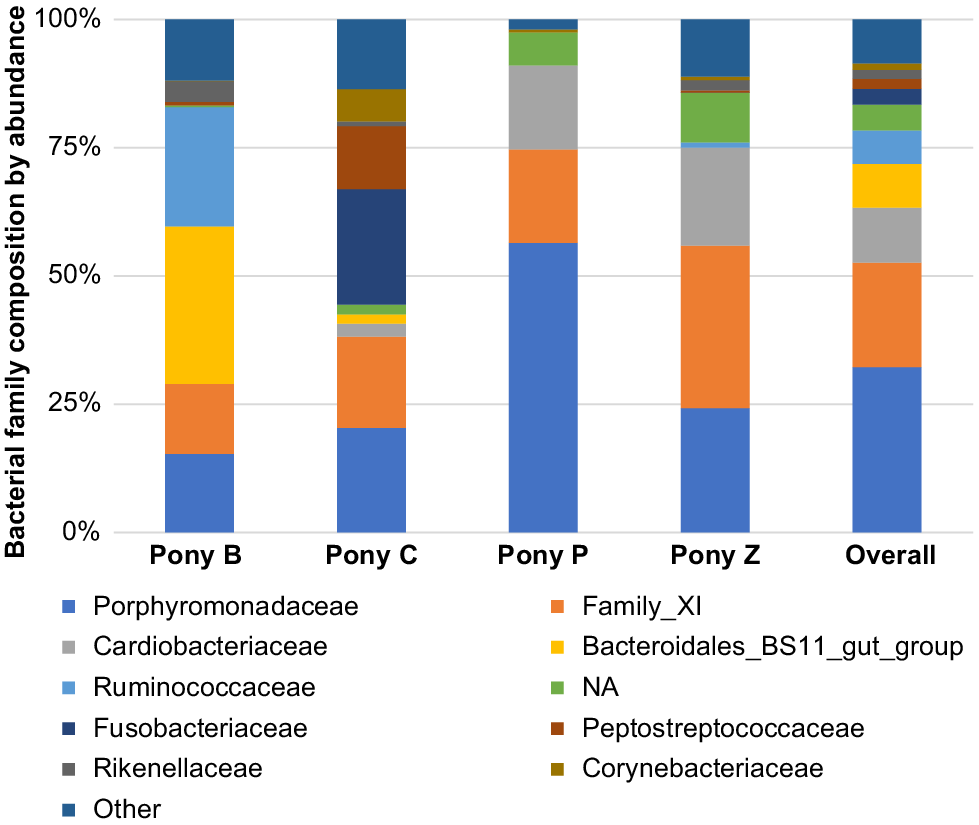
Family
The most abundant microbial families were Porphyromonadaceae, Family_XI, Cardiobacteriaceae, Bacteroidales_BS11_gut_group, Ruminococcaceae, Fusobacteriaceae, Peptostreptococcaceae, Rikenellaceae and Corynebacteriaceae. Of these, Porphyromonadaceae, Family_XI (Family XI Incertae sedis) and Cardiobacteriaceae predominated, comprising 62% (Fig. 5).
