

Sialyltransferase Inhibitors as Potential Anti-Cancer Agents*
Danielle Skropeta A B C , Christopher Dobie A , Andrew P. Montgomery A , Harrison Steele A , Rémi Szabo A and Haibo Yu A B
A B C , Christopher Dobie A , Andrew P. Montgomery A , Harrison Steele A , Rémi Szabo A and Haibo Yu A B
A Molecular Horizons and School of Chemistry and Molecular Bioscience, Faculty of Science, Medicine and Health, University of Wollongong, Wollongong, NSW 2522, Australia.
B Illawarra Health and Medical Research Institute, Wollongong, NSW 2522, Australia.
C Corresponding author. Email: skropeta@uow.edu.au
Australian Journal of Chemistry 74(11) 758-766 https://doi.org/10.1071/CH21195
Submitted: 12 August 2021 Accepted: 8 October 2021 Published: 22 November 2021
Abstract
Sialic acid occupies a privileged position at the terminus of the glycan chain of many cell-surface glycoconjugates. Owing to both their structure and location, charged sialic acid residues mediate numerous critical interactions in cell–cell communication including cell recognition, invasion, migration, receptor binding, and immunological responses. Sialyltransferases (STs) are the enzymes involved in the biosynthesis of sialylated glycans and are highly upregulated, up to 40–60 %, in a range of cancers, with tumour hypersialylation strongly correlated with both tumour progression and treatment resistance. Accordingly, inhibiting sialylation is currently being explored by several research groups worldwide as a potential new cancer treatment strategy. However, to progress small molecule ST inhibitors into the clinic, issues around selectivity, synthetic accessibility, and cell permeability need to be addressed. Using computationally guided design principles, we produced a leading series of ST inhibitors by replacing the cytidine nucleoside with uridine and substituting the charged phosphodiester linker with a carbamate or triazole moiety. Biological evaluation of the newly developed inhibitors was performed using commercially available human ST enzymes, with the Ki inhibition values of the lead compounds ranging from 1 to 20 µM. Compared with earlier generations of sialylation inhibitors, our inhibitors are non-toxic in a range of cell studies, with improved synthetic accessibility.
Keywords: organic synthesis, medicinal chemistry, molecular docking, enzyme inhibitors, sialyltransferase, sialic acid, cancer, metastasis.
Introduction
Aberrant glycosylation of the tumour cell surface is a hallmark of cancer contributing to tumour aggressiveness, metastasis, and drug resistance.[1–3] In particular, sialic acid located at the termini of most N- and O-glycans plays a key role in cell–cell communication, including interactions with immune cells and antibodies.[4–7] Hypersialylation in cancer is strongly linked to evasion of immunosurveillance, altered adhesion, and tumour growth and spread.[8] In humans, the biosynthesis of sialylated glycoconjugates is mediated by 20 different sialyltransferase (ST) enzymes, all of which utilise cytidine monophosphate N-acetylneuraminic acid (CMP-Neu5Ac) as the sialyl donor, defined as ST3, ST6 or ST8 subtypes based on the type of glycosidic linkage created and whether the acceptor is galactose, N-acetylgalactosamine or sialic acid (Fig. 1).[9,10]
