

Exploiting a fast neutron mutant genetic resource in Pisum sativum (pea) for functional genomics
Claire Domoney A , Maggie Knox A , Carol Moreau A , Mike Ambrose A , Sarah Palmer A B , Peter Smith B , Vangelis Christodoulou C , Peter G. Isaac C , Matthew Hegarty D , Tina Blackmore D , Martin Swain D and Noel Ellis D E FA Department of Metabolic Biology, John Innes Centre, Norwich Research Park, Norwich NR4 7UH, UK.
B Wherry and Sons Ltd, The Old School, High Street, Rippingale, Bourne, Lincolnshire PE10 0SR, UK.
C IDna Genetics Ltd, Norwich Research Park, Norwich NR4 7UH, UK.
D Institute of Biological, Environmental and Rural Sciences, Aberystwyth University, Ceredigion SY23 3EB, UK.
E Present address: CGIAR Research Program on Grain Legumes, c/o ICRISAT, Patancheru 502 324, Andhra Pradesh, India.
F Corresponding author. Email: noe2@aber.ac.uk
This paper originates from a presentation at the ‘VI International Conference on Legume Genetics and Genomics (ICLGG)’ Hyderabad, India, 2–7 October 2012.
Functional Plant Biology 40(12) 1261-1270 https://doi.org/10.1071/FP13147
Submitted: 19 May 2013 Accepted: 18 July 2013 Published: 28 August 2013
Journal Compilation © CSIRO Publishing 2013 Open Access CC BY-NC-ND
Abstract
A fast neutron (FN)-mutagenised population was generated in Pisum sativum L. (pea) to enable the identification and isolation of genes underlying traits and processes. Studies of several phenotypic traits have clearly demonstrated the utility of the resource by associating gene deletions with phenotype followed by functional tests exploiting additional mutant sources, from both induced and natural variant germplasm. For forward genetic screens, next generation sequencing methodologies provide an opportunity for identifying genes associated with deletions rapidly and systematically. The application of rapid reverse genetic screens of the fast neutron mutant pea population supports conclusions on the frequency of deletions based on phenotype alone. These studies also suggest that large deletions affecting one or more loci can be non-deleterious to the pea genome, yielding mutants that could not be obtained by other means. Deletion mutants affecting genes associated with seed metabolism and storage are providing unique opportunities to identify the products of complex and related gene families, and to study the downstream consequences of such deletions.
Additional keywords: fast neutron mutagenesis, genomic deletions, seed proteins.
Introduction
Fast neutron (FN) radiation is an effective mutagenesis technique for plants, giving rise to mutant populations that have been exploited to support gene discovery and isolation. DNA deletions that range in size from a few base pairs to several megabases have been described for a range of plant crop and model species, including Glycine max (L.) Merr. (Bolon et al. 2011), Glycine soja Siebold & Zucc. (Searle et al. 2003), Hordeum vulgare L. (Zhang et al. 2006), Arabidopsis thaliana (L.) Heynh. (Alonso et al. 2003; Belfield et al. 2012), Medicago truncatula Gaertn. (Rogers et al. 2009), Lotus japonicus (Regel) K. Larsen (Hoffmann et al. 2007) and Pisum sativum L. (Hofer et al. 2009). Although genomic deletions are expected to represent the majority of mutations identified in fast neutron mutagenised populations, it is clear that single base substitutions can occur at a higher frequency than deletion mutations with FN-induced G : C > A : T transitions being concentrated at pyrimidine dinucleotide sites (Belfield et al. 2012). Induced genetic rearrangements have also been discovered in deletion mutant screens (Rogers et al. 2009; Moreau et al. 2012).
An FN mutagenised population has been developed in Pisum sativum (pea), using as a parent the recombinant inbred line, JI 2822, a garden pea genotype with dwarf stature and early flowering traits that enable rapid cycling (Hofer et al. 2009). Studies that have exploited this population thus far have focussed on the genetics of phenotypic traits and have indicated that individual mutant lineages can segregate for several mutations, necessitating the backcrossing of lines for rigorous analysis of mutations. Several of these studies have been successful in identifying the genes underlying phenotypic mutations (Sainsbury et al. 2006; Wang et al. 2008; Hofer et al. 2009; Hellens et al. 2010; Chen et al. 2012; Couzigou et al. 2012; Moreau et al. 2012; Zhuang et al. 2012). The mutants referred to above, together with others described in the literature, are available from the John Innes Pisum germplasm collection (http://www.jic.ac.uk/germplasm/, accessed 31 July 2013). None of these studies of pea FN mutants has made a systematic use of next generation sequencing methods, but this approach would be expected to be successful for finding the gross sequence differences expected in comparisons between an FN mutant and its wild type progenitor. Here we describe the use of high-throughput DNA screening and sequencing methodologies to increase the utility of the population for both forward and reverse genetics and to further our knowledge of its genetic constitution. This resource, alongside the ethyl methane sulfonate-mutagenised population that is being exploited widely for reverse genetics (Dalmais et al. 2008), provides an important tool to underpin functional genomics in pea.