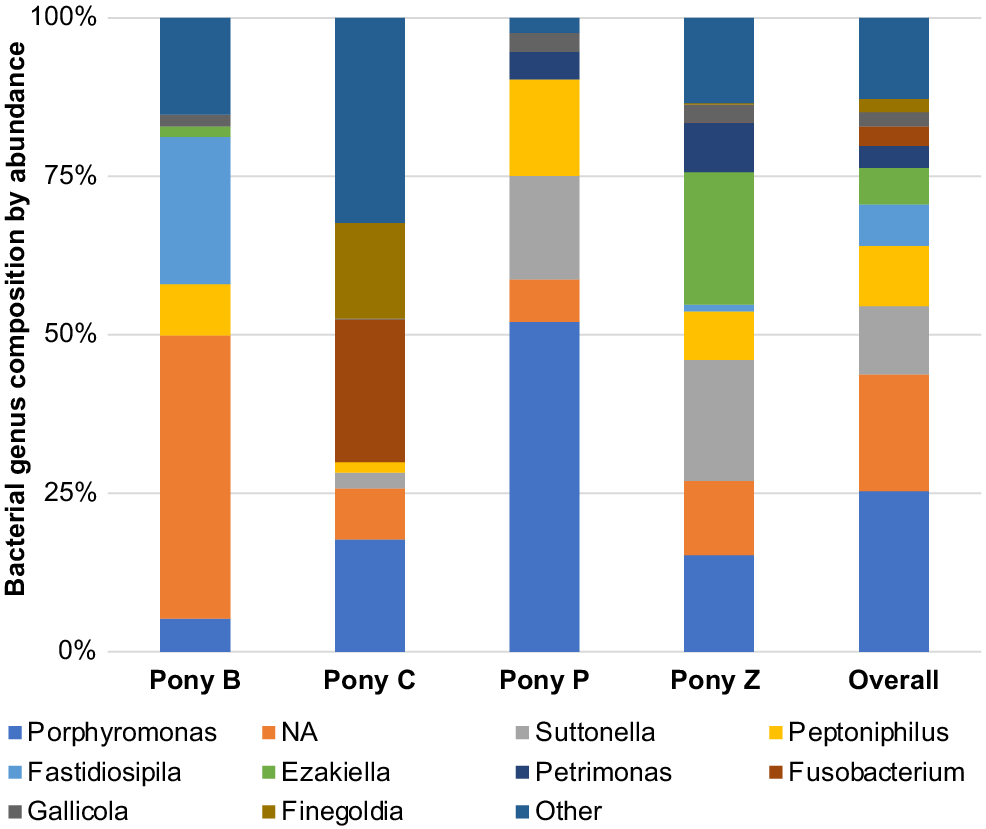
Genus
Within the taxonomical classification of microbial genus, Porphyromonas, Suttonella, Peptoniphilus, Fastidiosipila, Ezakiella, Petrimonas, Fusobacterium, Gallicola and Finegoldia were the most abundant genera identified in the semen. Of these, Porphyromonas, Suttonella, Peptoniphilus, Fastidiosipila, Ezakiella, Petrimonas and several undescribed taxa (NA) predominated (Fig. 6). In terms of the microbiota represented in individual ponies, some differences were observed in the abundance of the family and genus level classifications. For example, Pony B had greater abundance of Fastidiosipila than the other ponies, Pony C had greater abundance of Fusobacterium, Pony P had greater abundance of Suttonella and Pony Z had greater abundance of Ezakiella genus.
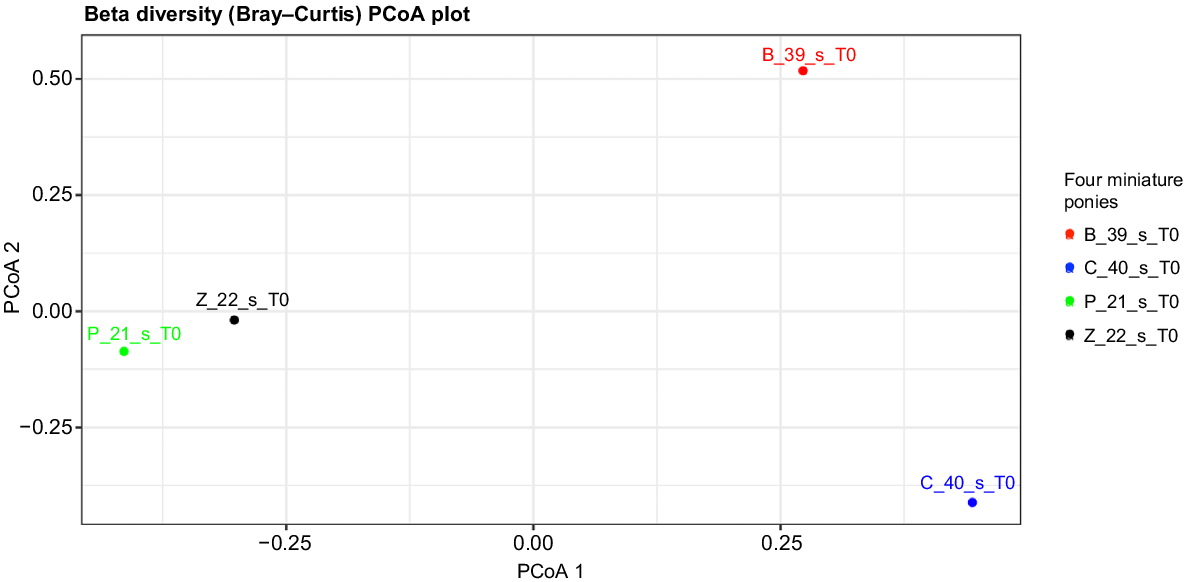
Beta diversity
Fig. 7 illustrates the beta diversity measure (microbial diversity between subjects). The microbial composition of Ponies P and Z was similar compared to Ponies B and C.
