

Gene families and evolution of trehalose metabolism in plants
John E. LunnA Max Planck Institute of Molecular Plant Physiology, Am Mühlenberg 1, 14424 Potsdam, Germany. Email: lunn@mpimp-golm.mpg.de
B This paper originates from a presentation at the 8th International Congress of Plant Molecular Biology, Adelaide, Australia, August 2006.
Functional Plant Biology 34(6) 550-563 https://doi.org/10.1071/FP06315
Submitted: 29 November 2006 Accepted: 11 January 2007 Published: 1 June 2007
Abstract
The genomes of Arabidopsis thaliana L., rice (Oryza sativa L.) and poplar (Populus trichocarpa Torr. & A.Gray) contain large families of genes encoding trehalose-phosphate synthase (TPS) and trehalose-phosphatase (TPP). The class I subfamily of TPS genes encodes catalytically active TPS enzymes, and is represented by only one or two genes in most species. A. thaliana is atypical in having four class I TPS genes, three of which (AtTPS2–4) encode unusual short isoforms of TPS that appear to be found only in members of the Brassicaceae family. The class II TPS genes encode TPS-like proteins with a C-terminal TPP-like domain, but there is no experimental evidence that they have any enzymatic activity and their function is unknown. Both classes of TPS gene are represented in the genomes of chlorophyte algae (Ostreococcus species) and non-flowering plants [Physcomitrella patens (Hedw.) Bruch & Schimp.(B.S.G.) and Selaginella moellendorffii (Hieron. in Engl. & Prantl.)]. This survey shows that the gene families encoding the enzymes of trehalose metabolism are very ancient, pre-dating the divergence of the streptophyte and chlorophyte lineages. It also provides a frame of reference for future studies to elucidate the function of trehalose metabolism in plants.
Additional keywords: trehalose-phosphate synthase, trehalose-phosphatase, trehalase.
Introduction
α,α-Trehalose (α-d-glucopyranosyl-1,1-α-d-glucopyranoside) is a non-reducing disaccharide that is commonly found in bacteria, fungi and some invertebrates (Elbein 1974). Within the past 10 years it has become clear that many, perhaps all, green plants can also synthesise trehalose, but the function of trehalose metabolism in plants is still something of a mystery. Trehalose has been found in several groups of non-vascular plants, including green algae and liverworts, as well as in lower vascular plants, such as spike-mosses (Selaginella species) and ferns (Anselmino and Gilg 1913; Elbein 1974; Kandler and Hopf 1980). Selaginella lepidophylla (Hook. and Grev.) Spring is a resurrection plant that can recover from almost complete desiccation, and it accumulates high levels of trehalose during dehydration. Trehalose is able to stabilise proteins and membranes in vitro by displacing water molecules and forming a glass-like structure. On the basis of these properties, it has been proposed that the accumulation of trehalose in S. lepidophylla, and some other resurrection plants, helps to protect intracellular structures from damage during anhydrobiosis. Yeast (Saccharomyces cerevisiae) is another desiccation-tolerant organism that accumulates large amounts of trehalose. However, a study of yeast mutants that cannot synthesise trehalose found that the cells were still able to survive desiccation, whereas induction of trehalose accumulation by osmotic stress did not improve desiccation tolerance (Ratnakumar and Tunnacliffe 2006). These results, and some previous observations on rotifers (Tunnacliffe and Lapinski 2003), suggest that trehalose accumulation is neither necessary nor sufficient on its own for desiccation tolerance in these organisms, and call into question its role in trehalose-accumulating resurrection plants. As an alternative function, trehalose accumulation has been shown to protect yeast cells from damage by reactive oxygen species (Benaroudj et al. 2001).
With the exception of a few resurrection plants, e.g. Myrothamnus flabellifolius Welw. and Sporobolus atrovirens Kunth (Drennan et al. 1993; Iturriaga et al. 2000), most angiosperms do not accumulate trehalose, and the trace amounts that have been found in some species were sometimes attributable to bacterial or fungal contamination (Kandler and Hopf 1980). In early studies on several desiccation-intolerant flowering plants – tobacco (Nicotiana tabacum L.), potato (Solanum tuberosum L.) and soybean [Glycine max (L.) Merr.] – trehalose levels in untreated plants were below the limits of detection of the assays used (Müller et al. 1995; Goddijn et al. 1997). Therefore, the discovery of genes encoding trehalose-phosphate synthase (TPS; EC 2.4.1.15) and trehalose-phosphatase (TPP; EC 3.1.3.12) in A. thaliana L. – another desiccation-intolerant species – was rather unexpected (Blázquez et al. 1998; Vogel et al. 1998). The complete sequencing of the A. thaliana genome (The Arabidopsis Initiative 2000) subsequently revealed a family of 11 genes (AtTPS1–11) encoding TPS or TPS-like proteins, and a family of 10 genes (AtTPPA–J) encoding TPP, whereas trehalase (EC 3.2.1.28) appears to be encoded by a single gene (AtTRE) (Leyman et al. 2001).
Phylogenetic analysis of the A. thaliana TPS genes showed that they cluster into two distinct subfamilies, designated class I (AtTPS1–4) and class II (AtTPS5–11) (Leyman et al. 2001). All of the proteins encoded by these genes contain a glucosyltransferase-like domain similar to the TPS enzymes from yeast (ScTPS1) and Escherichia coli (otsA) but only the class I isoforms from plants appear to have TPS activity. Expression of the AtTPS1 or S. lepidophylla TPS1 genes in the yeast tps1Δ (TPS–) mutant showed not only that they can restore trehalose synthesis and growth of the mutant on glucose, but also that they encode active TPS enzymes (Blázquez et al. 1998; Zentella et al. 1999; van Dijck et al. 2002). In contrast, AtTPS7 or AtTPS8 were unable to complement the yeast tps1Δ mutant (Vogel et al. 2001), and AtTPS5 shows no TPS activity (Harthill et al. 2006). The TPS1 proteins from both A. thaliana and S. lepidophylla contain an N-terminal extension compared to the yeast enzyme, and this extension appears to be specific to plant TPSs. The N-terminal extension of the S. lepidophylla TPS1 did not act as an intracellular targeting signal when expressed as a fusion protein with green fluorescent protein in tobacco cells, indicating that the S. lepidophylla TPS1 is likely to be cytosolic (van Dijck et al. 2002). Removal of the extension from the S. lepidophylla TPS1 and A. thaliana TPS1 dramatically increased the catalytic activity of the enzymes when expressed in yeast (van Dijck et al. 2002), suggesting that it acts as an autoinhibitory domain. The class I TPS2–4 isoforms from A. thaliana do not appear to possess this domain.
In addition to the TPS-like domain, the A. thaliana TPS class II isoforms contain a C-terminal region that resembles the phosphatase domain of the yeast TPP (encoded by the ScTPS2 gene), which also contains a non-catalytic, TPS-like domain at the N-terminus (Bell et al. 1998; Leyman et al. 2001). The phosphatase domain of all these proteins includes three amino acid motifs that are characteristic of the l-2-haloacid dehalogenase (HAD) superfamily of enzymes: motif I – DX(D/T/Y)X(T/V)(L/V/I), motif II – a Ser or Thr, generally in a hydrophobic context, and motif III – KX18–30(G/S)(D/S)X3(D/N). Motif I is the most highly conserved, and the invariant first Asp is the functional nucleophile that forms a phospho-acyl intermediate during catalysis (Fieulaine et al. 2005). The HAD superfamily embraces a wide range of phosphatases and hydrolases, including the analogous enzyme of sucrose synthesis, sucrose-phosphatase (Lunn et al. 2000), and crystal structures show that the three conserved motifs form the active site of HAD superfamily enzymes (Fieulaine et al. 2005; Burroughs et al. 2006). The presence of these motifs in the class II isoforms of TPS prompted speculation that they might have TPP activity (Leyman et al. 2001), but no such activity has been found. Neither AtTPS7 nor AtTPS8 was able to restore growth at 38.6°C, or trehalose synthesis after heat shock, when expressed in the yeast tps2Δ (TPP–) mutant (Vogel et al. 2001) and AtTPS5 showed no TPP activity (Harthill et al. 2006). Thus, the function of the class II isoforms of TPS in A. thaliana remains unresolved, and is one of the main puzzles in the enigma of trehalose metabolism in plants.