Materials and methods
Plant material
An FN-irradiated mutant population was constructed using the Pisum sativum L. accession JI 2822, a recombinant inbred line derived from a cross between JI 15 and JI 399 (Ellis et al. 1992). JI 2822 is characterised by its short stature (le), early flowering (lf) and wrinkled seeds (rb), derived from the JI 399 parent, and has wild-type flower colour (A, derived from the JI 15 parent and Ce, derived from the JI 399 parent). JI 2822 is a compact rapid cycling line, which can complete a generation within two months, and is suitable for mutagenesis (see Fig. S1, available as Supplementary Material to this paper). JI 2822 seeds were subjected to FN irradiation (either 20 or 25 Gy) generated from 252Cf at Oak Ridge National Laboratory, USA. The doses employed were somewhat lower than those reported for other studies, for example Glycine max L. (soybean, 32 Gy, Bolon et al. 2011) and Arabidopsis thaliana (L. Heynh.) (60 Gy, Belfield et al. 2012), but were based on these and related studies and a trial in the range from 15 to 25 Gy was conducted initially. The primary mutants (M1) were sown in 20 cm pots in John Innes Number 1 compost (http://www.jic.ac.uk/corporate/media-and-public/compost.htm, accessed 31 July 2013) supplemented with 30% chick grit. These individuals were generally mottled in appearance, but ~75% eventually set seed. Plants were screened for phenotypic mutations and several have been characterised (see ‘Introduction’). Seeds were collected separately from each individual M1 plant. M2 families of up to four plants per line were sown to give the M3 to M5 lineages, discussed here.
Individual plants from FN-mutagenised lines used for amplified fragment length polymorphism (AFLP) analysis, as described by Lu et al. (1996), are listed in Table 1.
|
The plants FN2122BC3S1, FN1889BC4S1 and FN2002BC4S1 are individuals corresponding to the selfed progeny of FN lines backcrossed to JI 2822 for three or four cycles as designated by BC number. The selfed plants were all homozygous for the recessive mutation of interest.
DNA assays
Two seeds of 2000 lines at M4 or M5 were sown for DNA preparation and targeted assays, using primers designed on genes affecting seed metabolism. Duplicate biological samples from individual lineages were considered an important control to minimise false positives, and enhance the chances of mutation detection among lineages where a mutation might be segregating. The relative measures of amplification efficiency and primer specificity were determined using unlabelled primers. For successful primer pairs, one was then designed to be fluorescently labelled. Multiplex PCR assays were used to give a range of discrete gene-specific products identifiable by label and size (amplicons in the range of 100–600 bases). The products were analysed by separation of fragments using capillary electrophoresis on an ABI3730 instrument, following addition of a size standard mix to every PCR sample. The resulting traces were converted to electropherograms and analysed by GeneMapper (Applied Biosystems/Life Technologies, Foster City, CA, USA). Missing amplicons were detected by eye, and also by a statistical analysis of the peak heights relative to the total peak height for the sample.
Standard PCR assays were employed to confirm missing amplicons, using the same primers or additional primers covering upstream and downstream regions of the gene targets.
Primers were designed on publically available sequences for the following pea gene or mRNA sequences (GenBank accession numbers in brackets): CD72/P54/sucrose binding protein (Y11207); lectin (Y00440); vicilin (VicB, X14076); convicilin (X06398, M73805.1); TCP transcription factor, PsCYC3 (EU574915 for k locus) (see Table S1, available as Supplementary Material to this paper). Where mRNA sequences alone were available, the positions of introns were deduced from orthologues identified in other species, chiefly M. truncatula and G. max.
Protein assays
Progeny seeds from mutant lines were used to obtain meal samples for analysis of seed composition. Approximately 3–5 mg of meal was extracted in LDS buffer (Invitrogen, Paisley, UK) at a ratio of 5 mg meal per mL buffer. Extracts were heated to 70°C for 10 min, and the supernatants removed following centrifugation. Aliquots were loaded onto gels (12% Bis-Tris gels, Invitrogen) and run in 1 × MOPS buffer at 140V for 1–2 h at room temperature, following addition of antioxidant (NuPAGE) to wells just before electrophoresis. Gels were stained using InstantBlue (Expedeon, Harston, UK).
Genetic mapping
The recombinant inbred population derived from a cross between JI 281 and JI 399 was exploited for mapping genes (Ellis and Poyser 2002), using primers based on the sequences listed above. The JI 281 × JI 399 population comprises 89 recombinant inbred lines bulked at F12 with a map based on 637 ± 22 markers. The AFLP protocol was as described by Lu et al. (1996) and based on PstI/MseI digestion.
DNA sequencing
Routine sequencing of amplicons was conducted at The Genome Analysis Centre, Norwich, UK. RAD (restriction site associated DNA) sequencing was performed according to work by Miller et al. (2007) and Baird et al. (2008), except that the rare cutting enzyme used was PstI. Because of the low complexity of the RAD library, multiple primers were used per genotype and these were collated.
