

New boron-based coumarin fluorophores for bioimaging applications†
Anita Marfavi A B , Jia Hao Yeo A , Kathryn G. Leslie A , Elizabeth J. New A B C and Louis M. Rendina A B *
A , Kathryn G. Leslie A , Elizabeth J. New A B C and Louis M. Rendina A B *
A School of Chemistry, The University of Sydney, Sydney, NSW 2006, Australia.
B The University of Sydney Nano Institute, Sydney, NSW 2006, Australia.
C Australian Research Council Centre of Excellence for Innovations in Peptide and Protein Science, The University of Sydney, Sydney, NSW 2006, Australia.
Australian Journal of Chemistry 75(9) 716-724 https://doi.org/10.1071/CH21320
Submitted: 3 December 2021 Accepted: 7 February 2022 Published: 29 March 2022
© 2022 The Author(s) (or their employer(s)). Published by CSIRO Publishing. This is an open access article distributed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License (CC BY-NC-ND)
Abstract
The synthesis and characterisation of five new boron-based coumarin fluorophores are reported, with key structural variations involving the linker at the C3-position (hydrazone or imine) of the 7-(diethylamino)-coumarin (7DEAC) core and the terminal boron moiety (i.e. boronic acid or closo-1,2-carborane). All the coumarin derivatives were found to display significant bathochromic shifts relative to the parent 7DEAC, with conjugate ICCb displaying the greatest overall shift. Confocal microscopy studies with A549 lung cancer cells showed clear differences in the observed intra-cellular distributions of the fluorophores. The polar boronic acid species (HCoBA, HCmBA and HCpBA) were found to localise in the endoplasmic reticulum. In contrast, the lipophilic closo-1,2-carborane derivatives (HCCb and ICCb) were found to localise within lipid droplets (LDs), showcasing the future potential for these probes to be utilised as stains for LD observations by means of confocal microscopy.
Keywords: bioimaging, boron, coumarin, endoplasmic reticulum, fluorescence microscopy, fluorescent probes, fluorophore, intramolecular charge transfer, lipid droplets, near‐infrared, Nile Red.
Introduction
Understanding the nature of complex biological events occurring in normal cellular physiology and pathology is fundamental to developing novel strategies for disease prevention, diagnosis and treatment. Fluorescence imaging, including confocal microscopy, has emerged as a landmark platform providing detailed cellular analysis with high sensitivity and selectivity.[1] These applications have also extended across an array of biomedical platforms, including non-invasive image guided surgery,[2,3] contrast agents, detection of tumours, tracking drug metabolism, monitoring biological processes (for example, thrombotic activity)[4] and for the detection of many types of metal ions.[5] In biological imaging, significant bathochromic shifts towards the near-infrared (NIR) region enables greater tissue penetration, reduced light scattering and minimal cellular autofluorescence.[6]
Significant drawbacks undermining existing organic biological probes include low aqueous solubility and small Stokes shifts. Coumarins are an important class of fluorophores that exhibit exceptional photophysical properties, with large Stokes shifts, high quantum yields, small molecular weights and good water solubility. Although their emission is relatively blue-shifted (410–470 nm), their ease of functionalisation can be exploited such that the photophysical properties may be improved to generate biologically useful fluorophores.[7,8] The most fundamental coumarin design strategy is to incorporate an electron-donating group at the 7-position (C7). This results in a ‘push–pull’ mechanism, contributing to an intramolecular charge transfer (ICT) during excitation via photon absorption. The ICT process is aided by the addition of electron-withdrawing or -donating groups (EWG or EDG, respectively) at the 3- or 4-positions (C3/C4) to red-shift the fluorescence spectra via resonance and inductive effects.[9] In particular, the incorporation of N-acylhydrazone derivatives (Schiff bases) has been well documented in coumarin derivatives that can detect metal ions.[7] In addition, an extended unsaturated π-conjugated bridge at C3 has led to extensive bathochromic shifts and more pronounced molar extinction coefficients.[10,11] Increasing the electron-donor strength at C7 (e.g. −N(C2H5)2 > −OH > −OCH3) also increases the red-shift in the fluorescence spectra.
Electron-deficient boron moieties, including boronic acids and C-substituted closo-1,2-carboranes, which can act as bioisosteres of phenyl rings, can be utilised as terminal EWGs to facilitate the ICT process and enhance the bathochromic shift of new fluorescent probes.[12,13] Boronic acids have been incorporated into fluorophores for the selective detection of metal ions for medicinal and environmental research.[14] Remarkably, carboranes have shown utility in the design of fluorescent probes with enhanced pharmacokinetics, stability and bathochromic shifts for applications in biological imaging and sensing.[15,16] We have previously demonstrated the lipophilic nature of closo-carboranes in the development of a carborane-containing coumarin which localises in cellular lipid droplets, as an alternative to the lipid-staining dye Nile Red.[13] The versatility of carboranes as luminescent sensors for fluoride anions has been reported,[17] with a donor–acceptor molecule comprised of an ortho-carborane unit and a bulky, tri-coordinate organoboron compound, a dimesitylboryl group (BMes2).
