

DNA methylation analysis using bisulphite-based amplicon sequencing of individuals exposed to maternal tobacco use during pregnancy, and offspring conduct problems in childhood and adolescence†
Alexandra J. Noble A * , John F. Pearson B , Alasdair D. Noble C , Joseph M. Boden D , L. John Horwood D , Martin A. Kennedy B and Amy J. Osborne A
A * , John F. Pearson B , Alasdair D. Noble C , Joseph M. Boden D , L. John Horwood D , Martin A. Kennedy B and Amy J. Osborne A
A School of Biological Sciences, University of Canterbury, Christchurch, New Zealand.
B Department of Pathology and Biomedical Sciences, University of Otago, Christchurch, New Zealand.
C AgResearch, Lincoln Research Centre, Christchurch, New Zealand.
D Department of Psychological Medicine, University of Otago, Christchurch, New Zealand.
Reproduction, Fertility and Development 34(7) 540-548 https://doi.org/10.1071/RD21108
Published online: 8 February 2022
© 2022 The Author(s) (or their employer(s)). Published by CSIRO Publishing. This is an open access article distributed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License (CC BY-NC-ND)
Abstract
Maternal tobacco smoking during pregnancy is a large driver of health inequalities and a higher prevalence of conduct problem (CP) has been observed in exposed offspring. Further, maternal tobacco use during pregnancy can also alter offspring DNA methylation. However, currently, limited molecular evidence has been found to support this observation. Thus we aim to examine the association between maternal tobacco use in pregnancy and offspring CP, to determine whether offspring CP is mediated by tobacco exposure-induced DNA methylation differences. Understanding the etiology of the association between maternal tobacco use and offspring CP will be crucial in the early identification and treatment of CP in children and adolescents. Here, a sub group of N = 96 individuals was sourced from the Christchurch Health and Development Study, a longitudinal birth cohort studied for over 40 years in New Zealand. Whole blood samples underwent bisulphite-based amplicon sequencing at 10 loci known to play a role in neurodevelopment, or which had associations with CP phenotypes. We identified significant (P < 0.05) differential DNA methylation at specific CpG sites in CYP1A1, ASH2L and MEF2C in individuals with CP who were exposed to tobacco in utero. We conclude that environmentally-induced DNA methylation differences could play a role in the observed link between maternal tobacco use during pregnancy and childhood/adolescent CP. However, larger sample sizes are needed to produce an adequate amount of power to investigate this interaction further.
Keywords: conduct disorder, developmental biology, DNA, DNA methylation, environmental epigenetics, epigenetics, pregnancy, tobacco exposure.
Introduction
Mothers who smoked tobacco during pregnancy have a higher prevalence of offspring developing a conduct problem (CP) phenotype compared to mothers who did not smoke (Wakschlag et al. 1997). This association has been proven in several different cohort studies and the observations have remained following adjustment for various other confounding factors, for example, socio economic status, maternal age, substance abuse, parental anti-social personality, and maladaptive parenting (Wakschlag et al. 1997; Joelsson et al. 2016). However, there is limited molecular evidence to suggest a link between in utero tobacco exposure and offspring conduct disorder, thus a direct link between in utero tobacco exposure and CP remains elusive. Previously we conducted a pilot study assessing differential DNA methylation in a small cohort of individuals who were exposed to tobacco in utero, with sub-groups of individuals defined as having high CP scores (Noble et al. 2021). We found nominally significant DNA methylation changes in several genes associated with neurodevelopment (Noble et al. 2021). Due to the limitations of using a small sample size combined with an array containing a large number of loci, results were underpowered, therefore observations were unable to reach genome wide significance. However, the biological relevance of these nominally significant CpG loci to the CP phenotype, combined with further research that has suggested an epigenetic link between in utero tobacco exposure and ADHD (Sengupta et al. 2017), implies that the link between DNA methylation and CP development in tobacco-exposed offspring should be investigated more fully.
Here, we further pursue this hypothesis, by exploring differential methylation in genes that have known roles during in utero neurodevelopment and CP phenotypes, to understand whether DNA methylation may help explain the relationship between in utero tobacco exposure and development of CP in offspring. We applied a targeted approach via bisulphite-based amplicon sequencing (BSAS) of regions of genes involved in neurodevelopment. Amplicon sequencing has the ability to interrogate a region of the genome, therefore specifically targeting consecutive CpG sites in a row. We then assessed differential methylation in the DNA of participants from the Christchurch Health and Development Study (CHDS) whose mothers consumed tobacco during pregnancy, with high and low CP scores, and compared this to controls who were not exposed. This approach allowed us to specifically ask whether DNA methylation at genes involved in neurodevelopment and CP phenotypes are specifically differentially methylated in the DNA of offspring with CP, who were exposed to tobacco in utero. A significant interaction here would provide further support of a role for DNA methylation in the link between in utero exposure and CP development, something thathas so far proved elusive.
Methods
Ethics declarations
All aspects of the study were approved by the Southern Health and Disability Ethics Committee, under application number CTB/04/11/234/AM10 ‘Collection of DNA in the Christchurch Health and Development Study’.