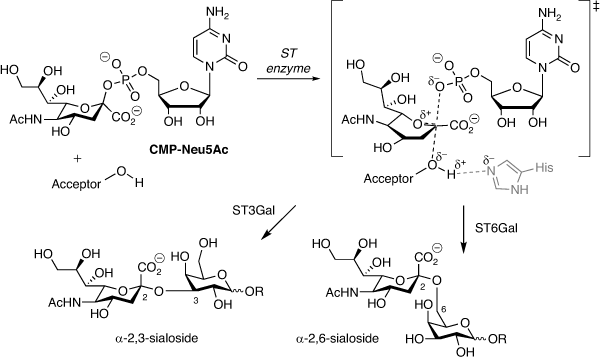
|
Tumour hypersialylation of 40–60 % occurs via upregulation of STs driven by oncogenes such as c-Myc that have been linked to increased ST transcription or by the alternative downregulation of neuraminidases.[11–16] Sialyl-mediated cancer progression occurs through multiple mechanisms including evading apoptosis and cell death via hypersialylation of the receptors such as Fas[17] and tumour necrosis factor leading to greater tumour cell survival (Fig. 2).[18] Increased cell surface sialylation leads to altered adhesion and invasion via integrin-mediated processes[19] in several cancers including colon, pancreatic, and lung cancer.[20,21] Higher expression of sialylated selectin ligands on tumours is also correlated with enhanced cancer metastasis. Relatedly, sialylated ligands for E-, P-, and L-selectins including sialyl Lewisa, sialyl Lewisx, and CD44, synthesised by the action of ST3Gal enzymes, were found to be present on the surface of circulating tumour cells.[22]

|
Many of the clinically used cancer biomarkers target tumour hypersialylation including CA19–9, the pancreatic cancer marker, and CA125 and CA15–3, sialylated mucin detection markers for ovarian and breast cancer, respectively.[23] Owing to the key role of sialylation in metastasis and tumour growth, inhibiting sialylation is a potential new cancer treatment strategy.[15,16] Herein is an account summarising our recent progress on the design, synthesis, and enzyme inhibition screening of synthetically accessible sialylation inhibitors.
Sialyltransferase Inhibitors
Natural products provide a wealth of bioactive compounds including those that inhibit protein sialylation, such as soyasaponin I and ginsenosides.[24,25] Another example is lithocholic acid and analogues, shown to reduce sialylation of α5, αv, and β1 integrins in A549 lung cancer cells, as well as markedly reducing the invasion and suppression of lung metastases from an in vivo model.[26]
In 2012, Paulson’s group reported the prodrug peracetylated 3Fax-Neu5Ac, which is metabolically converted into the active inhibitor CMP-3Fax-Neu5Ac utilising the cell’s biosynthesis pathways.[27,28] While greatly advancing the field of ST inhibitors, this strategy resulted in pan-ST inhibition and consequently in vivo testing of the compound in a murine model resulted in kidney and liver damage, underlining the need for selective ST inhibition targeting specific subtypes upregulated in cancer over pan-inhibition strategies.[29]