To the best of our knowledge, there are no reports providing a direct comparison of the intracellular distributions between various boron functional groups in coumarin fluorescent probes. Herein we report the synthesis and characterisation of new boron-based coumarins conjugated via hydrazone or imine linkers, with extensive consideration to structure–photophysical property relationships. Cell uptake and biodistribution of selected boronated fluorophores were also investigated to assess their potential as new biological stains for confocal microscopy.
Results and discussion
Probe design and synthesis
The main synthetic approach in the design of these novel coumarin fluorophores involved structural modifications of the parent 7-(diethylamino)-coumarin (7DEAC) scaffold. In this study, two main strategies were adopted: (a) the addition of an EWG at C3 (hydrazone linker) or (b) extension of the π-conjugation at C3 (imino linker) using a 3-formylcoumarin precursor (Fig. 1). These strategies were used in combination with the 7-diethylamino EDG at C7 of the coumarin framework and the addition of terminal boronic acid or closo-1,2-carborane moieties, to enhance the ICT process, increase the Stokes shift and ultimately generate a bathochromic shift in the excitation and emission maxima. Cumulatively, these structural modifications afforded four new hydrazone-coumarin conjugates and one imino-coumarin derivative, with two phenyl-containing analogues also included for comparison.

|
Synthesis of the hydrazone-coumarins initially involved a Knoevenagel condensation reaction between 4-(diethylamino)salicylaldehyde and diethyl malonate in the presence of a suitable base (piperidine).[18] The coumarin ester was readily converted into the carbohydrazide by a nucleophilic substitution reaction with hydrazine hydrate, according to the literature procedure.[19] Hydrazone formation occurred by means of a nucleophilic addition reaction between the carbohydrazide coumarin and formyl-functionalised benzaldehydes, such as 2, 3 or 4-formylphenylboronic acids, affording HCPh, HCoBA, HCmBA and HCpBA, respectively (Scheme 1). Mono-substituted formyl-ortho-carborane was prepared by means of a nucleophilic substitution with n-BuLi, proceeded by electrophilic functionalisation with methyl formate, and subsequent hydrazone formation (HCCb, Scheme 1) was achieved as above.
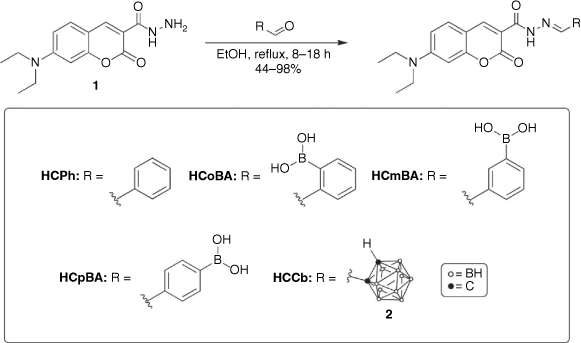
|
The first step in the synthesis of the imino-coumarin analogues was the formation of the parent 7DEAC, once again using a Knoevenagel condensation (see above), followed by acid reflux and decarboxylation by the addition of NaOH. The formation of 3-formylcoumarin (3) occurred in the following step, whereby the C3-position of the coumarin backbone is subjected to an electrophilic attack by a Vilsmeier–Haack reagent. Imine formation then occurred by means of an electrophilic attack between 3-formylcoumarin and either aniline or 1-amino-ortho-carborane to give ICPh and ICCb (Scheme 2).

|
Fluorescence studies
Preliminary fluorescence studies were performed to investigate the impact of substitution (with EWGs and π-conjugated bridges) at the C3-position of the parent 7DEAC. Coumarin fluorescence involves an ICT-induced mechanism, and perturbations in the ICT process can alter fluorescence properties. Evidently, the coumarins containing a hydrazone linker (HCPh, HCoBA, HCmBA, HCpBA and HCCb) exhibited similar excitation and emission maxima, which are both red-shifted relative to the parent coumarin (Table 1). As expected, due to the absence of a π-conjugated bridge, the hydrazone moiety does not appear to extensively enhance the bathochromic shift. The nature of the terminal EWGs in the hydrazone-coumarins also appears to have a minimal effect on their photophysical properties.

|
Conversely, a trend was established regarding the impact of π-conjugation and a comparison of phenyl and closo-1,2-carborane moieties, as observed for compounds ICPh and ICCb. The inclusion of a π-conjugated bridge with a terminal EWG leads to a significant red-shift in both the observed excitation and emission maxima. As expected, closo-1,2-carborane is more electron-withdrawing than a phenyl substituent, and this leads to a larger bathochromic shift (ca. λem increase by 45 nm (ICCb) and 30 nm (ICPh), as compared to 7DEAC). ICCb exhibited the most red-shifted emission maximum at 505 nm. Interestingly, the hydrazone-based coumarin derivatives showed higher quantum yields than the iminocoumarins, with the para-phenylboronic acid species HCpBA displaying the highest quantum yield (0.32). However, when compared to the parent, non-boronated 7DEAC, the quantum yields were found to be significantly reduced. In the imino-coumarin species (ICPh and ICCb) the ortho-1,2-carborane derivative displayed a ca. 4-fold increase in quantum yield compared to its phenyl counterpart. The variability in the quantum yields across both species suggests that both the linker and terminal EWG are crucial determinants in this particular photophysical property. Furthermore, the relative overall brightness was greatest in the hydrazone-coumarin derivatives, with HCpBA displaying the highest value.