Sample
A sub-group of individuals from the CHDS were selected for this study (Table 1). This longitudinal study originally included 97% of all the children (N = 1265) born in the Christchurch, New Zealand, urban region during a period in mid-1977 and has been studied at 24 time points from birth to age 40 (N = 987 at age 30). All participants were aged between 28 and 30 when blood samples and DNA was extracted.

|
For the subsets studied in this report, CHDS participants were chosen based on their in utero tobacco exposure status, their adult smoking status, and their CP scores. Group 1 consisted of individuals who were exposed in utero to tobacco smoke, and never smokers at the time blood samples were taken (N = 32). Group 2 consisted of individuals who were exposed in utero to tobacco smoke and were themselves regular smokers at the time the blood was taken (N = 32). Group 3 consisted of individuals who were not exposed to tobacco in utero, and never smokers at the time blood was taken (N = 32). In utero tobacco exposure was defined as 10+ cigarettes per day throughout pregnancy. Within each group of 32, 16 individuals were selected with a ‘high’ score on a measure of childhood CP at age 7–9 years and 16 with a ‘low’ score. Severity of childhood CP was assessed using an instrument that combined selected items from the Rutter and Conners child behaviour checklists (Conners 1969, 1970; Rutter et al. 1970; Fergusson et al. 1991) as completed by parents and teachers at annual intervals from 7–9 years. Parental and teacher reports were summed and averaged over the 3 years (Fergusson et al. 2005) to derive a robust scale measure of the extent to which the child exhibited conduct disordered/oppositional behaviours [mean (s.d.) = 50.1(7.9); range 41–97]. The behaviours sampled by the measures include many behaviours encompassed by the diagnostic classification, including violence toward peers and authority figures, fire setting, damage to the property of others, an unwillingness to follow rules or commands, and related behaviours. High conduct problem scores reflect the reporting of a larger number of these problems by parents and teachers. For the purposes of this report a ‘high’ score was defined as falling into the top quartile of the score distribution (scores > 53) and a ‘low’ score was defined as scores < 46.
Bisulphite-based amplicon sequencing
Bisulphite-based amplicon sequencing (BSAS) was carried out as described (Noble et al. 2021). Briefly, DNA was extracted from whole blood samples using the Kingfisher Flex System (Thermo Scientific, Waltham, MA, USA). DNA was quantified via nanodrop (Thermo Scientific). 500 ng of DNA underwent bisulphite treatment using the EZ DNA Methylation-Gold kit (Zymo Research, Irvine, CA, USA) as per the manufacturer’s instructions. DNA samples were then diluted to a final concentration of 100 ng/μL.
Amplicons for sequencing (Table 2 and Supplementary Table S1) were picked based upon several criteria: (i) previously published differential DNA methylation in response to in utero tobacco smoking; (ii) known associations with in utero brain development, and; (iii) known associations with CP phenotypes. Primers were then designed to flank the CpG sites of interest, ∼350 base pairs (bp) in total, or to amplify the promoter region of the gene if a specific CpG site was not known. Multiple pairs of primers were designed to amplify larger regions.

|
Bisulphite-converted DNA was amplified via PCR, using KAPA Taq HotStart DNA Polymerase (Sigma-Aldrich, St Louis, MO, USA) under the following conditions: 95°C for 10 min, 95°C for 30 s, 59°C for 20 s, 72°C for 7 min, and held at 4°C using the Mastercycler Nexus (Eppendorf, Macquarie Park, Australia). This was then cycled a total of 40 times. PCR products were purified with the Zymo DNA Clean & Concentrator Kit™ (Zymo Research, Irvine, CA, USA).
Following PCR, DNA was cleaned up with Agencourt® AMPure® XP beads (Beckman Coulter, Brea, CA, USA) and washed with 80% ethanol and allowed to air-dry. DNA was then eluted with 52.5 μL of 10 mM Tris pH 8.5 before being placed back into the magnetic stand. Once the supernatant had cleared, 50 μL was aliquoted for the experiment. DNA samples were quantified using the Quant-iT™ PicoGreen™ dsDNA Assay kit (Thermo Fisher) using the FLUROstar® Omega (BMG Labtech, Mornington, Australia). Samples were processed using the Illumina MiSeq™ 500 cycle Kit V2 and sequenced on the Illumina MiSeq™ system by Massey Genome Service (Palmerston North, New Zealand). Illumina MiSeq™ sequences were trimmed using SolexaQA++ software (Cox et al. 2010) and aligned to FASTA bisulphite converted reference sequences using the package Bowtie2 (ver. 2.3.4.3) Each individual read was then aligned to all reference sequences using the methylation-specific package Bismark (Krueger and Andrews 2011).
Statistics
Differential DNA methylation was assessed using the package edgeR (Chen et al. 2017). Coverage level was set to greater or equal to ‘8’ across unmethylated and methylated counts, as recommended by (Chen et al. 2017). Two models were used – the first was a bivariate model, to assess differences between the in utero exposed to tobacco compared to the non-exposed control group (model 1).
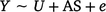
The second was a multiple regression to assess the interaction term in utero maternal smoke exposure and offspring conduct problem score (high or low, model 2).
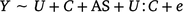
where Y is defined as the methylation M ratio, U is the exposed/unexposed in utero to maternal smoking, C is conduct problem score with high conduct problem score < 53 and low conduct problem core < 46, AS is adult smoker/non-smoker and e is the unexplained variation or error term.