The proteins encoded by the 10 A. thaliana TPP genes also contain the three HAD motifs, and two members of the gene family (AtTPPA and AtTPPB) encode active TPP enzymes that complement the yeast tps2Δ mutant (Vogel et al. 1998), as do two homologues from rice and maize (Pramanik and Imai 2005; Satoh-Nagasawa et al. 2006). The maize TPP (RA3) is of particular interest, because a lesion in the gene encoding this enzyme in the ramosa3 (ra3) mutant leads to increased inflorescence branching (Satoh-Nagasawa et al. 2006). It was proposed that the RA3 gene product could either interfere with a sugar signal that modifies the developmental fate of axillary shoot meristems, or that it acts directly as a transcriptional regulator (Satoh-Nagasawa et al. 2006).
This finding adds to the growing evidence that trehalose metabolism has an important function in plants, even in species that do not accumulate large amounts of this sugar. Loss of TPS1 gene function in A. thaliana is embryo lethal, and leads to growth arrest at the torpedo stage of embryo development (Eastmond et al. 2002). The tps1 mutant can be rescued through embryogenesis by inducible expression of TPS, but the resulting plants show retarded vegetative growth and are unable to flower when TPS expression is no longer induced (van Dijken et al. 2004). Expression of yeast or bacterial TPS or TPP genes in plants also gives rise to striking morphological and biochemical phenotypes (Goddijn et al. 1997; Romero et al. 1997; Pilon-Smits et al. 1998; Schluepmann et al. 2003). The contrasting phenotypic effects of TPS v. TPP or phosphotrehalase overexpression in A. thaliana suggested that these were caused by changes in the level of trehalose-6-phosphate (Tre6P) – the intermediate of trehalose synthesis – rather than trehalose itself (Schluepmann et al. 2003). The level of Tre6P is correlated with the amount of sucrose in A. thaliana rosette leaves, and addition of 15 mm sucrose to sugar-starved seedlings induced a 26-fold increase in Tre6P (Lunn et al. 2006), consistent with a role for Tre6P in sugar signalling. Increases in Tre6P were also correlated with redox activation of ADPglucose pyrophosphorylase (Lunn et al. 2006), supporting the hypothesis that one of the functions of Tre6P is to mediate sugar-dependent regulation of starch synthesis (Kolbe et al. 2005). However, this is unlikely to be the only function of Tre6P in plants, because embryos of the A. thaliana tps1 mutant show relatively normal synthesis of starch before their growth is arrested, despite the lack of Tre6P (Gómez et al. 2006). Also, the complex morphological phenotypes of TPS and TPP overexpressing plants suggest that changes in Tre6P levels have a far-reaching influence on growth and development, which would be difficult to explain by effects on starch metabolism alone.
Although much current research on trehalose metabolism in plants is focussed on discovering the role of Tre6P, we cannot dismiss the possibility that trehalose itself also has a function in plants. Inhibition of trehalase activity in vivo by treatment with validamycin A led to accumulation of trehalose, and reduced levels of sucrose and starch in flowers, leaves and stems of A. thaliana (Müller et al. 2001), and in root nodules of soybean (G. max) and cowpea (Vigna unguiculata (L.) Walp.) (Müller et al. 1995). Exogenous application of trehalose to plants can induce both abiotic and biotic stress responses, including transcription of genes involved in protection against oxidative stress and pathogen defence (Reignault et al. 2001; Bae et al. 2005a, 2005b). Exogenous trehalose derived from pathogenic and symbiotic microbes also appears to modify the metabolism of their host plants (Müller et al. 1998; Brodmann et al. 2002).
To understand the role of trehalose metabolism in plants we need to answer two questions: (1) what factors influence the synthesis and breakdown of Tre6P and trehalose in plants? and (2) what are the downstream effects of changes in Tre6P and trehalose? The large diversity of TPS and TPP isoforms in A. thaliana complicates efforts to understand how trehalose synthesis is controlled in this species. In particular, we do not know the significance of the unusual class I isoforms of TPS (TPS2–4), or the function(s) of the class II isoforms (TPS5–11) and the need for so many isoforms of TPP. There is also no clear picture of how far this diversity is present in plants other than A. thaliana. The recent completion of genome sequencing for rice (International Rice Genome Sequencing Project 2005) and poplar (Tuskan et al. 2006), and the release of genomic sequences from other species, offer a new opportunity to address some of these questions. This paper reports a survey of genome sequences from angiosperms and non-flowering plants to trace the origins and evolution of trehalose metabolism in the plant kingdom, with the aim of helping us to understand its function in plants.
Materials and methods
Materials
The tomato (Solanum lycopersicum L.) TPS1 cDNA clone (cTOD3K4) was a gift from Dr Alisdair Fernie (Max Planck Institute of Molecular Plant Physiology, Potsdam, Germany). The Physcomitrella patens (Hedw.) Bruch & Schimp.(B.S.G.) cDNA clones (PpTPS1, pphn27108; PpTPS5, pphb36122; PpTPP1, pphb35p21 and PpTRE1, pphn36m16) (Nishiyama et al. 2003) were obtained from the RIKEN BioResource Center, Japan (http://www.brc.riken.jp/inf/en/, accessed 18 February 2007).
DNA sequencing
Sequencing was carried out on both strands by the dideoxy chain termination method by Big Dye chain terminator chemistry (Applied Biosystems, Foster City, CA, USA). The sequences of cDNA clones reported in this paper have been deposited in the NCBI GenBank database with the following accession numbers: EF151131 – S. lycopersicum TPS1, EF151132 – P. patens TPS1, EF151133 – P. patens TPS5, EF151134 – P. patens TPP1 and EF151135 – P. patens TRE1.
Genomic sequence assembly and gene annotation
The rice (Oryza sativa L. subsp. japonica) and poplar (black cottonwood, Populus trichocarpa Torr. & A.Gray) genome sequences were searched for genes with similarity to the TPS, TPP and TRE genes of A. thaliana with the BLASTN and TBLASTN algorithms (Altschul et al. 1990). Each matching sequence was manually curated by checking for consistency with available cDNA and EST sequences, and by comparison of the translated sequence with related protein sequences from A. thaliana and other species. The gene identifiers of sequences that agree with the current annotations of the rice (RAP1; Ohyanagi et al. 2006) and poplar (Release 1.1; http://www.jgi.doe.gov/, accessed 18 February 2007) genomes are shown in Table S1 (see supplementary material available on the Functional Plant Biology website). Missing or improved gene models are also provided in the supplementary material. Putative TPS, TPP and TRE genes were identified in the partially assembled and annotated sequences of two prasinophyte green algae, Ostreococcus tauri and Ostreococcus lucimarinus (Derelle et al. 2006; http://www.jgi.doe.gov/), by BLAST and TBLASTN searches.