Results
Several forward genetic screens of the FN population generated in JI 2822 have contributed to the identification of novel alleles for several classical genetic markers in pea, as summarised in Table 2. These studies, together with the results presented here, indicate that a range of deletion sizes exists in the population and that individual lines could carry more than one mutation. For the deletion alleles of the mutants listed in Table 2, one rearrangement (Moreau et al. 2012) that appears to result from a non-homologous exchange involving an Ogre element was found, and two were small deletions, one of 22 bp (Hellens et al. 2010) and the other of 1.4 kb (Moreau et al. 2012); the remaining 25 alleles were all larger than the gene being characterised.

|
The number of mutations per line was estimated in an AFLP screen; for this purpose 16 FN lines were selected, eight had a clearly observable mutant phenotype and eight had no obvious mutation (Table 1). These were screened so that one PstI primer (with selective nucleotides AA) was analysed in conjunction with all selective (three nucleotide) primers at the frequent cutting MseI site. This generated 2638 distinguishable amplicons of which seven were confirmed to be lost in at least one FN individual. The number of band losses observed for mutant and apparently wild-type individuals was similar (3 vs 4) and the number of individuals with missing bands was also similar across the two groups (2 vs 1), suggesting that there is no strong evidence that the apparently wild-type individuals have escaped mutagenesis.
With the full set of 16 PstI selective primers we would expect ~100 lost amplicons in these 16 lineages and, as the whole FN population is ~3000 lines, we would expect ~20 000 independent deletions with each line carrying an average of seven independent deletions. This is far from complete coverage but, if available PstI sites are preferentially associated with coding sequences as would be expected from the methylation sensitivity of the enzyme, then we might expect a reasonable fraction of genes to have at least one mutant allele in the population. If the PstI sites are preferentially associated with genic DNA, then this is approaching a 0.6× gene coverage (given a gene number estimate in Medicago of 34 000).
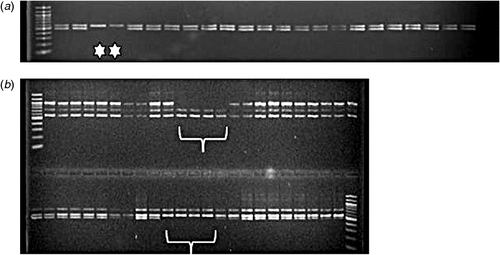
These indications as to mutation frequency and size have now been investigated further using reverse genetic DNA screens for several seed-expressed gene classes. Deletions of portions of genes encoding a putative sucrose-binding protein with antimicrobial properties (SBP), a class of vicilin polypeptide (VicB), lectin (LecA) and convicilin (Cvc) were identified in PCR multiplex assays shown in Fig. 1a–d. Targets were in the range from 101 to 681 base pairs and were amplified clearly in the wild-type lines. In Fig. 1, the deletions shown are of three types affecting the PCR products obtained for four wild-type genes (controls, arrowed). Simple deletions for single amplicons predicted for one gene are evident in Fig. 1a (101 bp product of SBP) and Fig. 1c (477 bp of LecA). In contrast, Fig. 1b shows a set of complex amplicons corresponding to the VicB gene class, where additional amplicons to that predicted are evident within the multiplex and all of these are absent from the mutants identified. Fig. 1d shows an example of the amplicons predicted for two related Cvc genes evident in wild type lines, where both are deleted from mutant lines. These data suggest that, where gene targets are known to be representative of a clustered group of closely related genes, the deletions may include a set of genes known to be very closely linked genetically.

|
Simplex PCR screens have validated the deletions shown in Fig. 1a–d and suggest that regions upstream and downstream of the targets shown in Fig. 1 are missing from deletion mutants. Fig. 2a shows the loss of a LecA amplicon from mutants, while more detailed analysis of the VicB gene class (Fig. 1b) has indicated that, in the mutants, all related genes have been deleted, based on assays that target both 5′ and 3′ genic regions (Fig. 2).
|
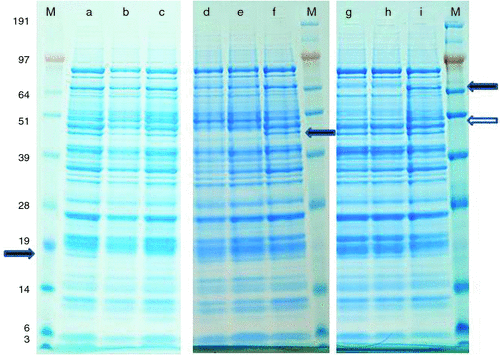
Analyses of seed extracts by protein gel electrophoresis have been used to extend these results. Fig. 3 shows examples of the total protein profiles obtained for some of the deletion mutants. Polypeptides of relative molecular mass (Mr) 18 000, 45 000 and 70 000, which are missing from LecA, SBP and Cvc mutants, respectively, are evident (Fig. 3a–i). Although allowing for signal peptides, these polypeptides are within or close to the Mr range of those predicted from gene sequences, the additional polypeptide missing from Cvc mutants (~Mr 50 000) is somewhat smaller than expected (Fig. 3g, h). This latter may reflect either the Cvc deletion or an unlinked second mutation.