Alpha diversity
Table 1 presents the summary statistics for alpha diversity measures of the semen microbiota of the four subjects. Variations in the species richness (number of unique species within the sample) had the highest variation as reflected by the large standard deviation (s.d.). The mean and median were the same for evenness and Shannon diversity indicating an approximately even distribution. A skewed distribution was observed for microbial richness of the semen samples as reflected by the 11.5 points difference in mean and median.
Discussion
This exploratory study has described the microbial composition of the semen microbiome of healthy miniature pony stallions. The overall findings indicate that several microbiota are possibly characteristic of the semen microbiome of healthy miniature pony stallions within each taxonomical ranking, with some inter-individual variations. While the microbial phyla and families present in equine semen have previously been described (Al-Kass et al. 2020a; Quiñones-Pérez et al. 2021), here we have described additional taxonomical levels and alpha and beta diversity thus expanding this area of knowledge.
Semen microbial phyla and families
Quiñones-Pérez et al. (2022) evaluated the phyla and families present in the semen of 12 healthy Arab, Anglo-Arab and Andalusian stallions (aged 7–24 years) located at the Spanish Army’s breeding centre in Seville, Spain. Like Quiñones-Pérez et al. (2021), we also found that Bacteroidetes and Firmicutes predominated equine semen. However, Actinobacteria was the third most abundant phylum present in the semen of the Spanish stallions, whereas Proteobacteria was the third most abundant phylum in the miniature ponies’ semen. These Gram-negative bacteria play an important metabolic, energy-generating role in the equine intestinal microbiome (Li et al. 2022). Proteobacteria have also been reported as being more represented in the gastrointestinal tract of horses with equine grass sickness (Garber et al. 2020) and colitis (Ayoub et al. 2022). Somewhat contrary to this, in a separate study, Proteobacteria colonies were reported to be less represented in the faeces of horses affected by diarrhoea (Li et al. 2022). The presence of Proteobacteria in the semen was not associated with these conditions in the miniature ponies studied, which were of good health prior to and throughout the study period, with no reports of diarrhoea or other illness. Interestingly, many of the less abundant phyla, including Fusobacteria, Spirochaetes, Synergestes, Tenericutes and Chloroflexi, were present in similar amounts in both studies. Such similarities across demographic locations signal there may be a characteristic semen microbiome of stallions that is independent of environmental factors.
At the taxonomic classification of family, Quiñones-Pérez et al. (2021) identified 69 families of microbes in their stallions’ semen, of which only 22 families were present in proportions greater than 1% and only nine were common to all samples (Quiñones-Pérez et al. 2021). The microbial family represented in the Spanish stallion’s semen were predominantly Porphyromonadaceae, which was similar in preponderance to our miniature ponies. Of interest to fertility research, Porphyromonadaceae is reported as a predominant family present in fertile stallion semen (Quiñones-Pérez et al. 2021), where it is found to be more dominant than in human semen (Hou et al. 2013). In contrast, the Spanish stallions had a much greater abundance of Corynebacteriaceae in the semen compared to the miniature ponies in this study, who had more Family XI present. The Corynebacteriaceae family, which has also been associated with fertility in stallions, was not highly represented in our cohort of miniature ponies.
Differences in phylum and family identification between the studies are not readily attributed to the chosen methods. In many regards, the methods in both studies were similar, involving similar collection processes during the spring season, to obtaining one semen specimen and using the 16S rRNA gene sequencing. One distinction which may account for the variations between the two studies was that the Spanish study used the 16S rRNA gene hypervariable V3 region for microbial detection. However, in our study full length (hypervariable regions 1–9) 16S rRNA gene amplicon sequencing was used to detect microbial taxa. In addition, using different DNA extraction protocols could influence sequencing outcomes and the bio-informatic pipelines employed by both studies were different, which may have influenced the respective findings. Differences between the Spanish study and the findings reported here are more likely to relate to breed, diet, geographical location and associated environmental factors (Theelen et al. 2021). The stallions studied by Quiñones-Pérez et al. (2022) in Spain were fed alfalfa hay and oats, rather than lucerne hay, which may have produced a different semen microbial profile to that of our stallions. Although, it could be argued that dietary variations are more likely to be associated with differences observed in the gastrointestinal rather than extraintestinal microbial communities. The faecal microbiome of ponies and horses in good health is known to be impacted by environmental factors, such as air exposure, farm location (region and country), season and access to pasture (Theelen et al. 2021), but these influences are yet to be demonstrated in relation to extraintestinal microbial communities such as semen.