This model was fitted with both ANOVA parameters and with contrasts between in utero exposure groups (exposed–non-exposed) within CP score levels. Top tables were constructed using the topTags function in edgeR, Log fold change, average log counts per million, and in some cases F statistic and were calculated and nominal significance was given for P < 0.05, these were then corrected using FDR. Scatter plots with the inclusion of confidence intervals were constructed from log transformed normalised methylated and unmethylated counts. Differential methylation was also assessed for adult tobacco smoking status, this was determined by using a linear model with just AS and e (Table S3).
Results
Here we assessed DNA methylation within 10 separate genes (Table 2). DNA sequence data for 15 amplicons from these 10 genes (Table S1) was generated, comprising a total of 280 CpG sites. These CpG sites included a combination of sites previously identified as differentially methylated, as well as amplification of all CpGs within the promoter region of genes associated with in utero neurodevelopment and CP phenotypes (Table 2). Differential methylation across these CpG sites was calculated to address whether any were specifically differentially methylated in individuals with CP, in response to in utero tobacco exposure.
Quantification of DNA methylation at previously reported CpG sites in response to in utero exposure to tobacco
Initially, we attempted to validate in our cohort (age ∼28–30 years) five CpG sites that have been previously reported to be differentially methylated in the DNA of cord blood from newborns, and whole blood from children and adolescents (ages newborn to 17) in response to in utero tobacco exposure (Table 1). Data were partitioned into those individuals exposed in utero, and those who were not (model 1), to assess whether or not BSAS could detect previously reported CpG sites (Table 3).

|
Aryl-hydrocarbon receptor repressor (AHRR) (cg05575921) displayed a 3.1% decrease in DNA methylation between exposed and non-exposed individuals, at a nominal P value of 0.02. This site has previously been identified as hypomethylated in adult tobacco smokers, as well as in postnatal cord blood samples between in utero tobacco-exposed and non-exposed individuals. Differential methylation found between adult smokers compared to non-smoking controls in this study are found in Table S3. The probe cg05549655 in the gene Cytochrome P450, family 1, subfamily A (CYP1A1) displayed a 5.19% increase in DNA methylation in the in utero exposed group, however, this site did not reach nominal statistical significance in our cohort. Cg09935388 and cg09662411 in the gene, Growth Factor Independent 1 (GFI1) were unable to be replicated as differentially methylated between the exposed and the non-exposed groups (no significant change in β values). Both CpG sites show hypomethylation, supporting previous observations of differential methylation within this gene. Contactin-associated protein-like 2 (CNTNAP2) (cg2594950) was similarly unable to be validated in our cohort using the method BSAS. Results of model 1 contain all CpG sites analysed in the 10 gene regions using BSAS are found in Table S2.
Differentially methylated CpGs under the interaction of in utero tobacco exposure and CP
Differential methylation dependent on both in utero exposure and CP score was found at 10 loci in six genes at nominal significance level, none were significant after correcting for false discovery rate (Table 4).

|
Of these CpG sites, five of the 10 CpG sites were found in the following genes: CYP1A1, GFI1, ASH2 like histone lysine methyltransferase complex subunit (ASH2L) and Glutamate Inotropic Receptor NMDA Type Subunit 2B (GRIN2b). Differential methylation was observed between in utero exposed and non-exposed associated with high conduct scores. No nominal significance from the interaction was observed in association with low conduct scores. The top three CpG sites with nominal significance under the interaction are displayed in Fig. 1. Here, differential methylation is found in response to high CP score and no differences are seen between the exposed and non-exposed low CP groups (Fig. 1).

|
Discussion
In utero tobacco exposure is known to alter DNA methylation at the genome-wide level in offspring (Joubert et al. 2012; Richmond et al. 2015; Joubert et al. 2016). The later-life implications of these tobacco-induced DNA methylation changes are unclear, however, an association between in utero tobacco exposure and CP has previously been observed (Sengupta et al. 2017). Given the complex etiology of CP phenotypes (Acosta et al. 2004; Beaver et al. 2007; Salvatore and Dick 2018) and the vast array of socioeconomic variables associated with tobacco use (Lantz et al. 1998), proving a causal link between maternal smoking and offspring CP is inherently challenging. Previously we quantified tobacco-induced DNA methylation changes that associate with CP phenotypes in offspring exposed to tobacco in utero (via maternal smoking) using the Illumina EPIC array, with results indicating that methylation was altered at the gene Fast Kinase Domain 1 (FASTKD1), which may have roles in neurodevelopment and CP phenotypes. However, due to a combination of a comparatively small sample size relative to the number of loci on the array, only nominal significance was observed. Thus, since the array data suggested a role for DNA methylation in the link between in utero tobacco exposure and CP, here we identified a panel of genes with known roles in neurodevelopment and CP phenotypes, and sought to determine whether DNA methylation is specifically altered at phenotypically relevant loci. Our previous research indicated that BSAS is an accurate tool through which to target amplicon-specific differential methylation, so here we used targeted BSAS to quantify differential methylation that is specific to the interaction between high CP score and in utero tobacco exposure.