|
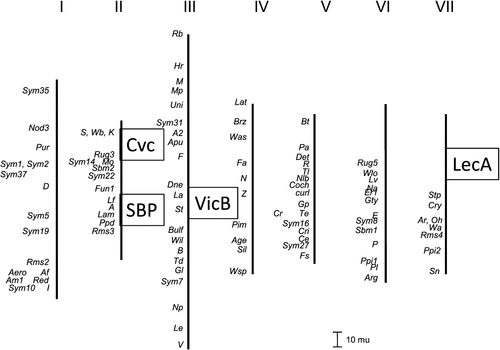
The genetic map positions of the seed protein mutations discussed in this paper are summarised in Fig. 4. The positions of Cvc (Matta and Gatehouse 1982; Ellis et al. 1992) and the gene encoding SBP (referred to as CD72 in Domoney and Casey 1990; Hellens et al. 2010) have been determined previously to be on the pea linkage group II. In both of these cases, a morphological marker affecting flower phenotype exists nearby (k, controlling keeled wings, close to Cvc, and A, controlling pigmentation in seed coats, flowers and leaf axils, close to CD72 encoding SBP). The SBP FN mutants have wild-type (coloured) flowers, implying that the deletion does not impinge on the genetic locus A. However, the Cvc mutants all have keeled flowers implying a deletion spanning the Cvc-k genetic region. Therefore we investigated the genetic distance affected by this latter deletion. Polymorphisms in the Cvc and k (PsCYC3) gene alleles in JI 281 and JI 399 were used to map both loci in recombinant inbred lines derived from this cross; out of 80 progeny lines, no recombinants were found (data not shown). The loss of both Cvc gene amplicons from mutants (Fig. 1d) and several VicB amplicons in others (Fig. 1b) is further indication of large deletions affecting entire loci comprising closely linked related genes (Ellis et al. 1986).
|
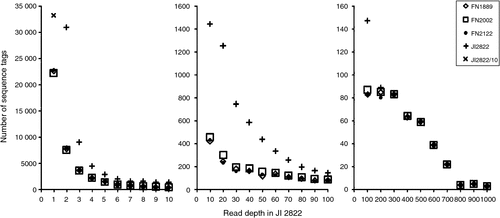
These results with reverse genetic screens, and the descriptions of mutant alleles previously isolated from this FN population, suggest that the majority (but not all) of the mutations correspond to deletions that are large with respect to the size of a gene. This suggests that a genome scan that samples sequences distributed throughout the genome would be likely to identify many of the sequences present in wild type but absent from a corresponding mutant. To determine whether this was the case we obtained sequence data for 41 bp immediately adjacent to PstI sites using a minor modification of the RAD procedure (Miller et al. 2007). Sequence tags adjacent to PstI sites were generated for four genotypes: JI 2822, FN2122BC3S1, FN1889BC4S1 and FN2002BC4S1. The FN lines were in the JI 2822 background (see above) and the individuals were selfed plants from the third or fourth backcross to JI 2822. FN2122BC3S1 is a stipules reduced (st) mutant (Pellew and Sverdrup 1923) and the other two are creep (Sidorova 1975) mutants. Sequence data were collected from two lanes of an Illumina (San Diego, CA, USA) HiSeq machine and ~110 million reads were collected, but these were not equally distributed; the wild type JI 2822 had approximately twice the number of reads as the other samples. Sequence reads were collated into unique tags with corresponding read depth using FASTX collapser (downloaded from http://hannonlab.cshl.edu/fastx_toolkit/, accessed 17 August 2012) and then tags identical to JI 2822 sequences were identified for each genotype using a Python script. Sequences absent from JI 2822 have been ignored. The abundance of sequence tags at different read depths for the four genotypes is indicated in Fig. 5. From this it is clear that there are many tags with low read depth that are unique to the JI 2822 sample. At least some of these are sequence variants of tags with higher read depth, and so are likely to be sequencing errors; such errors are likely to be unique to a sample. Above a read depth of ~150 (in the JI 2822 sample) the difference in the number of tags identified for each genotype declines considerably so, at this range, a sequence absence is sufficiently likely not to be a consequence of sequencing error or sampling variation as to be worth investigating. Nevertheless, chi-square tests show an excess of tags missing from more than one FN genotype than would be expected by chance alone (Table 3). The excess of missing tags from pairs of samples is not greatest when comparing the two creep lines, suggesting that most of this excess is due to sampling and or sequencing errors.