Semen microbial genera
The aforementioned factors may also explain the differences in genera identified in equine semen by Al-Kass et al. (2020a), who examined the microbial composition of semen specimens from seven healthy stallions (aged 7–17 years) on a commercial stud in Stockholm, Sweden. Characterising the microbiota from one ejaculate per animal to genus level (Al-Kass et al. 2020a), they identified almost twice as many genera in the semen in comparison to those discovered in our cohort, with only Porphyromonas and Peptoniphilus being common to both cohorts. The four most common genera found in the Swedish study were Porphyromonas, Corynebacterium, Finegoldia and Peptoniphilus, followed by Mobiluncus, Chondromyces, Suttonella, Treponema, Actinetobacter, Campylobacter and a group of ‘other’. In comparison, the three most abundant genera in the semen of our miniature ponies were Porphyromonas, Suttonella and Peptoniphilus, followed by Fastidiosipila, Ezakiella, Petrimonas, Fusobacterium, Gallicola, Finegoldia and ‘other’. No Ureaplasma, Spirochaeta, Prevotellaceae, Fibrobacter, Acinetobacter, Chondromyces or Clostridium were identified in our miniature ponies. Like the Al-Kass study, our miniature ponies displayed predominantly Porphyromonas genus, an organism also common to healthy human semen (Hou et al. 2013; Liu et al. 2014). Consistent with other equine studies, we did not find any Lactobacillus genus in our samples, a bacterium which has been associated with good sperm quality in humans (Baud et al. 2019).
The methodology and technology employed in the aforementioned Swedish study were similar in many regards to those employed in our study i.e. semen specimens were collected in spring from one ejaculate, and in the same regular collection routine and manner (using a Missouri AV without mare contact). Although the stallions of the Swedish study were engaged in an artificial insemination program, the procedure for collecting and storing each aliquot of semen before its transport to the laboratory for genomic sequencing was identical to that employed by our Australian stallion handlers, hence one would not expect to find an effect of stallion management on semen microbiome composition to influence the results of these two studies.
Soil bacteria may contact the stallion’s urogenital tract from its environment. A miniature pony stallion’s erect penis can more easily contact the ground than that of a full-sized stallion – thus giving rise to extrinsic contamination and differences in microbes detected. Petrimonas genus, found in the semen of our cohort of miniature pony stallions, resides in soil, where it is able to effectively degrade hazardous compounds (Zhao and Zhang 2021) to innocuous acetic acid and carbon dioxide (Grabowski et al. 2005). Fastidiosipila, a Gram-positive anaerobe previously unrecognised in equine semen, which was identified in the miniature stallions, has also been identified in stored waste of landfill (Wang et al. 2017), hence may have been acquired from the soil by these ponies. These soil-dwelling organisms were not isolated by Al-Kass et al. (2020a) from their full-size breeds. Gallicola genus was also unique in the semen of the miniature stallions. Very little exists in the literature about these microbes, other than that they are Gram-positive, strictly anaerobic cocci associated with dark fermentation of food waste (Shi et al. 2021) and may be a faecal contaminant of ground water sourced from chicken manure (Naphtali et al. 2019).
The seminovaginal microbiome
The influence of the vaginal microbiome on the composition of the semen microbiome is important to consider. The stallion urethral and possibly semen microbiomes may be contaminated by the mare’s vaginal microbiota following mating, and although there are no detailed descriptions of a shared complementary equine seminovaginal microbiome, one study has found microbiome network links between semi-feral Welsh Mountain pony stallions and mares (Antwis et al. 2018). Porphyromonas is the most abundant genus in Arabian mares’ vaginal microbiomes (approximately 14–17%) and Bacteroidales is the highest abundance by order (approximately 31%) (Barba et al. 2020). By comparison, our stallions’ semen contained 25% Porphyromonas and 42% Bacteroidales, although they had not had genital contact with mares, so perhaps the colonisation of the mare’s vaginal microbiome arises from the stallion’s penis, which harbours larger abundances of these genera. At phylum level, the core species of the vaginal microbiome of the mare are comparative in ranking to those phyla found in miniature pony stallion semen (Firmicutes, Bacteroidetes, Proteobacteria and Actinobacteria) (Barba et al. 2020). Microbes from the Corynebacteriaceae family are found in low abundance on equine skin (Costa et al. 2016; Kaiser-Thom et al. 2021) as well as in semen, which might suggest a degree of cross-contamination of this bacterium that occurs during specimen collection.