Validation of previously identified differentially methylated CpG’s from in utero tobacco exposure
First, we asked whether differentially methylated CpGs that have been previously associated with in utero tobacco exposure were supported by this cohort. Here, we present validation of differential methylation of a CpG site within the gene AHRR (cg05575921). AHRR is a well-defined tobacco smoking gene, which is consistently represented in tobacco methylation data. AHRR has previously been found to be differentially methylated in response to in utero tobacco exposure (Joubert et al. 2012; de Vocht et al. 2015; Richmond et al. 2015). In both of our analyses we included adult smoking as a covariate in our models. It is, however, very difficult to interpret the exact cause of differential methylation as we do also find several sites in AHRR to be differentially methylated when we assess adult tobacco smoking compared to controls (Table S3).
Four other CpG sites investigated here due to previous association with in utero tobacco exposure were not differentially methylated in our data. However, the direction of methylation change was supported at all five sites investigated (Rotroff et al. 2016; Tehranifar et al. 2018; Rauschert et al. 2019). We suggest that further investigation in a larger cohort may lead to nominal significance at the sites in CYP1A1, CNTNAP2 and GFI1.
Identification of in utero exposure-related differentially methylated CpG sites that are specific to individuals with high CP scores
Epidemiological data suggest that there is an increased association between in utero tobacco exposure and behavioural disorder in children and adolescents (Mick et al. 2002; Carter et al. 2008). Thus, here, we investigated DNA methylation changes induced by in utero tobacco exposure as a potential molecular mechanism of dysfunction that could link the phenotypic trait of CP to maternal tobacco use during pregnancy. We therefore analysed DNA methylation patterns within our gene panel in response to in utero tobacco exposure and its interaction with CP status. A total of 10 CpG sites in seven genes were found to display nominal significance in DNA methylation in response to in utero tobacco exposure and CP in this cohort (Table 4).
In the 10 CpG sites we identified under the interaction, CYP1A1 showed greater magnitude differential methylation in high CP scores (exposed in utero vs non-exposed with high CPS), with reduced, reversed or no evidence of differential methylation at the same sites with low CP score. This indicates that within the observed nominal methylation changes the interaction was being driven in the high CP score group. One gene ASH2L, contained three nominally significantly differentially methylated CpG sites, and CYP1A1 and Myocyte enhancer factor 2C (MEF2C) both had two.
CYP1A1 is a well-established marker for in utero tobacco smoke exposure (Lee et al. 2015; Richmond et al. 2015, 2018; Tehranifar et al. 2018). Neither of the two sites we observed in this study have probes at these locations on the Illumina array system, thus emphasising a benefit of amplicon sequencing compared to an array-based method. Variant differences in CYP1A1 have previously been associated with child behavioural problems at age 2, from prenatal maternal environmental tobacco smoke (Hsieh et al. 2010). This highlights the need for this gene to be further investigated for its role in the development of conduct problems following in utero tobacco exposure.
Three CpG sites from the gene ASH2L showed in consistent levels of differential methylation in response to in utero tobacco exposure and CP, with two displaying hyper- and one hypomethylation. ASH2L has been found to interact with MEF2C to mediate changes in histone 3 lysine 4 trimethylation (H3K4me3) (Jung et al. 2016). Here, we detected nominal significance at two CpG sites within MEF2C (chr5, 88179596 and 88179541). Both of these sites were associated with a greater level of hypomethylation in participants who were exposed to tobacco in utero with high CP scores in this cohort, although not at the FDR significance level. MEF2C plays a role in neural crest formation during development, where tissue-specific inactivation of the gene results in embryonic lethality (Verzi et al. 2007). Further, MEF2 interacts with oxytocin, which is affiliated with prosocial behaviours (Kosfeld et al. 2005; Zak et al. 2007). Alterations to oxytocin have been shown to change the morphology of neurons via MEF2A (Meyer et al. 2018, 2020). Functional roles of the gene in relation to early neuronal development still remain unclear, however it is thought to play a crucial role (Harrington et al. 2016). Recent research in animal models suggests that nicotine-dependent induction of the ASH2L and MEF2C complex during development induces alterations that could lead to fundamental changes in the brain.
There are several limitations in this study thatshould be addressed. Firstly, the lack of significance after adjustment for multiple testing impacted our ability to draw firm conclusions around the association between in utero tobacco exposure and CP. However, these data indicated that this association is worth pursuing in a larger cohort. An additional subgroup of individuals who were not exposed to tobacco in utero, but who are smokers in adulthood, would add strength to the study, but were not available to us at the time the study was undertaken. Assessing a brain-related phenotype using whole blood to sample can be a problem due to cellular heterogeneity, however as with most retrospective human studies of DNA methylation, this was unable to be acquired in this instance.
Our study design is limited by the ∼21-year age discrepancy between when CP diagnosis (age 7–9) occurred and when whole blood sampling was undertaken (age 28–30), furthermore, as in most retrospective studies on exposure-induced DNA methylation changes in humans, a more biologically relevant tissue was not available or feasible. Both of these factors may have impacted our findings, and ideally this study would have been better suited to be undertaken at CP diagnosis (age 7–9). However, investigating the association between exposure, methylation and CP in adults is still intriguing as this indicates the stability of some developmentally induced methylation changes (e.g. AHRR), which prior to this study had been demonstrated
Thus, while we cannot assert causality in this study, our targeted approach shows that in utero tobacco exposure may be altering methylation at CpG sites associated with neural phenotypes thatpersist into adulthood, and are associated with increased risk of CP.