|

|
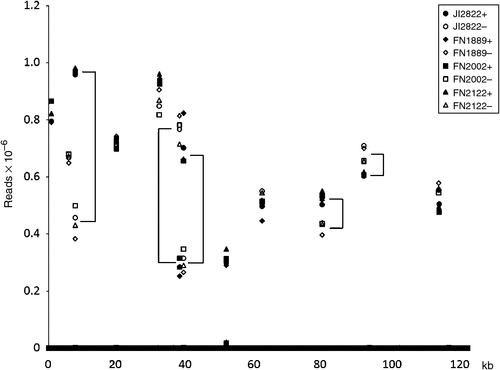
The sequence tags necessarily correspond to pairs of sequences either side of a PstI site, so if tags missing from both sides behave independently this can help with the identification of deleted sequences. To test this sequence tags were aligned to the pea (cv. Feltham First) chloroplast DNA sequence (HM029370) and the read depth of the corresponding tags (max e value 3.00E-16, min 97.62% identity) is shown in Fig. 6. Several features of this plot are surprising at first sight; first there are very many tags that map throughout the sequence and have low read depth. Those that are 100% identical have a read depth of 91 ± 31 (mean ± s.e.) and those with one mismatch have a read depth of 314 ± 200. This suggests the two types of sequence have different origins. The perfect matches may correspond to broken molecules whereas the sequences with a mismatch may include sequencing errors. Two tags have read depths of ~20 000 and may correspond to cpDNA from another cellular compartment. The minimum proportion of these sequence variants corresponding to the abundant tags is 0.03%, but more typically it is 1–3% (1.87 ± 0.12%). The higher value probably overestimates the error rate due to the presence of cpDNA in other cellular compartments but, if this is taken as an upper estimate of the rate of sequence errors, then for read depths of ~200 these sequence errors will contribute to tags with very low read depths.
|
A second feature is that not all sites have pairs of sequences, presumably due to sequence differences with respect to cv. Feltham First. A third feature of the plot is that for some pairs of tags (either side of a site) the read depth is significantly different, and consistently so for the different genotypes.
Together these observations suggest that read depths of ~150 or more should prove reliable at identifying deleted sequences.
A BLAST search of the tags with a read depth of at least 100 in JI 2822 against a transcriptome database (http://www.coolseasonfoodlegume.org/, accessed 20 November 2012) identified 4688 pairs, orientated and spaced as expected for sequences either side of a PstI site. These had a maximum of one nucleotide internal mismatch, and a minimum span of 80 nucleotides including the PstI site (4208/4688 spanned the expected 88 nucleotides).
Discussion
Forward and reverse genetic screens performed on a FN-mutagenised population of pea have identified a range of phenotypic mutants affecting plant and flower morphology, as well as seed protein profiles. In combination with analyses based on AFLPs, we conclude that the resource harbours a wealth of valuable mutants for future studies.
The data presented here illustrate the application of deletion mutants in identifying the products of complex gene families encoding seed proteins, which frequently undergo post-translational processing and modification (Casey and Domoney 1999). As an example, the function of the (putative) sucrose binding protein (SBP) is unknown and its product had not been identified previously in mature seeds. SBP is related to vicilin (Domoney and Casey 1990), being classified as a cupin, but is also closely related to a Vicia faba putative sucrose-binding protein and to M. truncatula vicilin-like antimicrobial peptides (86 and 75% identity respectively). Although it had been shown that the primary translation product of the pea (SBP) mRNA was of apparent Mr 68 000 on SDS-gel electrophoresis (Domoney and Casey 1990), the protein predicted from the gene sequence (GenBank CAA72090.1) is Mr 54 662. Removal of a signal peptide of 27 amino acids (http://www.expasy.org/tools/, accessed 12 March 2013) from this predicted protein suggests a mature protein of Mr 51 738, somewhat larger than the apparent Mr 45 000 in Fig. 3f (arrowed). However, amino acids 50–60 of the predicted mature protein constitute a repetitive QE motif that either may be cleaved (giving a ~Mr 45 000 for the rest of the polypeptide) or may contribute to anomalous behaviour on gels, as observed for some legumin proteins in particular (Heim et al. 1994). Sucrose-binding activity has not been demonstrated for either pea or V. faba SBP, the nomenclature deriving from studies of an orthologous gene in G. max; however, in V. faba, strong labelling of SBP mRNA was found over seed coat vascular strands and the embryo epidermal transfer cell layer that is reminiscent of sucrose transporter localisation (Heim et al. 2001).
A second example of processing is suggested by the Cvc mutants, where more than one polypeptide is missing (Fig. 3i, arrowed). Processing of Cvc polypeptides has not been reported previously and the predicted polypeptides (Mr ~70 000) are normally evident in total seed protein extracts (Casey and Domoney 1999). Further proteomic analysis will be necessary after backcrossing the mutant line to establish that the smaller polypeptide is derived from Cvc and not a second mutation affecting seed composition.
It is clear that the Cvc deletion mutation affects two closely linked genes (Ellis et al. 1986) and that these lines carry an additional morphological mutation, k. So far, our data indicate that the Cvc and k loci are very closely linked with no recombination among 80 recombinant inbred lines, in contrast to an earlier study which reported recombination between these (3.2 ± 1.1 crossover %, Matta and Gatehouse 1982). The latter study was based on F2 populations of over 100 individuals and protein mobility shifts in convicilin. Because heterozygotes were present in the populations used in this earlier work, the difficulty of scoring single polypeptides on gels was increased, and recombination may have been overestimated; alternatively the larger number of gametes examined may have facilitated the detection of recombinants. In a population of 80 recombinant inbred lines (RILs) a map distance of 3.2 cM corresponds to 4.7 ± 2.1 (mean ± s.e.) recombinants expected, so finding no recombinants is close to 2 s.d. units from the mean and therefore expected in ~5% of such tests. In the Cvc mutant described in this paper, the closely linked s (sticky seeds) and wb (waxless foliage) loci are unaffected (data not shown). These loci are positioned on the opposite side of Cvc to k, being ~18 map units from Cvc (Matta and Gatehouse 1982). The existence of the Cvc/k double deletion mutant suggests that a large genomic region has been deleted with no apparent detriment to the viability of the resulting plants. The tlx allele is a previously described large deletion (Gorel et al. 1994) that reduces the recombination fraction between flanking markers and is lethal in the homozygous state. The loss of several VicB amplicons in a FN mutant line (Fig. 1b) is further evidence for non- deleterious deletions affecting an entire locus comprising closely linked related genes (Ellis et al. 1986).