Whole genome shotgun sequences from P. patens, Selaginella moellendorffii and Sorghum bicolor L. Moench from the DOE Joint Genome Institute (http://www.jgi.doe.gov/) were extracted from the NCBI Trace archive (http://www.ncbi.nlm.nih.gov/, accessed 18 February 2007) with the Mega BLAST and discontiguous Mega BLAST search algorithms (Zhang et al. 2000). Maize genomic sequences (Chandler and Brendel 2002) were obtained from the NCBI High Throughput Genomic Sequence database. Contiguous genomic sequences (>97% identity) were assembled with GeneDoc (Nicholas and Nicholas 1997), with corresponding cDNA (EST) sequences used to bridge any gaps in the genomic sequence where possible. Protein coding sequences were deduced from the corresponding cDNA or EST sequences and by comparison of the translated sequence with known TPS, TPP or trehalase protein sequences. All protein sequences that are not available from public databases are provided in the supplementary material.
Phylogenetic analysis
Protein sequences were aligned with the PILEUP program from the GCG Wisconsin Package (Version 10.3; Accelrys Inc., San Diego, CA, USA). Minor corrections and other editing of the alignments were done with GeneDoc (Nicholas and Nicholas 1997). Phylogenetic analyses were carried out on full length sequences (with and without gaps), and regions corresponding to individual domains (without gaps), with the heuristic tree searching method of PAUP* (version 4.0.0d55 for Unix; Sinauer Associates, Inc., Sunderland, MA, USA), with optimal trees selected using minimum distance or maximum parsimony criteria. Maximum likelihood trees were constructed with PHYML with 100 bootstraps, implementing the JTT or DCMut evolutionary models of amino acid substitution (Guindon et al. 2005). Trees were displayed with TreeView (Page 1996).
Results and discussion
Trehalose-phosphate synthase gene families
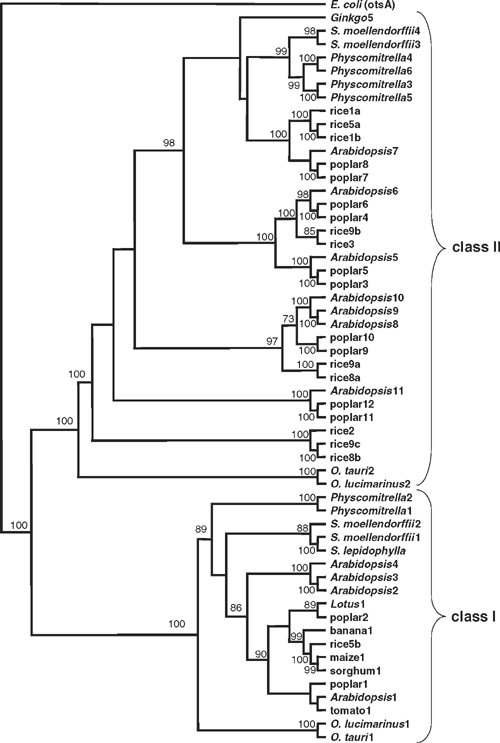
The rice and poplar genomes contain 11 and 12 TPS genes, respectively. In agreement with Leyman et al. (2001), phylogenetic analyses consistently split the plant TPS protein sequences into two distinct classes (Fig. 1), irrespective of the protein region analysed or the method used to select the optimal tree – maximum parsimony, minimum distance or maximum likelihood. In streptophyte plants (Streptophyta), this fundamental dichotomy is supported by clear differences in gene structure, with the class I genes containing at least 16 exons within the coding region, whereas the class II genes are much simpler with only 3–4 exons. Among the chlorophyte algae (Chlorophyta), two Ostreococcus species (Prasinophyceae) have single class I and class II TPS genes. The genomes from these prasinophyte algae have relatively little non-coding sequence compared with other eukaryotic genomes (Derelle et al. 2006), and both of the TPS genes lack introns. However, this is not a general feature of green algal TPS genes, because the incomplete class I and class II TPS gene sequences available from Chlamydomonas reinhardtii (Chlorophyceae) both have multiple introns.
|
In contrast to A. thaliana, which has four class I genes (AtTPS1–4), this class is represented by a single gene in rice (OsTPS5b), and only two in poplar (PtTPS1 and PtTPS2). Among other members of the Poaceae (grasses), sorghum also appears to have just a single class I gene (SbTPS1), whereas maize has two, possibly three. Only one full-length gene (ZmTPS1) could be assembled from the available genomic sequences from maize, but two non-overlapping fragments (ZmTPS2 and ZmTPS3) were also assembled. These fragments have >93% nucleotide sequence identity (>96% amino acid identity) with the 5′- and 3′-ends of the ZmTPS1 gene, respectively. Most of the class I TPS ESTs from maize are derived from the ZmTPS1 gene, with the ZmTPS2 and ZmTPS3 gene fragments accounting for the remaining 5′- and 3′-end ESTs. Thus, it seems likely that the ZmTPS2 and ZmTPS3 fragments come from opposite ends of a single gene, making a total of two class I TPS genes in maize. Single class I TPS genes were also identified from the unfinished genome sequences of Lotus japonicus (Regel) K.Larsen and banana (Musa acuminata Colla). A full-length TPS cDNA clone from tomato (S. lycopersicum) was sequenced, and this sequence shows 97–100% identity (nucleotide) with all of the available TPS class I ESTs from this species. From this analysis it appears that flowering plants from a wide range of taxonomic groups have only a single class I TPS gene, or two closely related genes in the ancient tetraploid species – poplar and maize – and that A. thaliana is somewhat exceptional in having four class I TPS genes.
All of the class I protein sequences from the other species have a long (85–128 amino acids) N-terminal extension, and they are more closely related to AtTPS1 than to the shorter class I isoforms (AtTPS2–4) from A. thaliana (Fig. 1). There is considerable sequence variation between species and isoforms in this N-terminal region. However, a short (12 amino acids) Arg/Leu-rich region close to the N-terminus, which includes the Arg17 and Leu27 residues that are implicated in autoinhibition of activity in AtTPS1 (van Dijck et al. 2002), appears to be conserved in the banana TPS1 and all of the eudicot sequences except poplar TPS1, which has Gln26 in place of the conserved Leu residue. It is difficult to align the TPS class I sequences from the Poaceae – rice, maize and sorghum – convincingly with the other sequences in the region of the N-terminal extension, but the former do contain a 10-aa Arg/Leu-rich region, which is perfectly conserved within the Poaceae, near to the N-terminal end of the glucosyltransferase domain. From these comparisons, it seems likely that the N-terminal extensions of the eudicot and banana class I TPSs, and possibly those from the Poaceae, have an autoinhibitory function, and the absence of this extension from the AtTPS2–4 isoforms in A. thaliana appears to be an unusual feature among the TPS class I sequences from flowering plants.
The short class I isoforms of TPS may be unique to the Brassicaceae
To investigate the origins of the short isoforms of TPS in A. thaliana, the search for class I TPS genes was extended to non-flowering plants and algae. The genomes of the prasinophyte algae, O. lucimarinus and O. tauri, contain single class I TPS genes (OlTPS1and OtTPS1) that encode proteins with long N-terminal extensions (136 and 141 amino acids, respectively). The moss P. patens has two class I isoforms (PpTPS1 and PpTPS2), which also have N-terminal extensions, although these are only 46 amino acids long. The very low similarity of the N-terminal extensions from these lower plants with the angiosperm sequences makes it difficult to identify any conserved residues in this region with certainty. In contrast, the TPS1 from the spike-moss S. lepidophylla is known to have an N-terminal extension that inhibits catalytic activity, and the two closely related sequences from S. moellendorffii (SmTPS1 and SmTPS2) also have N-terminal extensions containing the putative autoinhibitory domain. These observations show that the plant-specific, N-terminal extension of class I TPSs appeared very early in the evolution of plants, but it may only have acquired its autoinhibitory function after the divergence of vascular (Tracheophyta) and non-vascular plants. It can also be inferred that the lack of an N-terminal extension in the AtTPS2–4 isoforms is a derived feature resulting from loss of this region.