Conclusion
Here we have presented preliminary data to suggest that the association between maternal tobacco use during pregnancy and the development of CP in children and adolescents may in part be mediated by altered DNA methylation, induced by in utero tobacco exposure during development, at genes that have roles in in utero brain development and CP phenotypes. We acknowledge the limitations of this study described above, however, the data presented here are suggestive of a role for DNA methylation in the link between in utero tobacco exposure and offspring CP. Our findings should stimulate further study using larger sample sizes.
Supplementary material
Supplementary material is available online.
Data availability
Upon request.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Declaration of funding
Funding for this study came from the Maurice and Phyllis Paykel Trust. CHDS was funded by the Health Research Council of New Zealand (Programme Grant 16/600). The Canterbury Medical Research Foundation supplied funding for the manuscript to be written.
Author contributions
AJN-molecular lab work, data analysis, and major contributor to manuscript. JFP-study design, data analysis, and major contributor to manuscript. ADN-data analysis. JMB and LJH study design, provided DNA samples via CHDS. MAK-study design and over view. AJO-study design, molecular lab work, major contributor to manuscript and source of funding. All authors read and approved the final manuscript.
References
Acosta, MT, Arcos-Burgos, M, and Muenke, M (2004). Attention deficit/hyperactivity disorder (ADHD): complex phenotype, simple genotype? Genetics in Medicine 6, 1–15.| Attention deficit/hyperactivity disorder (ADHD): complex phenotype, simple genotype?Crossref | GoogleScholarGoogle Scholar | 14726804PubMed |
Beaver, KM, Wright, JP, DeLisi, M, Walsh, A, Vaughn, MG, Boisvert, D, and Vaske, J (2007). A gene × gene interaction between DRD2 and DRD4 is associated with conduct disorder and antisocial behavior in males. Behavioral and Brain Functions 3, 30.
| A gene × gene interaction between DRD2 and DRD4 is associated with conduct disorder and antisocial behavior in males.Crossref | GoogleScholarGoogle Scholar | 17587443PubMed |
Carter, S, Paterson, J, Gao, W, and Iusitini, L (2008). Maternal smoking during pregnancy and behaviour problems in a birth cohort of 2-year-old Pacific children in New Zealand. Early Human Development 84, 59–66.
| Maternal smoking during pregnancy and behaviour problems in a birth cohort of 2-year-old Pacific children in New Zealand.Crossref | GoogleScholarGoogle Scholar | 17499944PubMed |
Chen, Y, Pal, B, Visvader, JE, and Smyth, GK (2017). Differential methylation analysis of reduced representation bisulfite sequencing experiments using edger. F1000Res 6, 2055–55.
| Differential methylation analysis of reduced representation bisulfite sequencing experiments using edger.Crossref | GoogleScholarGoogle Scholar | 29333247PubMed |
Conners, CK (1969). A teacher rating scale for use in drug studies with children. American Journal of Psychiatry 126, 884–888.
| A teacher rating scale for use in drug studies with children.Crossref | GoogleScholarGoogle Scholar |
Conners, CK (1970). Symptom patterns in hyperkinetic, neurotic, and normal children. Child Development 41, 667–82.
| Symptom patterns in hyperkinetic, neurotic, and normal children.Crossref | GoogleScholarGoogle Scholar |
Cox, MP, Peterson, DA, and Biggs, PJ (2010). SolexaQA: at-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinformatics 11, 485.
| SolexaQA: at-a-glance quality assessment of Illumina second-generation sequencing data.Crossref | GoogleScholarGoogle Scholar | 20875133PubMed |
De Vocht, F, Simpkin, AJ, Richmond, RC, Relton, C, and Tilling, K (2015). Assessment of offspring DNA methylation across the lifecourse associated with prenatal maternal smoking using Bayesian mixture modelling. International Journal of Environmental Research and Public Health 12, 14461–14476.
| Assessment of offspring DNA methylation across the lifecourse associated with prenatal maternal smoking using Bayesian mixture modelling.Crossref | GoogleScholarGoogle Scholar | 26580635PubMed |
Demontis, D, Walters, RK, Martin, J, Mattheisen, M, Als, TD, Agerbo, E, Baldursson, G, Belliveau, R, Bybjerg-Grauholm, J, Bækvad-Hansen, M, and Cerrato, F (2019). Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nature Genetics 51, 63–75.
| Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder.Crossref | GoogleScholarGoogle Scholar | 30478444PubMed |
Fergusson, DM, Horwood, LJ, and Lloyd, M (1991). Confirmatory factor models of attention deficit and conduct disorder. Journal of Child Psychology and Psychiatry, and Allied Disciplines 32, 257–274.
| Confirmatory factor models of attention deficit and conduct disorder.Crossref | GoogleScholarGoogle Scholar | 2033107PubMed |
Fergusson, DM, John Horwood, L, and Ridder, EM (2005). Show me the child at seven: the consequences of conduct problems in childhood for psychosocial functioning in adulthood. Journal of Child Psychology and Psychiatry, and Allied Disciplines 46, 837–849.