The most successful ST inhibitors to date are those based on mimicking the transition state of the sialylation process based on the proposed activated form of CMP-Neu5Ac (Fig. 1), as pioneered by Schmidt et al.[30,31] A 2,3-didehydro-2-deoxyneuraminic acid-based analogue (1) was one of the early inhibitors showing excellent activity (with an inhibition constant Ki = 29 nM, rat liver α-2,6-ST),[32] along with an aryl-based phosphodiester analogue 2 (Ki = 70 nM, rat liver α-2,6-ST and Ki = 19 nM against human ST6Gal I; Fig. 3).[33,34] The latter compound (2) is still one of the most potent agents reported and widely used as a parent compound in further ST inhibitor development. Several CMP-based ST inhibitors have been explored with a range of activities and selectivities,[35] including the cyclopentyl derivative 3 (Ki = 28 nM against human ST6Gal I)[34] and benzamide derivative 4 (Ki = 16 nM against human ST6Gal I; Fig. 3).[36]
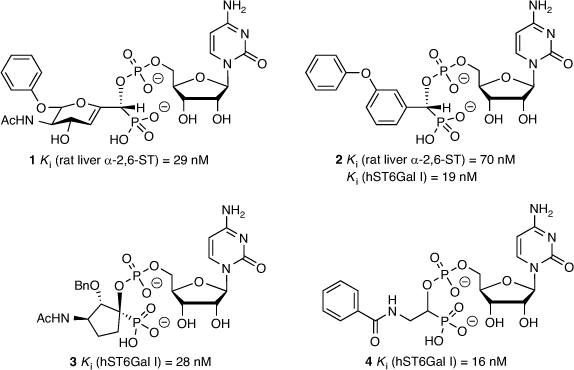
|
While the phosphodiester-based ST inhibitors show potent nanomolar activity, there are potential bioavailability issues with these charged compounds in terms of cell penetration and potential hydrolysis in vivo by phosphatase enzymes.[37] In order to improve on the structures, we replaced the phosphodiester moiety with a neutral linker, which has the effect of both reducing the charge on the inhibitors and simplifying their overall synthesis.[37–41] The first generation of inhibitors we explored included a neutral carbamate as the linker, prepared using alkoxy carbonylating agents as reported in Montgomery et al. (Fig. 4).[42]
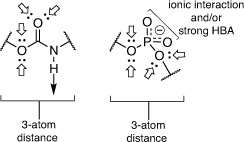
|
The carbamate functionality is widely used as a peptide bond isostere with good chemical and proteolytic stability and cell permeability.[42] Importantly for us, it invokes comparable hydrogen-bonding interactions with the phosphodiester, including a similar distance between the nucleoside component and the sialyl mimic (Fig. 4). We used molecular docking and molecular dynamics to show that carbamate-based analogues participate in similar interactions within the hST6Gal I active site (Protein Database PDB 4JS2) to the analogous phosphodiester-linked compounds. Free energy perturbation (FEP) studies also showed how the derivatives can emulate the phosphodiester linker with similar binding affinities via an enthalpy–entropy compensation.
While the cytidine moiety is often considered essential for ST inhibitory activity, we explored replacing this with uridine as it would improve synthetic access and yield ST inhibitors in fewer steps overall (Fig. 5). Gratifyingly, FEP calculations comparing analogous cytidine and uridine-based carbamate inhibitors with hST6Gal I (PDB ID: 4JS2)[43] gave a ΔΔGb value of −1.2 ± 0.3 kcal mol−1 (1 kcal mol−1 = 4.186 kJ mol−1) for the transformation of cytidine to uridine, suggesting this replacement would not impact binding to hST6Gal I to any significant extent, and could even be preferable.
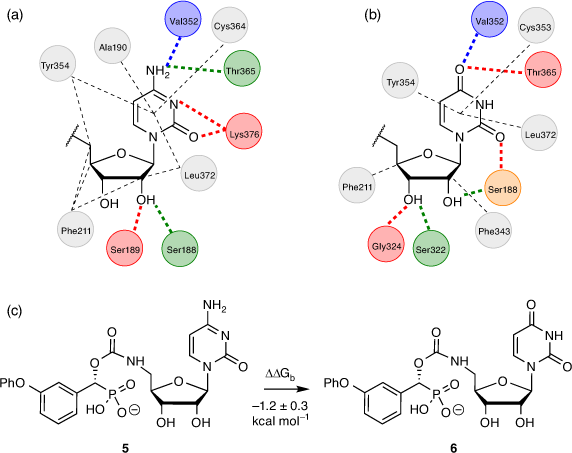
|