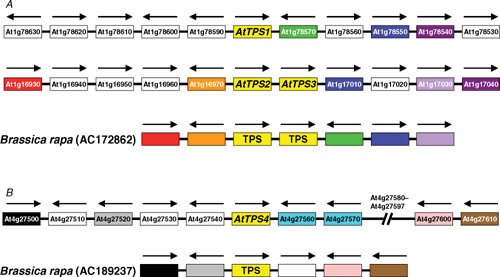
The AtTPS2 (At1g16980) and AtTPS3 (At1g17000) genes are adjacent to each other on chromosome 1 of A. thaliana (Fig. 2) (n.b. there is no At1g16990 gene), and at least two nearby genes (At1g17010 and At1g17040) downstream of these have homologues downstream of the AtTPS1 gene (At1g78580) (Fig. 2). This co-linearity suggests that the AtTPS2 and AtTPS3 genes could have arisen from a segmental duplication of the AtTPS1 gene region or a whole genome duplication, followed by a tandem duplication giving rise to the AtTPS2–AtTPS3 pair. Close inspection of the AtTPS3 gene suggests that it is unlikely to encode a functional TPS enzyme because two regions corresponding to exons 2 and 13 of the AtTPS2 coding sequence appear to be corrupted. There is no evidence from available ESTs that AtTPS3 is expressed, whereas the AtTPS2 gene is represented by an EST (BE523335) from developing seeds, and microarray data from the AtGeneExpress study (Schmid et al. 2005) confirm that this gene is expressed in developing seeds/siliques. The region of chromosome 4 in the immediate vicinity of the AtTPS4 gene does not show any obvious colinearity with the AtTPS1 or AtTPS2/3 regions on chromosome 1 (Fig. 2), suggesting that the AtTPS4 gene might have arisen from a more ancient duplication, or a transposition event that only conserved microsynteny within the TPS gene itself. The AtTPS4 gene is represented by a single EST (EG462462) from a mixed tissue sample, and the AtGeneExpress data indicate that expression is essentially restricted to developing seeds/siliques, as with AtTPS2 (Schmid et al. 2005).
|
The only close homologues of the AtTPS2–4 genes in the NCBI EST and genomic sequence databases were found in genomic sequences from Chinese cabbage [Brassica rapa L. subsp. pekinensis (Lour.) Kitam.] and Boechera stricta (Graham) Al-Shehbaz (GenBank accession number DU693992), both of which, like A. thaliana, belong to the Brassicaceae family. One of the genomic sequences from Chinese cabbage (AC172862) also includes a tandem pair of TPS genes, and shows substantial colinearity with the chromosomal region containing the AtTPS2 and AtTPS3 genes (Fig. 2). Interestingly, the Chinese cabbage sequence includes a homologue of the RHM1 gene (At1g78590) that lies just upstream of the AtTPS1 gene, but is absent from the AtTPS2–AtTPS3 gene region, and was presumably lost after the duplication event that gave rise to this region. The second genomic sequence from Chinese cabbage (AC189237) shows co-linearity with the AtTPS4 region on chromosome 4 (Fig. 2). No cDNA or EST sequences are available to determine the exact 5′-end of the coding regions of the putative TPS genes in Chinese cabbage, but analysis of potential open reading frames suggested that the genes encode short isoforms of TPS without N-terminal extensions, similar to AtTPS2–4. Two AtTPS1-like gene fragments (AJ856246 and DX046489) were also identified from Chinese cabbage, indicating the presence of at least one AtTPS1-like isoform in this species, in addition to the three shorter isoforms.
On current evidence it appears that the duplication or transposition events that gave rise to the AtTPS2–3 and AtTPS4 genes may have occurred after the divergence of the Brassicaceae lineage from other flowering plants, but predated the divergence of the Arabidopsis and Brassica lineages, which is estimated to have occurred ~20 million years ago (Rana et al. 2004). However, this tentative conclusion may need to be revised as more sequence data become available from other families within the order Brassicales, which are at present less well represented in the NCBI GenBank sequence database than the Brassicaceae. Of particular interest will be the genome sequence of papaya (Carica papaya) (order Brassicales, family Caricaceae), which is nearing completion (http://cgpbr.hawaii.edu/papaya, accessed 18 February 2007).
The functions of the short TPS isoforms in A. thaliana and other members of the Brassicaceae are unknown. Although transcripts from both AtTPS2 and AtTPS4 are present in developing seeds/siliques (Schmid et al. 2005), loss of AtTPS1 activity in the tps1 mutant causes arrest of embryo development at the torpedo stage (Eastmond et al. 2002), showing that AtTPS2 and AtTPS4 cannot compensate for the loss of AtTPS1 gene function. In contrast, expression of the E. coli otsA gene in heterozygous TPS1/tps1 plants could rescue tps1/tps1 embryos (van Dijken et al. 2004), showing that restoration of Tre6P and/or trehalose synthesis is sufficient to complement the mutation. Further work is needed to establish if the AtTPS2 and AtTPS4 genes are expressed in the same cells of developing seeds/siliques as AtTPS1, and if so, whether the transcripts are translated and the resulting proteins have TPS activity. Phenotypic analysis of knockout mutants, particularly tps2/tps4 double knockouts, could also help to reveal the functions of these unusual isoforms of TPS.
Trehalose-phosphate synthase gene families – class II
The rice and poplar genomes both contain 10 class II TPS genes, slightly more than in A. thaliana, which has seven (AtTPS5–11). All of these were clearly separated from the class I sequences in every phylogenetic tree (Fig. 1), but no consistent tree topology was found within the class II subfamily, reflected in the low bootstrap values for several of the branch points within this subfamily (Fig. 1). Separate analyses of the class II sequences alone also resulted in multiple trees with different topologies, depending on which region of the protein sequences was used for the analysis, and the method used for selecting the optimal tree – parsimony, distance, maximum likelihood or Bayesian inference (data not shown). Nevertheless, the following clusters were observed in nearly all trees: (1) AtTPS5/poplar3/poplar5, (2) AtTPS6/poplar4/poplar6/rice3/rice9b, (3) AtTPS7/poplar7/poplar8/rice1a/rice1b/rice5a, (4) AtTPS8–10/poplar9/poplar10/rice8a/ rice9a, (5) AtTPS11/poplar11/poplar12 and (6) rice2/rice8b/rice9c (Fig. 1, and data not shown). In two out of five equally parsimonious trees, constructed with the C-terminal phosphatase domain, the sequences within the AtTPS8–10 group (cluster 4) were split along species lines. However, this grouping was supported in the other three parsimony trees, and in the distance and maximum likelihood trees from the same region, as well as in all trees constructed using the full-length sequences or the N-terminal glucosyltransferase-like region. The AtTPS5 (1) and AtTPS6 (2) groups were always joined together, and this joint AtTPS5/AtTPS6 group was also clustered with the AtTPS7 (3) group in all trees except those constructed with the C-terminal phosphatase domain only. The AtTPS11/poplar11/poplar12 (5) and rice2/rice8b/rice9c (6) groups were clustered together only in trees constructed with the phosphatase domain (data not shown).