Density functional theory calculations
Density functional theory (DFT) calculations (B3LYP/6-31G**) were performed for all the new coumarin derivatives prepared in this work (Table 2, see below). The results showed the π-conjugated imino-coumarin derivatives (ICPh and ICCb) exhibited the lowest HOMO–LUMO energy gap (ΔE). As expected, the imino linker at C3 significantly improves the red-shift in the spectroscopic profiles, by lowering the energy of the electronic transitions thereby generating longer emission wavelengths. Furthermore, while an EWG such as hydrazone can enhance the ICT process, a π-conjugated bridge provides a more substantial contribution to the photophysical properties. A similar trend was observed between the calculated wavelengths (λ, Table 2) and the excitation wavelengths measured for each coumarin probe (Table 1), whereby the imino-coumarin derivatives (ICPh and ICCb) displayed a greater bathochromic shift.
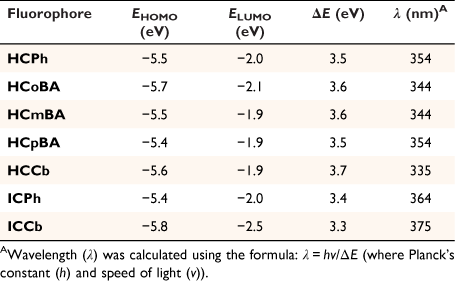
|
Confocal microscopy
One key aim of this work was to develop novel probes for bioimaging applications. To assess whether the compounds are suitable for imaging studies, human alveolar basal epithelial (A549) cells were investigated. A549 cells were incubated with the coumarin fluorophores (10 μM, 20 min) and imaged using a confocal microscope (Fig. 2, Supplementary Fig. S1).
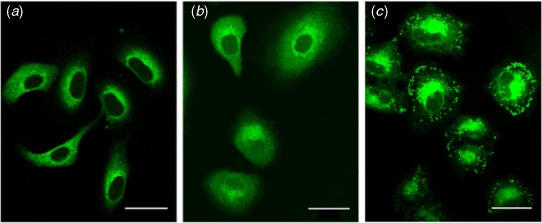
|
Preliminary imaging studies revealed variations in sub-cellular distribution across the set of compounds, according to the polarity of the boron or phenyl moiety (boronic acid > phenyl > closo-1,2-carborane, Fig. 2). Coumarins bearing hydrophobic moieties (i.e. closo-1,2-carborane) exhibited punctate fluorescent staining, consistent with lipid droplets, as previously observed (Fig. 2c).[13] Despite the bioisosteric relationship between phenyl and closo-1,2-carborane, the cytoplasmic and weakly diffusive fluorescence from the phenyl derivatives is markedly different from the punctate fluorescence staining of closo-1,2-carborane compounds (between HCPh and HCCb, Fig. 2; ICPh and ICCb, Supplementary Fig. S1). Such differences had not previously been observed for coumarin dyes functionalised with phenyl or closo-1,2-carborane at the 3-position.[13]
To further confirm the localisation of the probes in cells, co-localisation studies were performed using commercialised probes that target specific organelles. Co-localisation studies were first conducted against Nile Red, which marks lipid droplets in A549 cells.[20,21] Lipid droplet homeostasis is critical for cell signalling and the production of inflammatory mediators, with lipid droplet accumulation associated in cancer, cardiovascular diseases and insulin-resistance.[22] Co-staining studies of the highly lipophilic ortho-1,2-carborane derivatives (HCCb and ICCb) with Nile red revealed clear co-localisation (Fig. 3, Pearson’s correlation coefficients (PCC) of R = 0.83 ± 0.01 (HCCb) and R = 0.73 ± 0.06 (ICCb). This indicates a high selectivity for lipid droplets, in comparison to the phenyl derivatives (PCC of R < 0.5 for both HCPh and ICPh). The other coumarin probes (HCoBA, HCmBA and HCpBA) did not show lipid droplet staining. Evidently, modifications to the linker group had little effect on the sub-cellular localisation of these probes; it is the terminal EWG that provides the greatest distinction.