Following from this computational study, a library of carbamate-linked uridyl compounds were prepared for biological evaluation, along with a related set of cytidine-based compounds. The desired compounds were synthesised in seven steps overall from two key building blocks, those being a library of α-hydroxyphosphonates such as 7 as the sialyl mimic and the alloc-protected form of 5′-amino-5′-deoxynucleoside (8) (Scheme 1). The α-hydroxyphosphonates were produced from the corresponding aldehydes that were reacted with dibenzyl phosphite in the presence of triethylamine to give a range of desired α-hydroxyphosphonates in high yields as racemic mixtures, using benzyl protecting groups on the phosphonate for ease of removal later under catalytic hydrogenation conditions (Scheme 1).[44,45]
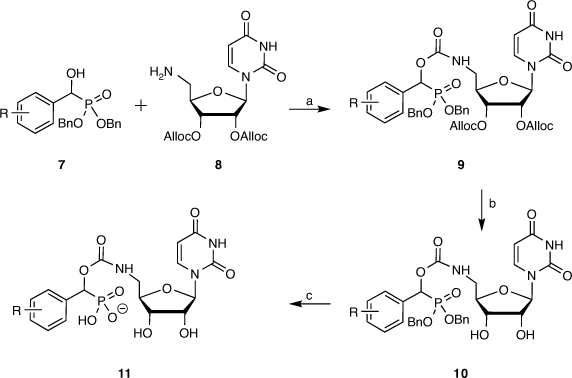
|
Selective azidonation of the primary alcohol of uridine via a Mitsunobu reaction, followed by diol protection and reduction of the 5′-azide moiety gave the alloc-protected 5′-aminouridine 8 in an overall yield of 61 % from uridine (Scheme 1), as described by Montgomery et al.[44] The α-hydroxyphosphonates 7 were coupled with the protected uridyl 8 with 4-nitrophenylchloroformate to give the target compounds 9 in good yields. This was followed by alloc removal to give the partially deprotected compound 10, followed by debenzylation and separation of the diastereomers by preparative reverse-phase HPLC. In the final step, the compounds underwent ion-exchange over IR120 resin to give the final compound 11 as a sodium salt (Scheme 1). Overall, the synthesis is seven steps from available starting materials and readily scalable for further biological evaluation and enzyme crystallography studies.
The analogous cytidine derivative was prepared from α-hydroxyphosphonate 12 and benzyloxycarbonyl (Cbz)-protected 5′-amino-5′-deoxycytidine 13. Benzyloxycarbonyl groups were used to facilitate global deprotection, to give the coupled product 14 and cytidine-based inhibitor 15 after deprotection, purification, and ion exchange (Scheme 2).[44]
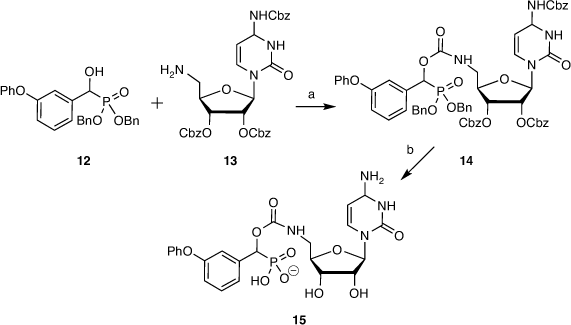
|
ST inhibition is determined using screening tools that range from traditional HPLC-based assays relying on quantification of transferred sialic acid onto a UV-labelled acceptor to more rapid microplate assays.[46] The different assays employed across the field use varying incubation times, enzyme purity, and detection methods (e.g. HPLC, mass spectrometry, fluorescence). They are also performed on both bacterial and non-human mammalian ST enzymes, with markedly different sequence homology preventing a comparison of inhibition values across studies.
We determined ST inhibitory activities using the highly sensitive CMP-Glo™ assay from Promega as detailed by Das et al.[47] CMP-Neu5Ac was used as the sialyl donor and unlabelled N-acetyllactosamine (LacNAc) as the acceptor glycan. We generated a series of 26 different carbamate-linked compounds in total, as described in Schemes 1 and 2, and tested these against recombinant hST6Gal I. All 26 were found to show some level of inhibitory activity, with the five lead compounds exhibiting Ki values in the range of 1–20 μM including 4-fluorophenyl (16), 3-fluorophenyl (17), 3-propoxyphenyl (18), 3-cyclopentoxyphenyl (19), and 2-benzothiophene (20) derivatives (Fig. 6).