The strong support for groups 1–5 in the phylogenetic analyses suggests that the genes within each group are orthologous. Each group includes a pair of closely related genes from P. trichocarpa, which may represent homeologues from the two ancestral genomes of this ancient tetraploid species. The presence of at least one rice gene as sister to the eudicot sequences in groups 2, 3 and 4 suggests that their common ancestor diverged from the other TPS genes before the monocot-eudicot split, which is estimated to have occurred ~200 million years ago (Mitchell-Olds and Clauss 2002). Group 1 appears to be eudicot-specific, resulting from a duplication within group 2 after the divergence of the monocots from the eudicots.
The prasinophyte algae are thought to represent one of the most basal lineages among the green plants (Derelle et al. 2006), so the presence of class I and class II TPS genes in the genomes of O. tauri and O. lucimarinus (Fig. 1) indicates that they were already present in the streptophyte lineage, which includes land plants, before it diverged from the chlorophyte algal lineage. BLAST searches of the NCBI GenBank database indicate that the plant TPS sequences are most closely related to those from fungi and other eukaryotes, e.g. red algae (Rhodophyta) and Dictyostelium sp. (Mycetozoa) (data not shown). This suggests a eukaryotic rather than prokaryotic origin for the plant TPS gene families. Further analyses including more green algae and other groups of photosynthetic eukaryotes (e.g. Rhodophyta – red algae) might reveal whether the class I and class II genes arose by divergence of a common ancestor within the plant lineage, or were acquired independently.
Within the streptophyte plant lineage, the moss P. patens has four closely related class II TPS genes, which account for all of the class II ESTs from this species (Fig. 1). Only two class II genes were found in the genomic sequences available from S. moellendorffii, and both of these cluster with those from P. patens. A single class II TPS sequence from the gymnosperm Ginkgo biloba L. usually clustered with this group as well. The relationship between these genes and those from flowering plants is unclear, as there was disagreement between the distance-based trees on the one hand, and the parsimony and maximum likelihood trees on the other. The former tended to place the sequences from non-flowering plants as a sister group to the AtTPS5/AtTPS6 cluster (data not shown), whereas the latter placed them with the AtTPS7 cluster (Fig. 1).
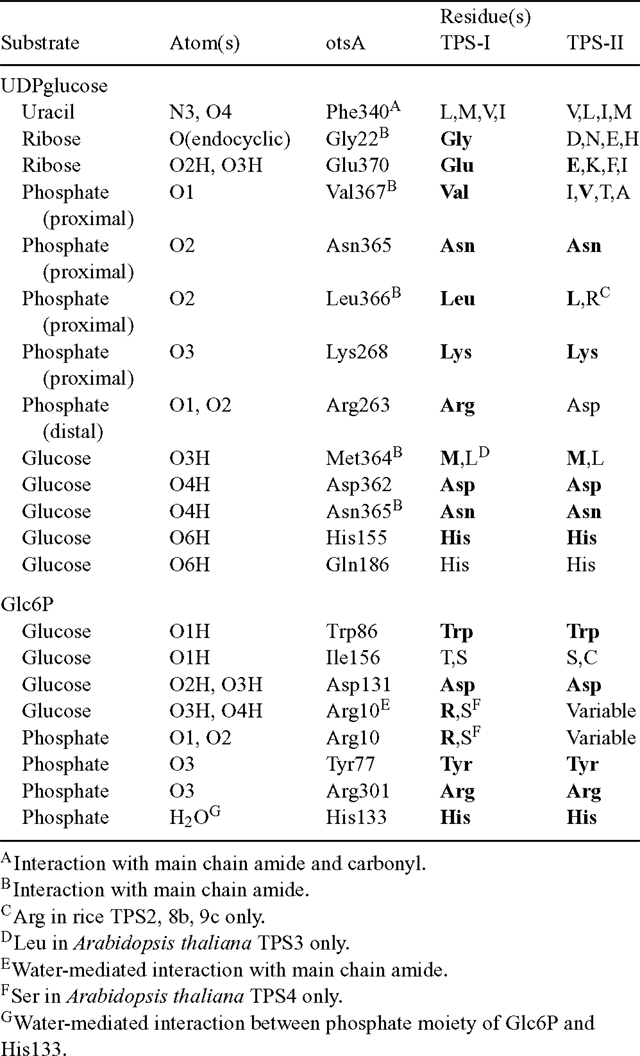
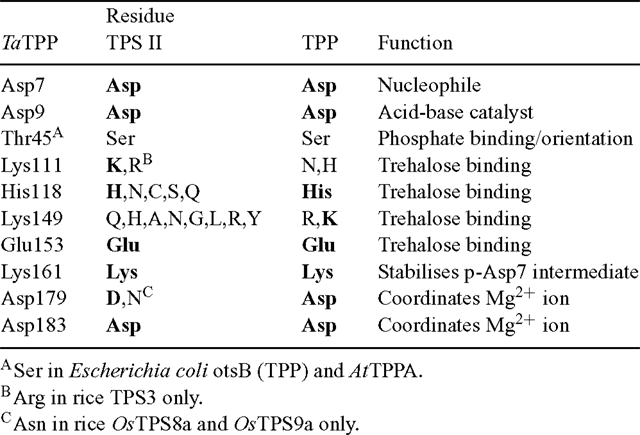
As noted previously, the function of the class II isoforms of TPS in plants is still unresolved. Comparison with crystal structures of the E. coli otsA (Gibson et al. 2002, 2004) shows that several of the TPS active site residues are less well conserved in the TPS class II proteins than in the class I proteins, which are known to have TPS activity (Table 1). In particular, residues involved in binding the ribosyl and distal phosphate moieties of UDPglucose differ in the class II proteins, as well as one of the residues involved in Glc6P-binding. The imperfect conservation of active site residues could explain the inability of these proteins to complement the yeast tps1Δ (TPS–) mutant, and their lack of detectable TPS activity (Vogel et al. 2001; Harthill et al. 2006). In contrast, their lack of TPP activity and inability to complement the yeast tps2Δ (TPP–) mutant are more surprising (Vogel et al. 2001; Harthill et al. 2006). Comparison with a crystal structure of the Thermoplasma acidophilum TPP, with Tre6P modelled in the active site (Rao et al. 2006), shows that most of the active site residues are highly conserved in the C-terminal phosphatase-like domain of the class II TPS proteins (Table 2). This includes the critical Asp residue (Asp7) that forms a phospho-acyl intermediate during the dephosphorylation reaction. All of the class II TPSs have a Ser in place of Thr45, which is involved in orientating the phosphate group of Tre6P in the correct position for nucleophilic attack by Asp7. However, a similarly conservative substitution is found in the E. coli otsB (TPP) and most of the plant TPP proteins, suggesting that this difference is unlikely to explain the lack of TPP activity. One of the residues involved in binding the trehalose moiety of Tre6P (His118) is conserved in some TPS class II sequences, but another residue (Lys149) is not conserved at all. For comparison, in plant TPPs, His118 is perfectly conserved and Lys149 is either conserved or, in most cases, conservatively substituted by Arg. This imperfect conservation of residues involved in binding the trehalose moiety of Tre6P might account for the lack of TPP activity in the class II TPS proteins, despite the conservation of the active site HAD motifs. A substitution of Asn for the first Asp (Asp179) residue in HAD motif III, as found in the rice OsTPS8a and OsTPS9a proteins, is associated with the absence of a metal ion cofactor in some members of the HAD superfamily (Burroughs et al. 2006).