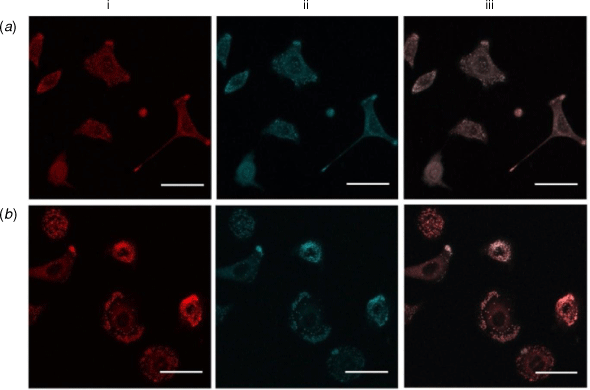
|
Since HCoBA, HCmBA and HCpBA showed punctate staining that was not consistent with the lipid droplets, further co-localisation studies were performed with MitoTracker Red CMXRos and LysoTracker Red DND-99 to probe for mitochondrial or lysosomal localisation, respectively. In neither case was there any significant overlap of the coumarin and the organelle marker. Endoplasmic reticulum (ER) localisation was confirmed by co-staining with an mCherry-ER construct expressed in human colon cancer (DLD-1) cells. While not every cell successfully incorporated the mCherry-ER plasmid, mCherry-stained cells showed colocalisation with HCoBA, HCmBA and HCpBA (PCC of R = 0.67 ± 0.03, 0.60 ± 0.07 and 0.73 ± 0.07, respectively; Supplementary Fig. S5). No other probes displayed ER localisation. Due to the significance of the ER in numerous pathologies (e.g. neurodegenerative disorders, inflammatory diseases and cancer) for the processing of transmembrane and secreted proteins,[23] cell-permeable fluorophores, such as HCoBA, HCmBA or HCpBA, could enable the delivery of small molecules to the ER or could prove to be useful small molecule ER markers.
In summary, all probes were observed to be taken up by human cancer cell lines, with significant variations in intracellular localisation owing to modifications in the polarity of the terminal EWG (phenyl, boronic acid and closo-carborane functional groups), which may lead to a diverse set of future applications.
Conclusion
Five novel boron-containing coumarin fluorophores (HCoBA, HCmBA, HCpBA, HCCb and ICCb) were successfully synthesised and characterised in this work. The influence of linker and terminal EWG modifications on the spectroscopic profile of the 7-(diethylamino)-coumarin fluorophore was demonstrated, whereby extension of the π-conjugated bridge at the C3-position provided the greatest overall red-shift. Confocal microscopy studies using A549 lung cancer cells displayed distinctive intra-cellular localisations of the new coumarin compounds. In particular, the exceptional selectivity of the closo-carboranes (HCCb and ICCb) towards lipid droplets was prevalent compared to their phenyl counterparts (HCPh and ICPh), and hence these dyes may be adapted for use as a green-emitting alternative to the standard Nile Red (a commonly used lipid-staining dye). Furthermore, the boronic acid fluorophores HCoBA, HCmBA and HCpBA exhibited accumulation in the ER, with potential applications in the design of ER-targeting therapeutics. Structure–photophysical property relationships revealed that the imino-coumarin derivative ICCb is an attractive candidate for further development as a NIR coumarin probe, both due to a π-conjugated linker and a strong terminal EWG (closo-carborane) that facilities the ICT process. Ultimately, the incorporation of boron-containing groups into fluorescent scaffolds may prove to be a useful strategy for tuning photophysical and cellular behaviour, with potential developments focussed on boron-based moieties leading to an expansion of exciting new small-molecule tools for biomedical applications.
Experimental
General
All reactions were performed under a nitrogen atmosphere using standard Schlenk technique unless otherwise indicated. All precursor chemicals were commercially available and were purchased from Sigma–Aldrich Co. Anhydrous solvents including acetonitrile, diethyl ether, methanol and dichloromethane were obtained from a solvent purification system, PureSolv MD 7 (Innovative Technology, Inc.). All other solvents were used without any further purification. Column chromatography was carried out using Merck silica gel 60 (230–400 mesh). Analytical thin layer chromatography (TLC) was performed using Merck TLC silica gel 60 F254 aluminium plates. Compounds were visualised under shortwave (254 nm) and/or longwave (365 nm) ultraviolet (UV) light. Carborane-containing compounds were visualised on TLC plates using an acidified PdCl2 (1%) stain and the plates heated by means of a heat gun. Compounds 1–4, HCPh and ICPh were synthesised as previously reported.[7,19,24–27]
Infrared absorption spectra were recorded using a Bruker ALPHA Fourier-transform infrared (FT-IR) spectrometer, with the data reported as wavenumbers (cm−1). All 1H, 11B{1H} and 13C{1H} NMR spectra were recorded at 300 K on either a Bruker AVANCE 300 (1H at 300 MHz, 13C at 75 MHz), Bruker AVANCE III 400 (1H at 400 MHz, 11B at 128 MHz and 13C at 100 MHz) or Bruker AVANCE III 500 (1H at 500 MHz, 11B at 160 MHz and 13C at 125 MHz) spectrometer. All NMR signals (δ) are reported in ppm. 1H and 13C NMR spectra recorded using CDCl3 were referenced to TMS. For all other solvents, the residual solvent peaks were used as internal standards. 11B spectra were referenced automatically by the spectrometer software according to the unified chemical shift scale via the lock frequency of the solvent.[28] Peak multiplicities have been abbreviated as: s (singlet), d (doublet), dd (doublet of doublets), t (triplet), dt (doublet of triplets), q (quartet), br (broad), and m (multiplet). Low resolution mass spectrometry (LRMS) was performed on a Bruker amazon SL operating in the positive ion mode using electrospray ionisation (ESI) or atmospheric pressure chemical ionisation (APCI). All MALDI mass spectrometric data were obtained using a Bruker Autoflex III matrix assisted laser desorptionionisation time-of-flight mass spectrometer (MALDI-TOF MS) with delayed extraction using both positive ion and reflector detection modes over the mass range of m/z 300–3000. For the hydrazone-coumarins (HCoBA, HCmBA, HCpBA and HCCb), stock solutions of trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB) (10 mg mL−1) in 50:50 DCM/MeCN were prepared. The samples were prepared at a concentration of 2 mg mL−1 in DCM. MALDI samples were prepared by combining 20 µL of matrix solution with 4 µL of sample solution. A 0.3 µL portion of this sample was deposited on a ground steel MALDI target plate and allowed to air dry.