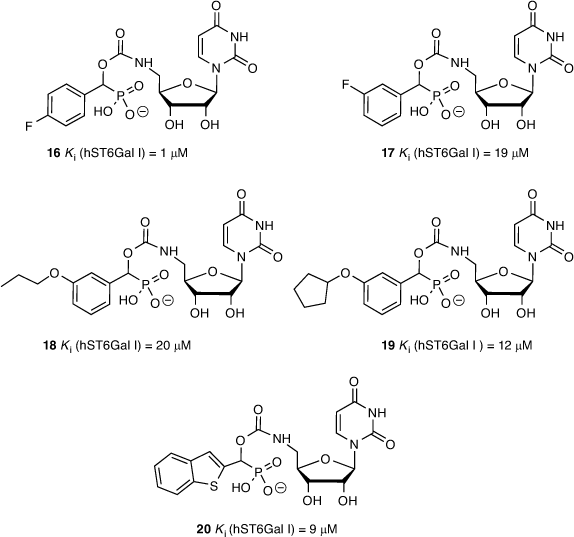
|
From the biological screening, the uridine-based compounds were more active than the analogous cytidine derivative (15), validating the earlier FEP calculations. While the overwhelming majority of ST inhibitors reported to date are cytidine-based derivatives, these results open up the opportunity to explore further alternatives to cytidine. The activity of the inhibitors in the low micromolar range is also comparable with other reported ST inhibitors. While less active than the parent phosphodiester compound (2), this series of carbamate-based compounds (11, 16–20) benefit from greater synthetic accessibility and reduced overall charge. To further explore phosphodiester replacements, we next prepared several ether-linked 1,2,3-triazole compounds, which gave us the opportunity to explore the result of increasing the linker length on ST inhibition, as described by Dobie et al.[45]
To this end, target molecules such as 24 were prepared by coupling a library of α-azidophosphonates with 5′-O-propargyluridine, the latter prepared in high yield by propargylation of acetonide-protected uridine followed by deprotection with catalytic indium triflate. The α-azidophosphonates (21) were synthesised by performing a Mitsunobu reaction on our earlier library of α-hydroxyphosphonates (7, 12). The two key azide 21 and alkyne 22 building blocks were reacted via a copper-catalysed azide–alkyne cycloaddition (CuAAC) using CuI generated in situ from Cu(OAc)2 and sodium ascorbate in 1:1 THF/H2O. Next, the partially protected triazole compounds (23) were debenzylated via catalytic hydrogenation over a Pd/C catalyst. The fully deprotected compounds (24) were purified by reverse-phase HPLC purification and transformed into sodium salts (Scheme 3).[45]
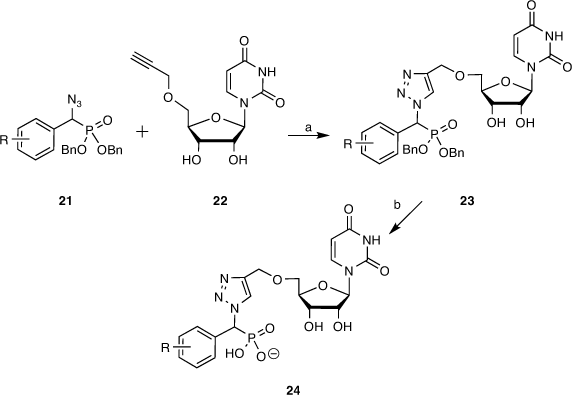
|
Fourteen diastereomeric pairs of the triazole-linked ST inhibitors of compound type 24 were evaluated against ST3/ST6Gal I, along with previously described reference compounds. From this series, the 3-phenoxy (25), 3-phenoxy-4-fluoro (26), and 3-(1,1,2,2-tetrafluoro)ethoxy (27) compounds were the most promising, showing activity at low micromolar concentrations (Fig. 7). All compounds were either mildly or entirely inactive against human ST3Gal I even when tested at 100 µM. The compounds from the current study showed reduced activity against hST3Gal I, which correlated with the reduced binding affinity observed against a human ST3Gal I homology model we developed based on the published structure for porcine ST3Gal, with no crystal structure available for human ST3Gal I at the time of writing (Fig. 8). Interestingly, the most active compounds against hST6Gal I also showed the highest activity against hST3Gal I, albeit a low range (e.g. 28–49 % inhibition at 100 uM), implying selectivity towards hST6Gal I.