|
|
With no experimental evidence of any enzymatic activity, we can only speculate on the function of the class II TPS isoforms. One possibility is that they act as regulatory subunits in a hetero-oligomeric complex with the class I TPS, analogous to the non-catalytic TPS3 and TSL1 subunits in the yeast trehalose-synthesising complex (Bell et al. 1998). Another possibility is that they are somehow involved in Tre6P signalling, perhaps as Tre6P-binding proteins. Although we do not know the function(s) of the class II TPS proteins, it seems clear that they are subject to a high degree of regulation. In A. thaliana leaves, the AtTPS5–11 genes all show strong diurnal cycles in transcript abundance, with AtTPS5 peaking at the end of the day, while the others peak at the end of the night (Bläsing et al. 2005). Expression of several members of the class II subfamily also responds dramatically to changes in sugar levels in seedlings; AtTPS5 is repressed during sugar starvation and induced by sucrose and glucose, whereas AtTPS8–10 are strongly induced by sugar starvation and repressed on sugar re-addition (Price et al. 2004; Osuna et al. 2007). These changes in transcript abundance suggest a high degree of regulation at the level of transcription. In addition, several of the class II TPS proteins are the targets of multi-site phosphorylation by sucrose-non-fermenting-1-related protein kinases and calcium-dependent protein kinases (Glinski and Weckwerth 2005; Harthill et al. 2006). AtTPS5, AtTPS6 and AtTPS7 also bind 14-3-3 proteins when phosphorylated, and in AtTPS5 this binding was shown to be dependent on phosphorylation of Ser5 and Thr32 (Harthill et al. 2006; n.b. the residue numbers referred to in this study, Ser22[= Ser5] and Thr49[= Thr32], are based on an earlier AtTPS5 gene model that has now been superseded). There is circumstantial evidence that phosphorylation of these residues and 14-3-3 protein binding could be linked to control of protein degradation via the 26S proteosome (Cotelle et al. 2000), but otherwise the significance of the transcriptional and post-translational regulation of the class II TPS proteins cannot be properly assessed until the function of these proteins has been elucidated.
Trehalose-phosphatase gene families
The rice and poplar genomes both contain 10 TPP genes, the same number as A. thaliana, whereas P. patens has two, S. moellendorffii has three and the prasinophyte algae (Ostreococcus species) each have only a single TPP gene. BLAST searches of the NCBI GenBank database indicate that the plant TPP sequences are most closely related to those from bacteria, especially proteobacteria (data not shown). This suggests that the plant TPP genes may have originated from the endosymbiotic ancestor of mitochondria, which is thought to have been similar to present day α-proteobacteria (Brown et al. 2001).
In phylogenetic analyses of the plant TPP protein sequences, no consensus tree topology was discovered when the sequences from non-flowering plants were included, although the angiosperm sequences were consistently divided into two major groups, except for poplar TPP9 and TPP10, which always clustered separately as outliers (Fig. 3). Separate analyses were carried out with the sequences from angiosperms only, and these provided robust support for a fundamental dichotomy within the angiosperm TPP family, with poplar TPP9 and TPP10 as outliers. In contrast to the clear differences in gene structure between the class I and class II TPS gene families, the position of introns is generally conserved in all of the TPP genes, although there appear to be several examples where introns have been lost. The broad conservation of gene structure, and the presence of A. thaliana, poplar and rice sequences within both of the major groups, indicate that the two subfamilies of TPP genes arose by duplication of a common ancestor before the separation of the monocot and eudicot lineages. There is also evidence of more recent duplications of TPP genes within each of the three species, for example, the A. thaliana TPPB/TPPD, TPPE/TPPG, TPPF/TPPH and TPPI/TPPJ isoforms were consistently paired together. Several pairs of A. thaliana and poplar sequences were always clustered together, e.g. AtTPPI/AtTPPJ and poplar TPP3/TPP4, suggesting that these represent orthologous groups, but it was more difficult to identify putative orthologues from rice (Fig. 3). The maize RA3 protein, an active TPP enzyme that is involved in control of inflorescence branching (Satoh-Nagasawa et al. 2006), was also included in the analyses, and belongs to a distinct clade of monocot sequences that includes the rice TPP3 and TPP7b (Fig. 3; see also Satoh-Nagasawa et al. 2006).
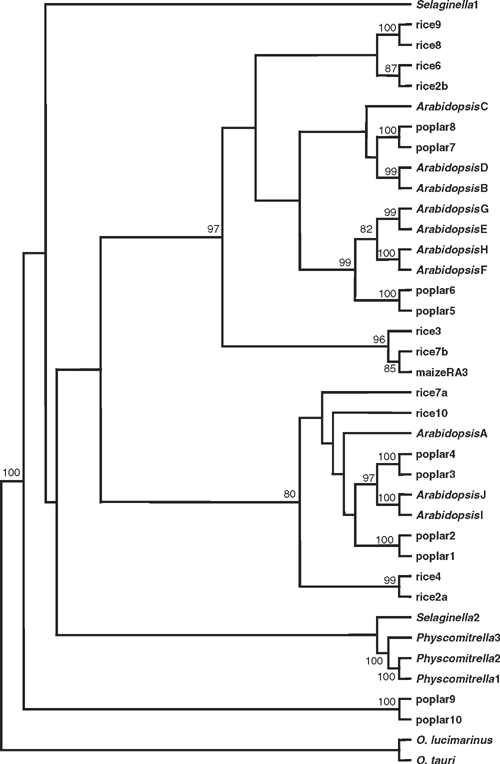
|
The AtTPPA and AtTPPB genes can complement the yeast tps2Δ (TPP–) mutant and have been shown to encode active TPP enzymes (Vogel et al. 1998), as have the rice TPP2a and maize RA3 (Pramanik and Imai 2005; Satoh-Nagasawa et al. 2006). All of the plant TPP proteins show perfect conservation of the three HAD motifs, and good conservation of the other residues involved in binding the trehalose moiety of Tre6P in the T. acidophilum TPP (Rao et al. 2006; Table 2). There is a conservative substitution of Lys149 by Arg in many of the plant TPPs, and Lys111 is substituted by Asn or His in the plant TPPs. However, a neighbouring Lys residue found in most of the plant TPPs, except those from Ostreococcus species (Met), S. moellendorffii TPP2 (Thr) and poplar TPP9–10 (Arg), might be expected to replace the function of Lys111 in the T. acidophilum TPP. The high degree of conservation of active site residues indicates that all of the plant TPP genes are likely to encode active TPP enzymes. In addition to the HAD phosphatase domain, the plant TPPs also have a highly variable N-terminal region of unknown function, although one possibility is that this region could be involved in intracellular targeting of the proteins.