Synthetic procedures
General procedure for the synthesis of the hydrazone-coumarin derivatives (Scheme 1)
A slight excess of a formyl reagent (benzaldehyde, 2-, 3- or 4-formylphenylboronic acid, or 2) was added to a stirred solution of 1 in absolute ethanol (5 mL). The reaction mixture was heated to reflux. Upon cooling to room temperature, the precipitate was filtered off and washed three times with cold ethanol (3 × 10 mL). NB: Acetonitrile was used as a solvent instead of ethanol in the synthesis of carboranyl hydrazone-coumarin (HCCb).
Synthesis of (E)-(2-((2-(7-(diethylamino)-2-oxo-2H-chromene-3-carbonyl)hydrazineylidene)methyl)phenyl)boronic acid (HCoBA, Scheme 1)
Formyl reagent: 2-formylphenylboronic acid (103 mg, 0.7 mmol), 1 (100 mg, 0.4 mmol). Reaction time: 18 h. Yield of HCoBA: 107 mg (72%) as a dark-yellow crystalline solid. FTIR (cm−1): 3306 (br, OH), 3241 (N–H), 1692 (C=O), 1615 (C=O). δH (300 MHz, DMSO-d6) 11.71 (s, 1H, NH), 8.74 (s, 1H, H4), 8.68 (s, 1H, H12), 8.48 (s, 2H, B(OH)2), 7.91 (d, J 7.2, 1H, H18), 7.74 (d, J 9.0, 1H, H5), 7.68 (d, J 3.6, 1H, H15), 7.48–7.37 (m, 2H, H16/17), 6.85 (dd, J 11.2, 6.9, 1H, H6), 6.67 (d, J 1.9, 1H, H8), 3.51 (q, J 6.9, 4H, (CH3CH2)2N), 1.15 (t, J 7.0, 6H, (CH3CH2)2N). δB (160 MHz, DMSO-d6) 30.1 (1B). δC (75 MHz, DMSO-d6) 161.6, 159.2, 157.4, 152.9, 150.0, 148.5, 137.5, 134.6, 131.9, 129.4, 129.0, 126.4, 110.4, 108.7, 108.7, 96.0, 44.5, 12.5. m/z (MALDI-TOF MS) calculated for [M + Na]+ 430.155, found 430.094.
Synthesis of (E)-(3-((2-(7-(diethylamino)-2-oxo-2H-chromene-3-carbonyl)hydrazineylidene)methyl)phenyl)boronic acid (HCmBA, Scheme 1)
Formyl reagent: 3-formylphenylboronic acid (103 mg, 0.7 mmol), 1 (100 mg, 0.4 mmol). Reaction time: 12 h. Yield of HCmBA: 129 mg (87%) as a yellow crystalline solid. FTIR (cm−1): 3436 (OH), 3100 (N–H), 1688 (C=O), 1641 (C=O). δH (400 MHz, DMSO-d6) 11.69 (s, 1H, NH), 8.70 (s, 1H, H4), 8.38 (s, 1H, H12), 8.21 (s, 2H, B(OH)2), 8.17 (s, 1H, H14), 7.84 (d, J 7.3, 1H, H16), 7.75 (d, J 7.8, 1H, H18), 7.67 (s, J 9.0, 1H, H5), 7.42 (t, J 7.5, 1H, H17), 6.75 (dd, J 9.0, 2.0, 1H, H6), 6.57 (d, J 1.9, 1H, H8), 3.43 (q, J 7.0, 4H, (CH3CH2)2N–), 1.11 (t, J 7.0, 6H, (CH3CH2)2N–). δB (160 MHz, DMSO-d6) 28.6 (1B). δC (75 MHz, DMSO-d6) 161.5, 159.0, 157.3, 152.7, 148.6, 148.4, 135.8, 133.2, 132.8, 131.8, 129.1, 127.8, 110.3, 108.6, 107.9, 95.9, 44.4, 12.3. m/z (MALDI-TOF MS) calc. for [M + H]+ 408.173, found 408.200.