Limitations of this study
Only one breed of stallion was recruited for this study, with single semen samples collected and profiled for each miniature pony stallion, thus reducing the generalisability of the findings. The ASVs display advantages over the more traditional approach of Operational Taxanomical Units (OTUs) as they represent the exact amplicon sequences without coarse graining into OTUs and can resolve even single nucleotide base differences. However, it should be considered that ASVs can sometimes over-split sequences, creating separate clusters for very similar sequences. This ‘over-splitting’ can lead to the separation of sequences that belong to the same bacterial genome, creating artificial diversity. (Schloss 2021).
Due to the sample size, detailed statistical analysis and comparison with various measures of sperm quality were not possible, thus associations between the composition of the semen microbiome and sperm quality could not be made. The detailed characteristics of the semen microbiome in miniature pony stallions may not be generalisable but provides preliminary data to inform such research.
Conclusion
The microbial composition of the semen of the four miniature pony stallions studied varied between horses, with some bacteria common to all ponies at various taxonomic classification ranks. The role of shared and unique microbiota in relation to the general health of the horse and fertility warrants further research. Future studies are encouraged to explore the richness, evenness or diversity of semen microbiome profiles and the potential for these microbiome characteristics to influence sperm quality biomarkers. More specifically, the findings of this study can be used to inform future research exploring the relationship between semen microbial composition and fertility of horses.
Data availability
The raw sequence data used in the analysis of this study will be made available in the NCBI – sequence read archive (SRA) following publication.
Author contributions
Study conceptualisation and design, C.G.C., J.H., K.S., C.G.G.; methodology, J.H., K.S and C.G.C., project administration, Z.G., C.G.C; bioinformatics processing, N.D.; statistical analysis design and data analysis, K.S.; original draft manuscript preparation, C.G.C., manuscript revisions and editing, J.H., C.G.G., Z.G., K.S., project supervision, J.H., C.G.G., Z.G. All authors have read and agreed to the published version of the manuscript.
Acknowledgements
The authors acknowledge the technical assistance provided by the Sydney Informatics Hub, a Core Research Facility of the University of Sydney.
References
Al Jassim RAM, Andrews FM (2009) The bacterial community of the horse gastrointestinal tract and its relation to fermentative acidosis, laminitis, colic, and stomach ulcers. Veterinary Clinics of North America: Equine Practice 25(2), 199-215.
| Crossref | Google Scholar | PubMed |
Al-Kass Z, Guo Y, Vinnere Pettersson O, Niazi A, Morrell JM (2020a) Metagenomic analysis of bacteria in stallion semen. Animal Reproduction Science 221, 106568.
| Crossref | Google Scholar | PubMed |
Al-Kass Z, Eriksson E, Bagge E, Wallgren M, Morrell JM (2020b) Microbiota of semen from stallions in Sweden identified by MALDI-TOF. Veterinary and Animal Science 10, 100143.
| Crossref | Google Scholar | PubMed |
Antwis RE, Lea JMD, Unwin B, Shultz S (2018) Gut microbiome composition is associated with spatial structuring and social interactions in semi-feral Welsh Mountain ponies. Microbiome 6(1), 207.
| Crossref | Google Scholar | PubMed |
Ayoub C, Arroyo LG, MacNicol JL, Renaud D, Weese JS, Gomez DE (2022) Fecal microbiota of horses with colitis and its association with laminitis and survival during hospitalization. Journal of Veterinary Internal Medicine 36(6), 2213-2223.
| Crossref | Google Scholar | PubMed |
Barba M, Martínez-Boví R, Quereda JJ, Mocé ML, Plaza-Dávila M, Jiménez-Trigos E, Gómez-Martín Á, González-Torres P, Carbonetto B, García-Roselló E (2020) Vaginal microbiota is stable throughout the estrous cycle in Arabian mares. Animals (Basel) 10(11), 2020.
| Crossref | Google Scholar |
Baud D, Pattaroni C, Vulliemoz N, Castella V, Marsland BJ, Stojanov M (2019) Sperm microbiota and its impact on semen parameters. Frontiers in Microbiology 10, 234.