| Show me the child at seven: the consequences of conduct problems in childhood for psychosocial functioning in adulthood.Crossref | GoogleScholarGoogle Scholar | 16033632PubMed |
Harrington, AJ, Raissi, A, Rajkovich, K, Berto, S, Kumar, J, Molinaro, G, Raduazzo, J, Guo, Y, Loerwald, K, Konopka, G, Huber, KM, and Cowan, CW (2016). MEF2C regulates cortical inhibitory and excitatory synapses and behaviors relevant to neurodevelopmental disorders. eLife 5, e20059.
| MEF2C regulates cortical inhibitory and excitatory synapses and behaviors relevant to neurodevelopmental disorders.Crossref | GoogleScholarGoogle Scholar | 27779093PubMed |
Hsieh, C-J, Jeng, S-F, Su, Y-N, Liao, H-F, Hsieh, W-S, Wu, K-Y, and Chen, P-C (2010). CYP1A1 modifies the effect of maternal exposure to environmental tobacco smoke on child behavior. Nicotine & Tobacco Research 12, 1108–1117.
| CYP1A1 modifies the effect of maternal exposure to environmental tobacco smoke on child behavior.Crossref | GoogleScholarGoogle Scholar |
Jiao, S-S, Shen, L-L, Zhu, C, Bu, X-L, Liu, Y-H, Liu, C-H, Yao, X-Q, Zhang, L-L, Zhou, H-D, Walker, DG, Tan, J, Götz, J, Zhou, X-F, and Wang, Y-J (2016). Brain-derived neurotrophic factor protects against tau-related neurodegeneration of Alzheimer’s disease. Translational Psychiatry 6, e907.
| Brain-derived neurotrophic factor protects against tau-related neurodegeneration of Alzheimer’s disease.Crossref | GoogleScholarGoogle Scholar | 27701410PubMed |
Joelsson, P, Chudal, R, Talati, A, Suominen, A, Brown, AS, and Sourander, A (2016). Prenatal smoking exposure and neuropsychiatric comorbidity of ADHD: a finnish nationwide population-based cohort study. BMC Psychiatry 16, 306.
| Prenatal smoking exposure and neuropsychiatric comorbidity of ADHD: a finnish nationwide population-based cohort study.Crossref | GoogleScholarGoogle Scholar | 27581195PubMed |
Joubert, BR, Håberg, SE, Nilsen, RM, Wang, X, Vollset, SE, Murphy, SK, Huang, Z, Hoyo, C, Midttun, Ø, Cupul-Uicab, LA, Ueland, PM, Wu, MC, Nystad, W, Bell, DA, Peddada, SD, and London, SJ (2012). 450K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environmental Health Perspectives 120, 1425–1431.
| 450K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy.Crossref | GoogleScholarGoogle Scholar | 22851337PubMed |
Joubert, BR, Felix, JF, Yousefi, P, Bakulski, KM, Just, AC, Breton, C, Reese, SE, Markunas, CA, Richmond, RC, and Xu, C-J (2016). DNA methylation in newborns and maternal smoking in pregnancy: genome-wide consortium meta-analysis. The American Journal of Human Genetics 98, 680–696.
| DNA methylation in newborns and maternal smoking in pregnancy: genome-wide consortium meta-analysis.Crossref | GoogleScholarGoogle Scholar | 27040690PubMed |
Jung, Y, Hsieh, LS, Lee, AM, Zhou, Z, Coman, D, Heath, CJ, Hyder, F, Mineur, YS, Yuan, Q, Goldman, D, Bordey, A, and Picciotto, MR (2016). An epigenetic mechanism mediates developmental nicotine effects on neuronal structure and behavior. Nature Neuroscience 19, 905–914.
| An epigenetic mechanism mediates developmental nicotine effects on neuronal structure and behavior.Crossref | GoogleScholarGoogle Scholar | 27239938PubMed |
Kosfeld, M, Heinrichs, M, Zak, PJ, Fischbacher, U, and Fehr, E (2005). Oxytocin increases trust in humans. Nature 435, 673–676.
| Oxytocin increases trust in humans.Crossref | GoogleScholarGoogle Scholar | 15931222PubMed |
Krueger, F, and Andrews, SR (2011). Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27, 1571–1572.
| Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications.Crossref | GoogleScholarGoogle Scholar | 21493656PubMed |
Lantz, PM, House, JS, Lepkowski, JM, Williams, DR, Mero, RP, and Chen, J (1998). Socioeconomic factors, health behaviors, and mortality results from a nationally representative prospective study of US Adults’. JAMA 279, 1703–08.
| Socioeconomic factors, health behaviors, and mortality results from a nationally representative prospective study of US Adults’.Crossref | GoogleScholarGoogle Scholar | 9624022PubMed |
Lee, KWK, Richmond, R, Hu, P, French, L, Shin, J, Bourdon, C, Reischl, E, Waldenberger, M, Zeilinger, S, Gaunt, T, McArdle, W, Ring, S, Woodward, G, Bouchard, L, Gaudet, D, Smith, GD, Relton, C, Paus, T, and Pausova, Z (2015). Prenatal exposure to maternal cigarette smoking and DNA methylation: epigenome-wide association in a discovery sample of adolescents and replication in an independent cohort at birth through 17 years of age. Environmental Health Perspectives 123, 193–199.