Synthesis of (E)-(4-((2-(7-(diethylamino)-2-oxo-2H-chromene-3-carbonyl)hydrazineylidene)methyl)phenyl)boronic acid (HCpBA, Scheme 1)
Formyl reagent: 4-formylphenylboronic acid (103 mg, 0.7 mmol), 1 (100 mg, 0.4 mmol). Reaction time: 8 h. Yield of HCpBA: 145 mg (98%) as a yellow crystalline solid. FTIR (cm−1): 3306 (br, OH), 3118 (N–H), 1702 (C=O), 1667 (C=O). δH (400 MHz, DMSO-d6) 11.74 (s, 1H, NH), 8.76 (s, 1H, H4), 8.43 (s, 1H, H12), 8.13 (s, 2H, B(OH)2), 7.86 (d, J 8.0, 2H, H15), 7.74 (d, J 9.1, 1H, H5), 7.70 (d, J 8.1, 2H, H14), 6.84 (dd, J 11.3, 6.8, 1H, H6), 6.66 (d, J 2.0, 1H, H8), 3.50 (q, J 7.0, 4H, (CH3CH2)2N–), 1.15 (t, J 7.0, 6H, (CH3CH2)2N–). δB (160 MHz, DMSO-d6) 31.7 (1B). δC (75 MHz, DMSO-d6) 161.5, 159.0, 157.4, 152.8, 148.5, 148.3, 135.6, 134.4, 131.8, 126.1, 110.4, 108.5, 107.9, 95.9, 44.4, 12.29. m/z (MALDI-TOF MS) calc. for [M + H]+ 408.173, found 408.210.
Synthesis of 7-(diethylamino)-2-oxo-3-((closo-1,2-carboranyl)carbohydrazide)-2H-chromene-2-one (HCCb, Scheme 1)
Formyl reagent: 2 (17 mg, 0.1 mmol), 1 (13.6 mg, 0.5 mmol). Yield of HCCb: 9.34 mg (44%) as an orange crystalline solid. FTIR (cm−1): 3371 (N–H), 2529 (BH), 1680 (C=O), 1649 (C=O). δH (400 MHz, DMSO-d6) 11.82 (s, 1H, NH), 8.71 (s, 1H, H4), 8.17 (s, 1H, H12), 7.74 (d, J 9.0, 1H, H5), 6.85 (d, J 9.1, 1H, H6), 6.65 (d, J 1.6, 1H, H8), 5.38 (br s, 1H, H14), 3.51 (q, J 7.0, 4H, (CH3CH2)2N–), 3.45–1.20 (br, B-H, 10H), 1.15 (t, J 7.0, 6H, (CH3CH2)2N–). δB (128 MHz, DMSO-d6) −3.1 (2B), −9.6 (4B), −11.9 (4B). δC (75 MHz, CDCl3) 162.8, 160.6, 158.2, 153.4, 149.6, 141.1, 131.7, 110.5, 108.5, 107.8, 96.6, 77.4, 57.1, 45.3, 12.4. m/z (MALDI-TOF MS) calc. for [M + H]+ 430.313, found 430.451.
Synthesis of (E)-7-(diethylamino)-3-((closo-1,2-carboranylimino)methyl)-2H-chromen-2-one (ICCb, Scheme 2)
Compound ICCb was prepared by refluxing 4 (20 mg, 0.1 mmol) with 3 (61.6 mg, 0.3 mmol) in MeCN for 14 h. Purification was achieved by means of preparative TLC (DCM/hexane, 70:30) to afford ICCb (22 mg, 57%), as an orange crystalline solid. FTIR (cm−1): 2574 (BH), 1707 (C=N), 1620 (C=O). δH (400 MHz, CDCl3) 8.59 (s, 1H, H11), 8.20 (s, 1H, H4), 7.34 (d, J 9.0, 1H, H5), 6.62 (dd, J 11.4, 6.6, 1H, H6), 6.48 (d, J 2.2, 1H, H8), 4.03 (br s, 1H, H13), 3.46 (q, J 7.1, 4H, (CH3CH2)2N–), 3.45–1.20 (br, 10H, B-H), 1.25 (t, J 7.1, 6H, (CH3CH2)2N–). δB (128 MHz, CDCl3) −3.71 (2B), −8.09 (2B), −10.23 (2B), −12.13 (2B), −14.04 (2B). δC (75 MHz, CDCl3) 161.8, 158.4, 153.1, 142.9, 131.6, 112.7, 110.2, 108.6, 97.3, 94.0, 77.4, 62.1, 45.3, 12.6. m/z (APCI MS) calc. for [M + H]+ 388.28, found 388.28.
Fluorescence spectroscopy
All fluorescence studies were performed in absolute ethanol at a concentration of 1 μM, measured on a Varian Cary Eclipse fluorescence spectrophotometer using 1 cm pathlength quartz cuvettes.
Quantum yields were measured and calculated using a PTI QuantaMaster 400 spectrofluorometer with an integrating sphere (IS) detector, using the absolute quantum yield method. These measurements were performed at room temperature with dilute fluorophore samples (conc. <1 μM). Excitation and emission correction were performed by the instrument.