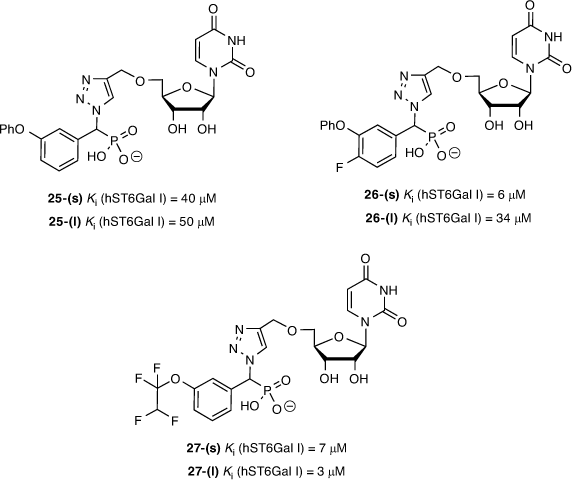
|

|
The differences in the NMR chemical shifts of the diastereomers were not large enough to reliably use computational methods to assign each stereoisomer. As such, the diastereomers are described here as (s) and (l), representing short and long HPLC retention times.[48,49] As previously reported, there is surprisingly little difference in activity between the diastereomers of the most active inhibitors against hST6Gal I, such as 27-(s) and 27-(l). This could be due to the size of the binding pocket of ST6Gal I, which needs to be large enough to accommodate both CMP-Neu5Ac and an acceptor molecule (see Fig. 8). Overall, the most potent inhibitor we found is the tetrafluoroethoxy derivative 27-(l) (Ki = 3.4 ± 0.6 µM; hST6Gal I), where the polyfluorine moiety may participate in similar binding interactions to the polyol chain on the natural sialic acid donor.[50] Other reported inhibitors with activity against ST enzymes in a similar range of 1–10 µM have shown in vivo efficacy in a murine cancer model, inhibiting tumour growth, angiogenesis, and metastasis of MDA-MB-231 breast cancer cells.[51]
There are a host of promising opportunities for targeting sialic-based interactions in cancer, including an exciting approach by Bertozzi et al. to reduce cancer cell sialylation using a sialidase from Vibrio cholera conjugated to Trastuzumab, a HER2-targeting antibody.[52] But there are also challenges including non-specific protein sialylation, remodelling of cell-surface glycans by extracellular STs,[53] and understanding the effects of diet, stress, and aging on protein sialylation.[54–57] While ST3Gal I, ST6Gal I, ST8Sia II, and ST8Sia IV are widely studied in cancer, some ST subtypes are still relatively unexplored. For ST inhibitors to advance to the clinic, it is imperative that they are selective for specific ST subtypes to limit any off-target effects. Until recently, crystal structure data were only available for three human STs (ST6Gal I,[58] ST8Sia III,[59] and ST6GalNAc I,[60]) along with porcine ST3Gal I.[61] However, the availability of predicted 3D structures through the artificial intelligence-based AlphaFold Protein Structure database has unlocked the chest, enabling wider evaluation of inhibitor selectivity across all STs subtypes.[62]
Selectivity is the key to moving ST inhibitors into preclinical models. The metabolic pan-ST inhibitor peracetylated 3Fax-Neu5Ac is commercially available and widely used in cell studies on sialylation; however, its clinical use is limited by toxicity issues. To address this, Bull et al. selectively delivered peracetylated 3Fax-Neu5Ac encapsulated into tumour-targeting nanoparticles, successfully reducing metastatic spread in a lung cancer model in mice.[63] To complement these strategies, the development of non-toxic, subtype-specific ST inhibitors is much needed.