| Prenatal exposure to maternal cigarette smoking and DNA methylation: epigenome-wide association in a discovery sample of adolescents and replication in an independent cohort at birth through 17 years of age.Crossref | GoogleScholarGoogle Scholar |
Li, L, Ruan, X, Wen, C, Chen, P, Liu, W, Zhu, L, Xiang, P, Zhang, X, Wei, Q, Hou, L, Yin, B, Yuan, J, Qiang, B, Shu, P, and Peng, X (2019). The COMPASS family protein ASH2L mediates corticogenesis via transcriptional regulation of wnt signaling. Cell Reports 28, 698–711.e5.
| The COMPASS family protein ASH2L mediates corticogenesis via transcriptional regulation of wnt signaling.Crossref | GoogleScholarGoogle Scholar | 31315048PubMed |
Meyer, M, Berger, I, Winter, J, and Jurek, B (2018). Oxytocin alters the morphology of hypothalamic neurons via the transcription factor myocyte enhancer factor 2A (MEF-2A). Molecular and Cellular Endocrinology 477, 156–162.
| Oxytocin alters the morphology of hypothalamic neurons via the transcription factor myocyte enhancer factor 2A (MEF-2A).Crossref | GoogleScholarGoogle Scholar | 29928931PubMed |
Meyer, M, Kuffner, K, Winter, J, Neumann, ID, Wetzel, CH, and Jurek, B (2020). Myocyte Enhancer Factor 2A (MEF2A) defines oxytocin-induced morphological effects and regulates mitochondrial function in neurons. International Journal of Molecular Sciences 21, 2200.
| Myocyte Enhancer Factor 2A (MEF2A) defines oxytocin-induced morphological effects and regulates mitochondrial function in neurons.Crossref | GoogleScholarGoogle Scholar |
Mick, E, Biederman, J, Faraone, SV, Sayer, J, and Kleinman, S (2002). Case-control study of attention-deficit hyperactivity disorder and maternal smoking, alcohol use, and drug use during pregnancy. Journal of the American Academy of Child & Adolescent Psychiatry 41, 378–385.
| Case-control study of attention-deficit hyperactivity disorder and maternal smoking, alcohol use, and drug use during pregnancy.Crossref | GoogleScholarGoogle Scholar |
Noble, AJ, Pearson, JF, Boden, JM, Horwood, LJ, Gemmell, NJ, Kennedy, MA, and Osborne, AJ (2021). A validation of Illumina EPIC array system with bisulfite-based amplicon sequencing. PeerJ 9, e10762.
| A validation of Illumina EPIC array system with bisulfite-based amplicon sequencing.Crossref | GoogleScholarGoogle Scholar | 33614276PubMed |
Rauschert, S, Melton, PE, Burdge, G, Craig, JM, Godfrey, KM, Holbrook, JD, Lillycrop, K, Mori, TA, Beilin, LJ, Oddy, WH, Pennell, C, and Huang, R-C (2019). Maternal smoking during pregnancy induces persistent epigenetic changes into adolescence, independent of postnatal smoke exposure and is associated with cardiometabolic risk. Frontiers in Genetics 10, 770.
| Maternal smoking during pregnancy induces persistent epigenetic changes into adolescence, independent of postnatal smoke exposure and is associated with cardiometabolic risk.Crossref | GoogleScholarGoogle Scholar | 31616461PubMed |
Richmond, RC, Simpkin, AJ, Woodward, G, Gaunt, TR, Lyttleton, O, McArdle, WL, Ring, SM, Smith, ADAC, Timpson, NJ, Tilling, K, Davey Smith, G, and Relton, CL (2015). Prenatal exposure to maternal smoking and offspring DNA methylation across the lifecourse: findings from the Avon Longitudinal Study of Parents and Children (ALSPAC). Human Molecular Genetics 24, 2201–2217.
| Prenatal exposure to maternal smoking and offspring DNA methylation across the lifecourse: findings from the Avon Longitudinal Study of Parents and Children (ALSPAC).Crossref | GoogleScholarGoogle Scholar | 25552657PubMed |
Richmond, RC, Suderman, M, Langdon, R, Relton, CL, and Davey Smith, G (2018). DNA methylation as a marker for prenatal smoke exposure in adults. International Journal of Epidemiology 47, 1120–1130.
| DNA methylation as a marker for prenatal smoke exposure in adults.Crossref | GoogleScholarGoogle Scholar | 29860346PubMed |
Riva, V, Battaglia, M, Nobile, M, Cattaneo, F, Lazazzera, C, Mascheretti, S, Giorda, R, Mérette, C, Émond, C, Maziade, M, and Marino, C (2015). GRIN2B predicts attention problems among disadvantaged children. European Child & Adolescent Psychiatry 24, 827–836.
| GRIN2B predicts attention problems among disadvantaged children.Crossref | GoogleScholarGoogle Scholar |
Rotroff, DM, Joubert, BR, Marvel, SW, Håberg, SE, Wu, MC, Nilsen, RM, Ueland, PM, Nystad, W, London, SJ, and Motsinger-Reif, A (2016). Maternal smoking impacts key biological pathways in newborns through epigenetic modification in utero. BMC Genomics 17, 976.