The need for so many isoforms of TPP is not immediately obvious. However, analysis of the AtGeneExpress data indicates that the TPP genes in A. thaliana have different spatial and temporal patterns of expression. For example, the transcript abundance of AtTPPA shows a strong diurnal rhythm in rosette leaves, peaking at the end of the night, whereas AtTPPH peaks in the middle of the day (Bläsing et al. 2005), and AtTPPE is highly expressed in flowers and developing seeds/siliques but hardly at all in rosette leaves (Schmid et al. 2005). The RA3 gene in maize shows very specific expression in localised domains subtending the axillary meristems in the inflorescence (Satoh-Nagasawa et al. 2006). In A. thaliana, salt stress induces expression of several TPP genes in roots (e.g. AtTPPA, AtTPPC, AtTPPE and AtTPPG), and low temperature induces expression of other TPP genes in both shoots and roots (e.g. AtTPPG-J). Chilling stress in rice has been shown to induce expression of the rice TPP2a gene, together with transient increases in both TPP activity and the level of trehalose in the roots (Pramanik and Imai 2005). However, at present we do not know if the stress-induced changes in the expression of TPP genes in A. thaliana lead to any change in TPP activity, or altered levels of Tre6P and trehalose. To understand the functions of the many isoforms of TPP will require more detailed expression analyses at the transcript and protein levels, coupled with measurements of TPP activity and the amounts of Tre6P and trehalose. Phenotypic characterisation of A. thaliana knockout mutants could also be informative, although the presence of several pairs of closely related TPP genes in this species suggests there may be considerable redundancy of function, and so double knockout mutants could be needed to see any phenotype.
Trehalase
In contrast to the large TPS and TPP gene families, trehalase is encoded by a single gene in all of the plant and green algal species examined except poplar and P. patens, which both have three TRE genes. Two of the three closely related (>92% identity) poplar genes, PtTRE1 and PtTRE2, are adjacent to each other on chromosome I, whereas PtTRE3 is located on chromosome III. It seems likely that the PtTRE1/PtTRE2 pair resulted from a relatively recent duplication, and that the ancestral gene was homeologous with PtTRE3. The trehalases from non-flowering plants cluster together in a separate clade from the flowering plant sequences, with the algal (Ostreococcus species) trehalases in yet another group (Fig. 4). There is a clear monocot-eudicot divide among the trehalases from flowering plants. The plant trehalases are most closely related to those from animals, indicating a eukaryotic origin for the plant TRE genes.
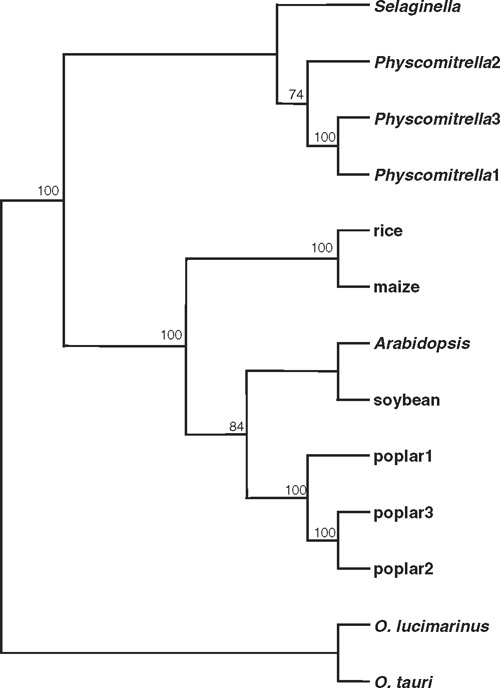
|
Trehalase activity has been detected in several plant species, including A. thaliana, and is particularly high in legume root nodules (Müller et al. 1995, 2001). Trehalase activity was also found to be strongly induced in roots and hypocotyls of A. thaliana plants infected with the clubroot pathogen Plasmodiophora brassicae (Brodmann et al. 2002). An apoplastic trehalase from soybean was reported to have a broad pH optimum and to hydrolyse only trehalose and maltose (Müller et al. 1992; Aeschbacher et al. 1999), whereas an acid trehalase from Phaseolus vulgaris L. root nodules was less specific, hydrolysing sucrose, melibiose, cellobiose and raffinose, in addition to trehalose and maltose (García et al. 2005). Inhibition of endogenous trehalase activity by validamycin A was reported to increase levels of trehalose in several plant species, indicating that the enzyme normally prevents accumulation of trehalose (Müller et al. 1995, 2001; Goddijn et al. 1997), but overexpression of trehalase had little effect on the phenotype of A. thaliana plants (Schluepmann et al. 2003). In A. thaliana, AtTRE transcripts are particularly abundant in flowers and developing seeds/siliques (Schmid et al. 2005). Expression of the gene appears to be lower in rosette leaves and shows no obvious diurnal rhythm (Bläsing et al. 2005).
In resurrection plants that accumulate large amounts of trehalose as they become dehydrated, trehalase has an obvious role to play in recycling the trehalose once water is restored to the plants. Studies on trehalase-deficient yeast cells suggest that removal of trehalose could be essential to allow cellular repair mechanisms, such as protein refolding by chaperonins, to operate effectively in resurrection plants once the drought stress is alleviated (Wera et al. 1999). Trehalase may also be involved in trehalose-mediated signalling processes in plants. As noted in the Introduction, trehalose released by pathogenic and symbiotic microbes appears to modify the metabolism of their host plants (Müller et al. 1998; Brodmann et al. 2002). It has been speculated that induction of trehalase in pathogen-infected tissues may be part of the plant’s defence response to prevent excessive accumulation of trehalose that could otherwise interfere with its metabolism (Brodmann et al. 2002). Trehalose produced by symbiotic nitrogen-fixing bacteria in legume root nodules appears to induce changes in the carbohydrate metabolism of the plant cells that favour the bacteroids, and the concomitant induction of trehalase in the plant cells may help to maintain a balance between the needs of the bacteroids and those of the host cell (Xie et al. 2003). Further work is needed to fully understand the importance of trehalose in these microbial–plant interactions, and whether it has any role in other plant partnerships such as those with mychorrhizal fungi or endophytic organisms (Secks et al. 1999).
Concluding comments
The discovery of TPS and TPP genes in A. thaliana less than 10 years ago (Blázquez et al. 1998; Vogel et al. 1998) has led to a complete reappraisal of the importance of trehalose metabolism in plants. Previously it was thought to be restricted to just a few specialised resurrection plants, but is now known to be widespread, perhaps universal, in plants. However, the functions of trehalose metabolism in plants are less well understood. There is growing evidence that Tre6P, the intermediate of trehalose synthesis, is a signalling molecule that influences both metabolic and developmental processes, but the details are still sketchy. Trehalose itself may also be a signal metabolite, especially in plant–microbe interactions.
Analysis of the A. thaliana genome sequence first showed the great diversity of genes encoding the enzymes of trehalose metabolism in plants, in particular the presence of two distinct subfamilies of TPS genes (Leyman et al. 2001). In the present study, the phylogenetic analysis has been extended to a much broader range of species, including monocots, non-flowering plants and green algae. This has revealed that the four main gene families related to trehalose metabolism in flowering plants – TPS class I, TPS class II, TPP and TRE – have very ancient origins, dating back to before the divergence of the streptophyte and chlorophyte lineages. Both classes of TPS genes appear to have a eukaryotic origin, whereas the TPP genes may be derived from the endosymbiotic bacterial ancestor of mitochondria. The domain structure of the plant class II TPSs closely resembles that of the yeast TPP enzyme (TPS2), and it is tempting to speculate that the plant class II TPSs might once have had TPP activity, but this became redundant after the acquisition of the prokaryotic type TPP, allowing the plant class II TPSs to evolve a new function.
Proliferation of TPS class II and TPP genes has occurred independently in several plant lineages, whereas the TPS class I and TRE gene families are usually much smaller, and often represented by only a single gene. In this respect, A. thaliana is atypical in having four TPS class I genes, three of which encode the unusual short isoforms of TPS that appear to be restricted to the Brassicaceae. Analysis of the TPP gene families from A. thaliana together with rice and poplar showed for the first time that the genes are divided into two subfamilies (Fig. 3), although the differences between the two groups are less distinctive than between the class I and class II TPS genes. This division suggests that there are fundamental differences in the properties and/or functions of the isoforms encoded by the two subfamilies of TPP genes, but at present we do not know what these might be.