Mammalian cell studies
Human cell lines
Human alveolar adenocarcinoma (A549) cells were used in this study. Cells were cultured at 37°C with 5% CO2 as monolayers in exponential growth. Cells were maintained in Advanced Dulbecco’s Modified Eagle Medium (ADMEM) supplemented with 2% foetal bovine serum (FBS) and 2 mM glutamine. Subculturing of cells were performed every 3–4 days when 80% confluency was reached. Trypsin–EDTA (0.25%) were used to facilitate dislodgement of cells from the flask during sub-culturing.
For determining co-localisation with ER, human colorectal adenocarcinoma (DLD-1) cells, previously transfected with a mCherry fluorescent protein tagged to the ER, were used. Cells were cultured at 37°C with 5% CO2 as monolayers at exponential growth. Cells were maintained in ADMEM supplemented with 2% FBS, 400 µg mL−1 gentamycin and 2 mM glutamine. Subculturing of cells were performed every 3–4 days when 80% confluency was reached. Trypsin–EDTA (0.25%) was used to facilitate dislodgement of cells from the flask during sub-culturing.
Confocal microscopy imaging
Approximately 100 000 cells were seeded on 35 mm MatTek dishes pre-coated with l-proline and allowed to adhere overnight. Cells were then dosed with a 10 μM coumarin compound in DMSO (final DMSO concentration < 1% (v/v)) in ADMEM (supplemented with 2% FBS, 2 mM glutamine) for 20 min at 37°C. Cells were then imaged in FluoroBrite DMEM media (1 mL, supplemented with 10% FBS, 2 mM glutamine).
Fluorescent micrographs were acquired using an Olympus FluoView FV1000 inverted microscope equipped with a UPLSAPO 60X water-immersion objective lens (N.A. = 1.15). Coumarin probes were excited at 458 nm using an Argon laser, and fluorescence emitted between 468 and 568 nm was acquired. Fluorescent images were collected using FV10-ASW viewer software v1.7 (Olympus) and processed using Image J® (Fig. 2, Supplementary Fig. S1).
Co-localisation studies with commercial fluorescent probes
For assessing co-localisation of probes in the various organelles, A549 cells were incubated concurrently with 10 μM coumarin dye and respective commercial fluorescent probes (Table 3) in ADMEM (supplemented with 2% FBS, 2 mM glutamine) for 20 min at 37°C. Excess probes were washed and imaged in FluoroBrite DMEM media (1 mL, supplemented with 10% FBS, 2 mM glutamine).

|
Co-localisation studies were performed using a Leica SP5 microscope equipped with a 63.0× (N.A. = 1.20) water-immersion objective lens. Fluorescence emitted from the coumarin probes was collected between 468 and 568 nm (458 nm laser excitation). Fluorescence from the respective sub-cellular localising commercial probes or plasmid are listed in Table 3, a 561 nm laser excitation was used. Images acquired were processed on Image J®. Co-localisation assessments were performed using the Coloc 2 plugin determined by the Pearson’s correlation coefficient. A R-value of less than 0.5 indicates no co-localisation, while a R-value of more than 0.7 is interpretated as co-localisation with the organelle assessed.[29,30]
Data availability
The data that support this study are available in the article and accompanying online supplementary material.
Conflicts of interest
The authors declare no conflicts of interest.
Declaration of funding
A. M. and K. G. L. acknowledge the Australian Government for a Research Training Program Scholarship. L.M.R. is grateful for past and present funding from the Australian Research Council, Cure Cancer Foundation of Australia, National Breast Cancer Foundation, and the Prostate Cancer Foundation of Australia. E. J. N. acknowledges the Australian Research Council (DP180101353, DP180101897, CE200100012) for funding.
Supplementary material
Supplementary figures, including additional confocal microscopy images, fluorescence, NMR and MS spectra are available online.
Acknowledgements
The authors acknowledge the support of the facilities and the scientific and technical assistance provided by the Australian Microscopy and Microanalysis Research Facility at the Australian Centre for Microscopy and Microanalysis (ACMM) at the University of Sydney, and thank Dr Samrat Dasgupta for providing the 1-amino-ortho-carborane derivative (4). This research was facilitated by access to Sydney Analytical, a core research facility at the University of Sydney.
References
[1] J Jonkman, CM Brown, J Biomol Tech 2015, 26, 54.| Crossref | GoogleScholarGoogle Scholar | 25802490PubMed |
[2] AL Vahrmeijer, M Hutteman, JR van der Vorst, CJH van de Velde, J V Frangioni, Nat Rev Clin Oncol 2013, 10, 507.
| Crossref | GoogleScholarGoogle Scholar | 23881033PubMed |
[3] SL Gibbs, Quant Imaging Med Surg 2012, 2, 177.
| 23256079PubMed |
[4] L Boni, G David, A Mangano, G Dionigi, S Rausei, S Spampatti, Surg Endosc 2015, 29, 2046.
| Crossref | GoogleScholarGoogle Scholar | 25303914PubMed |
[5] J Qin, Z Yang, Anal Methods 2015, 7, 2036.
| Crossref | GoogleScholarGoogle Scholar |
[6] S He, J Song, J Qu, Z Cheng, Chem Soc Rev 2018, 47, 4258.