The sequence of M. truncatula may be exploited to indicate the likely deletion size for some of the pea mutants; the positions of cupin (vicilin) genes in the former seem to be physically clustered mainly at a locus on Medicago chromosome 7, which is genetically syntenic to pea linkage group V (i.e. pea Vc-2). In contrast several distinct loci have been described for vicilins and convicilins in pea (Ellis et al. 1986; Domoney and Casey 1990; Casey and Domoney 1999). For the Medicago cupin gene cluster, five genes have been annotated within 31 000 bp that includes three hypothetical protein encoding regions: such an arrangement is in agreement with the close genetic linkage within individual loci in pea.
The deletion mutants reported here will facilitate the further study of proteins that appear to bridge the gap between storage, metabolic and defence functions during seed development and storage. Both SBP and lectin are implicated in plant defence and the latter has been postulated to play a role also in the signalling pathways that operate in roots during nodulation (van Rhijn et al. 2001); no naturally occurring lectin mutants in pea have been available hitherto to test this hypothesis.
The application of next generation sequencing methodologies enhances the prospects for more fully exploiting this valuable mutant resource, and the data indicate that a large number of already identified transcripts may be directly assayed by the RAD sequencing method. The resource has already contributed to the identification of genes underlying several phenotypic traits (Table 2) in an important legume crop that also serves as a model to underpin fundamental plant genetics.
Acknowledgements
This work was supported by: BBSRC (BB/J004561/1) and the John Innes Foundation, Defra (IF0147, Pulse Crop Genetic Improvement Network), and BBSRC/Defra/Technology Strategy Board (TS/J002852/1). NE, MH and TB acknowledge support from the BEAA strategic partnership between Aberystwyth and Bangor Universities and the BBSRC.
References
Alonso JM, Stepanova AN, Solano R, Wisman E, Ferrari S, Ausubel FM, Ecker JR (2003) Five components of the ethylene-response pathway identified in a screen for weak ethylene-insensitive mutants in Arabidopsis. Proceedings of the National Academy of Sciences of the United States of America 100, 2992–2997.| Five components of the ethylene-response pathway identified in a screen for weak ethylene-insensitive mutants in Arabidopsis.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXitVaiu7k%3D&md5=eb5dabfccf9edbc1840a692ac1c5e4edCAS | 12606727PubMed |
Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, Selker EU, Cresko WA, Johnson EA (2008) Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 3, e3376
| Rapid SNP discovery and genetic mapping using sequenced RAD markers.Crossref | GoogleScholarGoogle Scholar | 18852878PubMed |
Belfield EJ, Gan X, Mithani A, Brown C, Jiang C, Franklin K, Alvey E, Wibowo A, Jung M, Bailey K, Kalwani S, Ragoussis J, Mott R, Harberd NP (2012) Genome-wide analysis of mutations in mutant lineages selected following fast-neutron irradiation mutagenesis of Arabidopsis thaliana. Genome Research 22, 1306–1315.
| Genome-wide analysis of mutations in mutant lineages selected following fast-neutron irradiation mutagenesis of Arabidopsis thaliana.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhtVylurvL&md5=4f4d9d298f9926cebbbc549965387679CAS | 22499668PubMed |
Bolon Y-T, Haun WJ, Xu WW, Grant D, Stacey MG, Nelson RT, Gerhardt DJ, Jeddeloh JA, Stacey G, Muehlbauer GJ, Orf JH, Naeve SL, Stupar RM, Vance CP (2011) Phenotypic and genomic analyses of a fast neutron mutant population resource in soybean. Plant Physiology 156, 240–253.
| Phenotypic and genomic analyses of a fast neutron mutant population resource in soybean.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmsVWgt7k%3D&md5=9919110847bc6c1c3b71d23800b41c27CAS | 21321255PubMed |
Casey R, Domoney C (1999) Pea globulins. In ‘Seed proteins’. (Eds PR Shewry, R Casey) pp. 171–208. (Kluwer Academic Publishers: Dordrecht, The Netherlands)
Chen J, Moreau C, Liu Y, Kawaguchic M, Hofer J, Ellis N, Chen R (2012) Conserved genetic determinant of motor organ identity in Medicago truncatula and related legumes. Proceedings of the National Academy of Sciences of the United States of America 109, 11 723–11 728.