| Crossref | Google Scholar |
Böttger EC (1989) Rapid determination of bacterial ribosomal RNA sequences by direct sequencing of enzymatically amplified DNA. FEMS Microbiology Letters 65(1–2), 171-176.
| Crossref | Google Scholar |
Bull K, Davies G, Jenkins TP, Peachey LE (2021) The equine faecal microbiota undergoes step-wise compositional changes in accordance with increasing levels of domestication. Research Square
| Crossref | Google Scholar |
Costa MC, Silva G, Ramos RV, Staempfli HR, Arroyo LG, Kim P, Weese JS (2015) Characterization and comparison of the bacterial microbiota in different gastrointestinal tract compartments in horses. The Veterinary Journal 205(1), 74-80.
| Crossref | Google Scholar | PubMed |
Costa MC, Stämpfli HR, Allen-Vercoe E, Weese JS (2016) Development of the faecal microbiota in foals. Equine Veterinary Journal 48(6), 681-688.
| Crossref | Google Scholar | PubMed |
Garber A, Hastie P, Murray J-A (2020) Factors influencing equine gut microbiota: current knowledge. Journal of Equine Veterinary Science 88, 102943.
| Crossref | Google Scholar | PubMed |
Grabowski A, Tindall BJ, Bardin V, Blanchet D, Jeanthon C (2005) Petrimonas sulfuriphila gen. nov., sp. nov., a mesophilic fermentative bacterium isolated from a biodegraded oil reservoir. International Journal of Systematic and Evolutionary Microbiology 55(3), 1113-1121.
| Crossref | Google Scholar |
Heil BA, Paccamonti DL, Sones JL (2019) Role for the mammalian female reproductive tract microbiome in pregnancy outcomes. Physiological Genomics 51(8), 390-399.
| Crossref | Google Scholar | PubMed |
Henneke DR, Potter GD, Kreider JL, Yeates BF (1983) Relationship between condition score, physical measurements and body fat percentage in mares. Equine Veterinary Journal 15(4), 371-372.
| Crossref | Google Scholar | PubMed |
Hou D, Zhou X, Zhong X, Settles ML, Herring J, Wang L, Abdo Z, Forney LJ, Xu C (2013) Microbiota of the seminal fluid from healthy and infertile men. Fertility and Sterility 100(5), 1261-1269.e3.
| Crossref | Google Scholar | PubMed |
Hughes JP, Loy RG (1975) The relation of infection to infertility in the mare and stallion. Equine Veterinary Journal 7(3), 155-159.
| Crossref | Google Scholar | PubMed |
Julliand V, Grimm P (2017) The impact of diet on the hindgut microbiome. Journal of Equine Veterinary Science 52, 23-28.
| Crossref | Google Scholar |
Kaiser-Thom S, Hilty M, Axiak S, Gerber V (2021) The skin microbiota in equine pastern dermatitis: a case-control study of horses in Switzerland. Veterinary Dermatology 32(6), 646-e172.
| Crossref | Google Scholar | PubMed |
Kauter A, Epping L, Semmler T, Antao E-M, Kannapin D, Stoeckle SD, Gehlen H, Lübke-Becker A, Günther S, Wieler LH, Walther B (2019) The gut microbiome of horses: current research on equine enteral microbiota and future perspectives. Animal Microbiome 1(1), 14.
| Crossref | Google Scholar | PubMed |
Li Y, Lan Y, Zhang S, Wang X (2022) Comparative analysis of gut microbiota between healthy and diarrheic horses. Frontiers in Veterinary Science 9, 882423.
| Crossref | Google Scholar | PubMed |
Liu CM, Osborne BJW, Hungate BA, Shahabi K, Huibner S, Lester R, Dwan MG, Kovacs C, Contente-Cuomo TL, Benko E, Aziz M, Price LB, Kaul R (2014) The semen microbiome and its relationship with local immunology and viral load in HIV infection. PLoS Pathogens 10(7), e1004262.
| Crossref | Google Scholar | PubMed |
Madsen M, Christensen P (1995) Bacterial-flora of semen collected from danish warmblood stallions by artificial vagina. Acta Veterinaria Scandinavica 36(1), 1-7.