| Crossref | GoogleScholarGoogle Scholar | 29725670PubMed |
[7] G Niu, W Liu, H Xiao, H Zhang, J Chen, Q Dai, Chem Asian J 2016, 11, 498.
| Crossref | GoogleScholarGoogle Scholar | 26558738PubMed |
[8] J Chen, W Liu, B Zhou, G Niu, H Zhang, J Wu, J Org Chem 2013, 78, 6121.
| Crossref | GoogleScholarGoogle Scholar | 23705772PubMed |
[9] X Liu, JM Cole, PG Waddell, TC Lin, J Radia, A Zeidler, J Phys Chem A 2012, 116, 727.
| Crossref | GoogleScholarGoogle Scholar | 22117623PubMed |
[10] M Musa, J Cooperwood, MO Khan, Curr Med Chem 2008, 15, 2664.
| Crossref | GoogleScholarGoogle Scholar | 18991629PubMed |
[11] Y Nagasawa, AP Yartsev, K Tominaga, AE Johnson, K Yoshihara, J Am Chem Soc 1993, 115, 7922.
| Crossref | GoogleScholarGoogle Scholar |
[12] N DiCesare, JR Lakowicz, J Biomed Opt 2002, 7, 538.
| Crossref | GoogleScholarGoogle Scholar | 12421119PubMed |
[13] A Wu, JL Kolanowski, BB Boumelhem, K Yang, R Lee, A Kaur, Chem Asian J 2017, 12, 1704.
| Crossref | GoogleScholarGoogle Scholar | 28640518PubMed |
[14] Z Guo, I Shin, J Yoon, ChemComm 2012, 48, 5956.
| Crossref | GoogleScholarGoogle Scholar |
[15] F Issa, M Kassiou, LM Rendina, Chem Rev 2011, 111, 5701.
| Crossref | GoogleScholarGoogle Scholar | 21718011PubMed |
[16] J Kahlert, CJD Austin, M Kassiou, LM Rendina, Aust J Chem 2013, 66, 1118.
| Crossref | GoogleScholarGoogle Scholar |
[17] J Kahlert, L Böhling, A Brockhinke, H-G Stammler, B Neumann, LM Rendina, Dalton Trans 2015, 44, 9766.
| Crossref | GoogleScholarGoogle Scholar | 25939355PubMed |
[18] Y Ma, W Luo, PJ Quinn, Z Liu, RC Hider, J Med Chem 2004, 47, 6349.
| Crossref | GoogleScholarGoogle Scholar | 15566304PubMed |
[19] H Takechi, Y Oda, N Nishizono, K Oda, M Machida, Chem Pharm Bull 2000, 48, 1702.
| Crossref | GoogleScholarGoogle Scholar |
[20] R Chowdhury, B Jana, A Saha, S Ghosh, K Bhattacharyya, Medchemcomm 2014, 5, 536.
| Crossref | GoogleScholarGoogle Scholar |
[21] BB Boumelhem, C Pilgrim, VE Zwicker, JL Kolanowski, JH Yeo, KA Jolliffe, J Cell Sci 2022, 135, jcs258322.
| Crossref | GoogleScholarGoogle Scholar | 34114626PubMed |
[22] ALS Cruz, E Barreto, A de, NPB Fazolini, JPB Viola, PT Bozza, Cell Death Dis 2020, 11, 105.
| Crossref | GoogleScholarGoogle Scholar | 32029741PubMed |
[23] A Li, N-J Song, BP Riesenberg, Z Li, Front Immunol 2020, 10, 1.
| Crossref | GoogleScholarGoogle Scholar |
[24] P Dozzo, RA Kasar, SB Kahl, Inorg Chem 2005, 44, 8053.
| Crossref | GoogleScholarGoogle Scholar | 16241155PubMed |
[25] J-S Wu, W-M Liu, X-Q Zhuang, F Wang, P-F Wang, S-L Tao, Org Lett 2007, 9, 33.
| Crossref | GoogleScholarGoogle Scholar | 17192078PubMed |
[26] Z Zhou, N Li, A Tong, Anal Chim Acta 2011, 702, 81.
| Crossref | GoogleScholarGoogle Scholar | 21819863PubMed |
[27] Y Nie, Y Wang, J Miao, Y Li, Z Zhang, J Organomet Chem 2015, 798, 182.
| Crossref | GoogleScholarGoogle Scholar |
[28] MA Antoniades, G V Eleftheriades, Proc 5th Eur Conf Antennas Propagation, EUCAP 2011 2011, 73, 2406.
[29] S Bolte, FP Cordelières, J Microsc 2006, 224, 213.
| Crossref | GoogleScholarGoogle Scholar | 17210054PubMed |
[30] JS Aaron, AB Taylor, T-L Chew, J Cell Sci 2018, 131, jcs211847.
| Crossref | GoogleScholarGoogle Scholar | 29439158PubMed |
† Dedicated to Prof. Glen B. Deacon, on the occasion of his 85th birthday and in recognition of his many outstanding contributions to organometallic chemistry.