| Conserved genetic determinant of motor organ identity in Medicago truncatula and related legumes.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xht1Ciu7rK&md5=ee5f5fd894015220398d35de4df8c985CAS |
Couzigou J-M, Zhukov V, Mondy S, el Heba GA, Cosson V, Ellis THN, Ambrose M, Wen J, Tadege M, Tikhonovich I, Mysore KS, Putterill J, Hofer J, Borisov AY, Ratet P (2012) NODULE ROOT and COCHLEATA maintain nodule development and are legume orthologs of Arabidopsis BLADE-ON-PETIOLE genes. The Plant Cell 24, 4498–4510.
| NODULE ROOT and COCHLEATA maintain nodule development and are legume orthologs of Arabidopsis BLADE-ON-PETIOLE genes.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXpsVKmuw%3D%3D&md5=0217473d702e724a86c40e73f917cf31CAS | 23136374PubMed |
Dalmais M, Schmidt J, Le Signor C, Moussy F, Burstin J, Savois V, Aubert G, Brunaud V, de Oliveira Y, Guichard C, Thompson R, Bendahmane A (2008) UTILLdb, a Pisum sativum in silico forward and reverse genetics tool. Genome Biology 9, R43
| UTILLdb, a Pisum sativum in silico forward and reverse genetics tool.Crossref | GoogleScholarGoogle Scholar | 18302733PubMed |
Domoney C, Casey R (1990) Another class of vicilin gene in Pisum. Planta 182, 39–42.
| Another class of vicilin gene in Pisum.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3cXltFSgurw%3D&md5=84a2b393c0176e20dd417a31b1197756CAS |
Ellis THN, Poyser SJ (2002) An integrated and comparative view of pea genetic and cytogenetic maps. New Phytologist 153, 17–25.
| An integrated and comparative view of pea genetic and cytogenetic maps.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38Xns1yktw%3D%3D&md5=aa723a87d83c1f2dc8cf5a908d594a90CAS |
Ellis THN, Domoney C, Castleton J, Cleary W, Davies DR (1986) Vicilin genes of Pisum. Molecular & General Genetics 205, 164–169.
| Vicilin genes of Pisum.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2sXhtl2qug%3D%3D&md5=b32e5af5c7b2289c50ffd6b3b5ec31deCAS |
Ellis THN, Turner L, Hellens RP, Lee D, Harker CL, Enard C, Domoney C, Davies DR (1992) Linkage maps in pea. Genetics 130, 649–663.
Gorel FL, Berdnikov VA, Temnykh SV (1994) A deletion covering the Tl locus in Pisum sativum. Pisum Genetics 26, 16–17.
Heim U, Bäumlein H, Wobus U (1994) The legumin gene family: a reconstructed Vicia faba legumin gene encoding a high-molecular-weight subunit is related to type B genes. Plant Molecular Biology 25, 131–135.
| The legumin gene family: a reconstructed Vicia faba legumin gene encoding a high-molecular-weight subunit is related to type B genes.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2cXlslWrs7k%3D&md5=a900633ccd34768c2111df3ef8cd74e9CAS | 8003694PubMed |
Heim U, Wang Q, Kurz T, Borisjuk L, Golombek S, Neubohn B, Adler K, Gahrtz M, Sauer N, Weber H, Wobus U (2001) Expression patterns and subcellular localization of a 52 kDa sucrose-binding protein homologue of Vicia faba (VfSBPL) suggest different functions during development. Plant Molecular Biology 47, 461–474.
| Expression patterns and subcellular localization of a 52 kDa sucrose-binding protein homologue of Vicia faba (VfSBPL) suggest different functions during development.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXns1Gmsrg%3D&md5=38b32f44945a5c0893a032d51e16d958CAS |
Hellens RP, Moreau C, Lin-Wang K, Schwinn KE, Thomson SJ, Fiers MWEJ, Frew TJ, Murray SR, Hofer JMI, Jacobs JME, Davies KM, Allan AC, Bendahmane A, Coyne CJ, Timmerman-Vaughan GM, Ellis THN (2010) Identification of Mendel’s white flower character. PLoS ONE 5, e13230
| Identification of Mendel’s white flower character.Crossref | GoogleScholarGoogle Scholar | 20949001PubMed |
Hofer J, Turner L, Moreau C, Ambrose M, Isaac P, Butcher S, Weller J, Dupin A, Dalmais M, Le Signor C, Bendahmane A, Ellis N (2009) Tendril-less regulates tendril formation in pea leaves. The Plant Cell 21, 420–428.
| Tendril-less regulates tendril formation in pea leaves.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXksVKrtr4%3D&md5=44d3c91923c7bf3fbef66e90afccc883CAS | 19208900PubMed |
Hoffmann D, Jiang Q, Men A, Kinkema M, Gresshoff PM (2007) Nodulation deficiency caused by fast neutron mutagenesis of the model legume Lotus japonicus. Journal of Plant Physiology 164, 460–469.
| Nodulation deficiency caused by fast neutron mutagenesis of the model legume Lotus japonicus.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXkvFSqt7s%3D&md5=ff4240e88244de90a369ae7351b71940CAS | 17363108PubMed |
Lu J, Knox MR, Ambrose MJ, Brown JKM, Ellis THN (1996) Comparative analysis of genetic diversity in pea assessed by RFLP- and PCR-based methods. Theoretical and Applied Genetics 93, 1103–1111.