In conclusion, this survey of trehalose metabolism-related genes in the genomes of rice, poplar and other more primitive plants has provided new insights into the origins and evolution of trehalose metabolism in plants, and although it raises several new questions, it should provide a framework on which to base future studies into the function(s) of trehalose metabolism in plants.
Aeschbacher RA,
Müller J,
Boller T, Wiemken A
(1999) Purification of the Trehalase MTRE1 from soybean nodules and cloning of its cDNA. GMTRE1 is expressed at a low level in multiple tissues. Plant Physiology 119, 489–496.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
[Verified 18 February 2007]
Nishiyama T,
Fujita T,
Shin-I T,
Seki M, Nishide H , et al.
(2003) Comparative genomics of Physcomitrella patens gametophytic transcriptome and Arabidopsis thaliana: implication for land plant evolution. Proceedings of the National Academy of Sciences USA 100, 8007–8012.
| Crossref | GoogleScholarGoogle Scholar |

Ohyanagi H,
Tanaka T,
Sakai H,
Shigemoto Y,
Yamaguchi K, Habara T , et al.
(2006) The rice annotation project database (RAP-DB): hub for Oryza sativa spp. japonica genome information. Nucleic Acids Research 34, D741–D744.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Osuna D,
Usadel B,
Morcuende R,
Gibon Y,
Blasing OE, Hohne M , et al.
(2007) Temporal responses of transcripts, enzyme activities and metabolites after adding sucrose to carbon-deprived Arabidopsis seedlings. The Plant Journal 49, 463–491.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Page RD
(1996) TreeView: an application to display phylogenetic trees on personal computers. Computer Applications in the Biosciences 12, 357–358.
| PubMed |
Pilon-Smits EAH,
Terry N,
Sears T,
Kim H,
Zayed A,
Hwang S,
van Dun K,
Voogd E,
Verwoerd TC,
Krutwagen RWHH, Goddijn OJM
(1998) Trehalose-producing transgenic tobacco plants show improved growth performance under drought stress. Journal of Plant Physiology 152, 525–532.
Pramanik MHR, Imai R
(2005) Functional identification of a trehalose 6-phosphate phosphatase gene that is involved in transient induction of trehalose biosynthesis during chilling stress in rice. Plant Molecular Biology 58, 751–762.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Price J,
Laxmi A,
St. Martin SK, Jang J-C
(2004) Global transcription profiling reveals multiple sugar signal transduction mechanisms in Arabidopsis. The Plant Cell 16, 2128–2150.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Rana D,
van den Booqaart T,
O’Neill CM,
Hynes L,
Bent E,
Macpherson L,
Park JY,
Lim YP, Bancroft I
(2004) Conservation of microstructure of genome segments in Brassica napus and its diploid relatives. The Plant Journal 40, 725–733.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Rao NR,
Kumaran D,
Seetharaman J,
Bonanno JB,
Burley SK, Swaminathan S
(2006) Crystal structure of trehalose-6-phosphate phosphatase-related protein: Biochemical and biological implications. Protein Science 15, 1735–1744.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Ratnakumar S, Tunnacliffe A
(2006) Intracellular trehalose is neither necessary nor sufficient for desiccation tolerance in yeast. FEMS Yeast Research 6, 902–913.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Reignault P,
Cogan A,
Muchembled J,
Lounes-Hadj Sahraoui A,
Durand R, Sancholle M
(2001) Trehalose induces resistance to powdery mildew in wheat. New Phytologist 149, 519–529.
| Crossref | GoogleScholarGoogle Scholar |
Romero C,
Bellës JM,
Vayá JL,
Serrano R, Culiáñez-Maciá FA
(1997) Expression of the yeast trehalose-6-phosphate synthase gene in transgenic tobacco plants: pleiotropic phenotypes include drought tolerance. Planta 201, 293–297.
| Crossref |
Satoh-Nagasawa N,
Nagasawa N,
Malcomber S,
Sakai H, Jackson D
(2006) A trehalose metabolic enzyme controls inflorescence architecture in maize. Nature 441, 227–230.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Schluepmann H,
Pellny T,
van Dijken A,
Smeekens S, Paul M
(2003) Trehalose 6-phosphate is indispensable for carbohydrate utilization and growth in Arabidopsis thaliana. Proceedings of the National Academy of Sciences USA 100, 6849–6854.
| Crossref | GoogleScholarGoogle Scholar |
Schmid M,
Davison TS,
Henz SR,
Pape UJ,
Bemar M,
Vingron M,
Scholkopf B,
Weigel D, Lohmann JU
(2005) A gene expression map of Arabidopsis thaliana development. Nature Genetics 37, 501–506.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Secks ME,
Richardson MD,
West CP,
Marlatt ML, Murphy JB
(1999) Role of trehalose in desiccation tolerance of endophyte-infected tall fescue. Arkansas Agricultural Experimental Station Research Series 475, 134–140.
The Arabidopsis Initiative
(2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408, 796–815.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Tunnacliffe A, Lapinski J
(2003) Resurrecting van Leewenhoek’s rotifers: a reappraisal of the role of disaccharides in anhydrobiosis. Philosophical Transactions of the Royal Society of London Series B 358, 1755–1771.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Tuskan GA,
Difazio S,
Jansson S,
Bohlmann J,
Grigoriev I, Hellsten U , et al.
(2006) The genome of black cottonwood Populus trichocarpa (Torr. & Gray). Science 313, 1596–1604.
| Crossref |
PubMed |
Vogel G,
Aeschbacher RA,
Müller J,
Boller T, Wiemken A
(1998) Trehalose-6-phosphate phosphatases from Arabidopsis thaliana: identification by functional complementation of the yeast tps2 mutant. The Plant Journal 13, 673–683.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Vogel G,
Fiehn O,
Jean-Richard-dit-Bressel L,
Boller T,
Wiemken A,
Aeschbacher RA, Wingler A
(2001) Trehalose metabolism of Arabidopsis: occurrence of trehalose and molecular cloning and characterization of trehalose-6-phosphate synthase homologues. Journal of Experimental Botany 52, 1817–1826.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Wera S,
de Schrijver E,
Geyskens I,
Nwaka S, Thevelein JM
(1999) Opposite roles of trehalase activity in heat-shock recovery and heat-shock survival in Saccharomyces cerevisiae. Biochemical Journal 343, 621–626.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Xie Z-P,
Staehelin C,
Broughton WJ,
Wiemken A,
Boller T, Müller J
(2003) Accumulation of soluble carbohydrates, trehalase and sucrose synthase in effective (Fix+) and ineffective (Fix–) nodules of soybean cultivars that differentially nodulate with Bradyrhizobium japonicum. Functional Plant Biology 30, 965–971.
| Crossref | GoogleScholarGoogle Scholar |
Zentella R,
Mascorro-Gallardo JO,
van Dijck P,
Folch-Mallol J, Bonini B , et al.
(1999) A Selaginella lepidophylla trehalose-6-phosphate synthase complements growth and stress-tolerance defects in a yeast tps1 mutant. Plant Physiology 119, 1473–1482.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Zhang Z,
Schwartz S,
Wagner L, Miller W
(2000) A greedy algorithm for aligning DNA sequences. Journal of Computational Biology 7, 203–214.
| Crossref | GoogleScholarGoogle Scholar | PubMed |