| Maternal smoking impacts key biological pathways in newborns through epigenetic modification in utero.Crossref | GoogleScholarGoogle Scholar | 27887572PubMed |
Rutter M, Tizard J, Whitmore K (1970) ‘Education, health and behavior.’ (Longmans: London)
Rzehak, P, Saffery, R, Reischl, E, Covic, M, Wahl, S, Grote, V, Xhonneux, A, Langhendries, J-P, Ferre, N, Closa-Monasterolo, R, Verduci, E, Riva, E, Socha, P, Gruszfeld, D, and Koletzko, B Rzehak, P, Saffery, R, Reischl, E, Covic, M, Wahl, S, Grote, V, Xhonneux, A, Langhendries, J-P, Ferre, N, Closa-Monasterolo, R, Verduci, E, Riva, E, Socha, P, Gruszfeld, D, and Koletzko, B (2016). Maternal smoking during pregnancy and DNA-methylation in children at age 5.5 years: epigenome-wide-analysis in the European Childhood Obesity Project (CHOP)-study. PLoS ONE 11, e0155554.
| Maternal smoking during pregnancy and DNA-methylation in children at age 5.5 years: epigenome-wide-analysis in the European Childhood Obesity Project (CHOP)-study.Crossref | GoogleScholarGoogle Scholar | 27171005PubMed |
Salvatore, JE, and Dick, DM (2018). Genetic influences on conduct disorder. Neuroscience & Biobehavioral Reviews 91, 91–101.
| Genetic influences on conduct disorder.Crossref | GoogleScholarGoogle Scholar |
Sengupta, SM, Smith, AK, Grizenko, N, and Joober, R (2017). Locus-specific DNA methylation changes and phenotypic variability in children with attention-deficit hyperactivity disorder. Psychiatry Research 256, 298–304.
| Locus-specific DNA methylation changes and phenotypic variability in children with attention-deficit hyperactivity disorder.Crossref | GoogleScholarGoogle Scholar | 28662467PubMed |
Skogstrand, K, Hagen, CM, Borbye-Lorenzen, N, Christiansen, M, Bybjerg-Grauholm, J, Bækvad-Hansen, M, Werge, T, Børglum, A, Mors, O, Nordentoft, M, Mortensen, PB, and Hougaard, DM (2019). Reduced neonatal brain-derived neurotrophic factor is associated with autism spectrum disorders. Translational Psychiatry 9, 252.
| Reduced neonatal brain-derived neurotrophic factor is associated with autism spectrum disorders.Crossref | GoogleScholarGoogle Scholar | 31591381PubMed |
Suter, M, Abramovici, A, Showalter, L, Hu, M, Shope, CD, Varner, M, and Aagaard-Tillery, K (2010). In utero tobacco exposure epigenetically modifies placental CYP1A1 expression. Metabolism-Clinical and Experimental 59, 1481–1490.
| In utero tobacco exposure epigenetically modifies placental CYP1A1 expression.Crossref | GoogleScholarGoogle Scholar | 20462615PubMed |
Tehranifar, P, Wu, H-C, McDonald, JA, Jasmine, F, Santella, RM, Gurvich, I, Flom, JD, and Terry, MB (2018). Maternal cigarette smoking during pregnancy and offspring DNA methylation in midlife. Epigenetics 13, 129–134.
| Maternal cigarette smoking during pregnancy and offspring DNA methylation in midlife.Crossref | GoogleScholarGoogle Scholar | 28494218PubMed |
van Otterdijk, SD, Binder, AM, and Michels, KB (2017). Locus-specific DNA methylation in the placenta is associated with levels of pro-inflammatory proteins in cord blood and they are both independently affected by maternal smoking during pregnancy. Epigenetics 12, 875–885.
| Locus-specific DNA methylation in the placenta is associated with levels of pro-inflammatory proteins in cord blood and they are both independently affected by maternal smoking during pregnancy.Crossref | GoogleScholarGoogle Scholar | 28820654PubMed |
Verzi, MP, Agarwal, P, Brown, C, McCulley, DJ, Schwarz, JJ, and Black, BL (2007). The transcription factor MEF2C is required for craniofacial development. Developmental Cell 12, 645–652.
| The transcription factor MEF2C is required for craniofacial development.Crossref | GoogleScholarGoogle Scholar | 17420000PubMed |
Wakschlag, LS, Lahey, BB, Loeber, R, Green, SM, Gordon, RA, and Leventhal, BL (1997). Maternal smoking during pregnancy and the risk of conduct disorder in boys. Archives of General Psychiatry 54, 670–676.
| Maternal smoking during pregnancy and the risk of conduct disorder in boys.Crossref | GoogleScholarGoogle Scholar | 9236551PubMed |
Zak, PJ, Stanton, AA, and Ahmadi, S (2007). Oxytocin increases generosity in humans. PLoS ONE 2, e1128.
| Oxytocin increases generosity in humans.Crossref | GoogleScholarGoogle Scholar | 17987115PubMed |
† Pre print version available here, https://www.biorxiv.org/content/10.1101/2020.07.02.183285v2