| Comparative analysis of genetic diversity in pea assessed by RFLP- and PCR-based methods.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXpvFKmsQ%3D%3D&md5=39c4701c914b065fecd6542d23e47039CAS |
Matta NK, Gatehouse JA (1982) Inheritance and mapping of storage protein genes in Pisum sativum L. Heredity 48, 383–392.
| Inheritance and mapping of storage protein genes in Pisum sativum L.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL38XlsF2lur8%3D&md5=5dd5254612ef4d9940d4bfcff08d5b1fCAS |
Miller MR, Dunham JP, Amores A, Cresko WA, Johnson EA (2007) Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Research 17, 240–248.
| Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhsFKis7w%3D&md5=575a59f2fa513d841857561c549c9bb6CAS | 17189378PubMed |
Moreau C, Ambrose MJ, Turner L, Hill L, Ellis THN, Hofer JMI (2012) The b gene of pea encodes a defective flavonoid 3′,5′-hydroxylase, and confers pink flower color. Plant Physiology 159, 759–768.
| The b gene of pea encodes a defective flavonoid 3′,5′-hydroxylase, and confers pink flower color.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XovFamu7o%3D&md5=c94c343a3baca7f38270bdb8e90c891cCAS | 22492867PubMed |
Pellew C, Sverdrup A (1923) New observations on the genetics of peas. Journal of Genetics 13, 125–131.
| New observations on the genetics of peas.Crossref | GoogleScholarGoogle Scholar |
Rogers C, Wen J, Chen R, Oldroyd G (2009) Deletion-based reverse genetics in Medicago truncatula. Plant Physiology 151, 1077–1086.
| Deletion-based reverse genetics in Medicago truncatula.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVCjsbrK&md5=4f09eafe7f3078aec744aec69e968ef3CAS | 19759346PubMed |
Sainsbury F, Tattersall AD, Ambrose MJ, Turner L, Ellis THN, Hofer JMI (2006) A crispa null mutant facilitates identification of a crispa-like pseudogene in pea. Functional Plant Biology 33, 757–763.
| A crispa null mutant facilitates identification of a crispa-like pseudogene in pea.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XnsFCrsbw%3D&md5=f2c82d7acfc4846781567358c9bdebd7CAS |
Searle IR, Men AE, Laniya TS, Buzas DM, Iturbe-Ormaetxe I, Carroll BJ, Gresshoff PM (2003) Long-distance signaling in nodulation directed by a CLAVATA1-like receptor kinase. Science 299, 109–112.
| Long-distance signaling in nodulation directed by a CLAVATA1-like receptor kinase.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XpvVensLo%3D&md5=e3a125e1e9d2a134253299a94c21115aCAS | 12411574PubMed |
Sidorova KK (1975) Induced pea mutant with creeping stem. Pisum Newsletter 7, 57–58.
van Rhijn P, Fujishige NA, Lim PO, Hirsch AM (2001) Sugar-binding activity of pea lectin enhances heterologous infection of transgenic alfalfa plants by Rhizobium leguminosarum biovar viciae. Plant Physiology 126, 133–144.
| Sugar-binding activity of pea lectin enhances heterologous infection of transgenic alfalfa plants by Rhizobium leguminosarum biovar viciae.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXjslWjsLs%3D&md5=b91cfbf632f547b0d9d067cdee466b13CAS | 11351077PubMed |
Wang Z, Luo Y, Li X, Wang L, Xu S, Yang J, Weng L, Sato S, Tabata S, Ambrose M, Rameau C, Feng X, Hu X, Luo D (2008) Genetic control of floral zygomorphy in pea (Pisum sativum L.) Proceedings of the National Academy of Sciences of the United States of America 105, 10 414–10 419.
| Genetic control of floral zygomorphy in pea (Pisum sativum L.)Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXpsFKktbw%3D&md5=4bddb8c3e9ad0c2b31864e5e35f6a78dCAS |
Zhang L, Fetch T, Nirmala J, Schmierer D, Brueggeman R, Steffenson B, Kleinhofs A (2006) Rpr1, a gene required for Rpg1-dependent resistance to stem rust in barley. Theoretical and Applied Genetics 113, 847–855.
| Rpr1, a gene required for Rpg1-dependent resistance to stem rust in barley.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XotlChtLc%3D&md5=3aab5fc7be5adb94be19536b2e3d1cc6CAS | 16832646PubMed |
Zhuang L, Ambrose M, Rameau C, Weng L, Yang J, Hu X, Luo D, Li X (2012) LATHYROIDES, encoding a WUSCHEL-related homeobox1 transcriptional factor, controls organ lateral growth and regulates tendril and dorsal petal identities in garden pea (Pisum sativum L.). Molecular Plant 5, 1333–1345.
| LATHYROIDES, encoding a WUSCHEL-related homeobox1 transcriptional factor, controls organ lateral growth and regulates tendril and dorsal petal identities in garden pea (Pisum sativum L.).Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhslKntrrP&md5=70cc13dab63d0ee7ab7d2c951f3cb3dbCAS | 22888154PubMed |