| Crossref | Google Scholar | PubMed |
Mach N, Ruet A, Clark A, Bars-Cortina D, Ramayo-Caldas Y, Crisci E, Pennarun S, Dhorne-Pollet S, Foury A, Moisan M-P, Lansade L (2020) Priming for welfare: gut microbiota is associated with equitation conditions and behavior in horse athletes. Scientific Reports 10(1), 8311.
| Crossref | Google Scholar | PubMed |
Malaluang P, Wilén E, Lindahl J, Hansson I, Morrell JM (2021) Antimicrobial resistance in equine reproduction. Animals (Basel) 11(11), 3035.
| Crossref | Google Scholar |
Mignard S, Flandrois JP (2006) 16S rRNA sequencing in routine bacterial identification: a 30-month experiment. Journal of Microbiological Methods 67, 574-581.
| Crossref | Google Scholar | PubMed |
Naphtali P, Mohiuddin MM, Paschos A, Schellhorn HE (2019) Application of high-throughput 16S rRNA sequencing to identify fecal contamination sources and to complement the detection of fecal indicator bacteria in rural groundwater. Journal of Water and Health 17(3), 393-403.
| Crossref | Google Scholar | PubMed |
Ong CT, Ross EM, Boe-Hansen G, Turni C, Hayes BJ, Fordyce G, Tabor AE (2022) Adaptive sampling during sequencing reveals the origins of the bovine reproductive tract microbiome across reproductive stages and sexes. Scientific Reports 12(1), 15075.
| Crossref | Google Scholar | PubMed |
Paßlack N, Vahjen W, Zentek J (2020) Impact of dietary cellobiose on the fecal microbiota of horses. Journal of Equine Veterinary Science 91, 103106.
| Crossref | Google Scholar |
Quiñones-Pérez C, Hidalgo M, Ortiz I, Crespo F, Vega-Pla JL (2021) Characterization of the seminal bacterial microbiome of healthy, fertile stallions using next-generation sequencing. Animal Reproduction 18, e20200052.
| Crossref | Google Scholar |
Quiñones-Pérez C, Martínez A, Ortiz I, Crespo F, Vega-Pla JL (2022) The semen microbiome and semen parameters in healthy stallions. Animals (Basel) 12(5), 534.
| Crossref | Google Scholar |
Rota A, Calicchio E, Nardoni S, Fratini F, Ebani VV, Sgorbini M, Panzani D, Camillo F, Mancianti F (2011) Presence and distribution of fungi and bacteria in the reproductive tract of healthy stallions. Theriogenology 76(3), 464-470.
| Crossref | Google Scholar | PubMed |
Rowe M, Veerus L, Trosvik P, Buckling A, Pizzari T (2020) The reproductive microbiome: an emerging driver of sexual selection, sexual conflict, mating systems, and reproductive isolation. Trends in Ecology & Evolution 35(3), 220-234.
| Crossref | Google Scholar | PubMed |
Schloss PD (2021) Amplicon sequence variants artificially split bacterial genomes into separate clusters. mSphere 6(4), e0019121.
| Crossref | Google Scholar | PubMed |
Shi Z, Zhang L, Yuan H, Li X, Chang Y, Zuo X (2021) Oyster shells improve anaerobic dark fermentation performances of food waste: hydrogen production, acidification performances, and microbial community characteristics. Bioresource Technology 335, 125268.
| Crossref | Google Scholar | PubMed |
Theelen MJP, Luiken REC, Wagenaar JA, Sloet van Oldruitenborgh-Oosterbaan MM, Rossen JWA, Zomer AL (2021) The equine faecal microbiota of healthy horses and ponies in the netherlands: impact of host and environmental factors. Animals (Basel) 11(6), 1762.
| Crossref | Google Scholar |
Wang X, Cao A, Zhao G, Zhou C, Xu R (2017) Microbial community structure and diversity in a municipal solid waste landfill. Waste Management 66, 79-87.
| Crossref | Google Scholar | PubMed |
Woo PCY, Lau SKP, Teng JLL, Tse H, Yuen K-Y (2008) Then and now: use of 16S rDNA gene sequencing for bacterial identification and discovery of novel bacteria in clinical microbiology laboratories. Clinical Microbiology and Infection 14(10), 908-934.
| Crossref | Google Scholar | PubMed |
Zhao H, Zhang Q (2021) Performance of electro-Fenton process coupling with microbial fuel cell for simultaneous removal of herbicide mesotrione. Bioresource Technology 319, 124244.
| Crossref | Google Scholar | PubMed |