Conclusions
Herein, we have described the design, computational modelling, synthesis, and biological evaluation of our leading series of ST inhibitors. All our compounds are non-toxic (up to 200 uM) based on cell viability assays against various cancer cell lines. This is a positive outcome from the drug development perspective as inhibiting sialylation is expected to impact cell adhesion and migration properties rather than cell viability. Alongside selectivity, other important elements to address in order to further progress sialylation inhibitors as potential anti-cancer agents are membrane permeability and bioavailability. Currently, we are aiming to improve the cell permeability of our inhibitors further using neutral analogues along with masking the charge through a prodrug approach. In collaboration, we are screening our compounds against a broader panel of ST subtypes in clinically relevant models of cancer metastasis and chemo- and radiotherapy resistance.
Data Availability Statement
Data sharing is not applicable as no new data were generated or analysed during this study.
Conflicts of Interest
The authors declare no conflicts of interest.
Declaration of Funding
We acknowledge an Australian Government PhD Scholarship for A.M. We also thank Phil Clingan, Maxine Stewart and Illawarra Cancer Carers for support, along with an Australian Research Council’s Discovery Project (DP170101773), Future Fellowship for H.Y. (FT110100034), Australian Institute of Nuclear Science (AINSE) funding, and NCI National Facility and Computational Merit Allocation Scheme.
References
[1] J. Munkley, D. J. Elliott, Oncotarget 2016, 7, 35478.| Crossref | GoogleScholarGoogle Scholar | 27007155PubMed |
[2] H. Läubli, L. Borsig, Front. Immunol. 2019, 10, 2120.
| Crossref | GoogleScholarGoogle Scholar | 31552050PubMed |
[3] K. F. Boligan, C. Mesa, L. E. Fernandez, S. von Gunten, Cell. Mol. Life Sci. 2015, 72, 1231.
| Crossref | GoogleScholarGoogle Scholar | 25487607PubMed |
[4] H. Steele, A. J. Tague, D. Skropeta, Curr. Med. Chem. 2021, 28, 5251.
| 33593248PubMed |
[5] M. Cohen, A. Varki, OMICS 2010, 14, 455.
| Crossref | GoogleScholarGoogle Scholar | 20726801PubMed |
[6] A. Varki, Trends Mol. Med. 2008, 14, 351.
| Crossref | GoogleScholarGoogle Scholar | 18606570PubMed |
[7] D. Skropeta, Bioorg. Med. Chem. 2009, 17, 2645.
| Crossref | GoogleScholarGoogle Scholar | 19285412PubMed |
[8] O. M. Pearce, H. Läubli, Glycobiology 2016, 26, 111.
| Crossref | GoogleScholarGoogle Scholar | 26518624PubMed |
[9] A. Harduin-Lepers, V. Vallejo-Ruiz, M. A. Krzewinski-Recchi, B. Samyn-Petit, S. Julien, P. Delannoy, Biochimie 2001, 83, 727.
| Crossref | GoogleScholarGoogle Scholar | 11530204PubMed |
[10] R. Szabo, D. Skropeta, Med. Res. Rev. 2017, 37, 219.
| Crossref | GoogleScholarGoogle Scholar | 27678392PubMed |
[11] T. Miyagi, K. Takahashi, K. Hata, K. Shiozaki, K. Yamaguchi, Glycoconj. J. 2012, 29, 567.
| Crossref | GoogleScholarGoogle Scholar | 22644327PubMed |
[12] E. Rodrigues, M. S. Macauley, Cancers 2018, 10, 207.
| Crossref | GoogleScholarGoogle Scholar |
[13] C. Büll, M. A. Stoel, M. H. den Brok, G. J. Adema, Cancer Res. 2014, 74, 3199.
| Crossref | GoogleScholarGoogle Scholar | 24830719PubMed |
[14] A. Peixoto, M. Relvas-Santos, R. Azevedo, L. L. Santos, J. A. Ferreira, Front. Oncol. 2019, 9, 380.
| Crossref | GoogleScholarGoogle Scholar | 31157165PubMed |
[15] C. Dobie, D. Skropeta, Br. J. Cancer 2021, 124, 76.
| Crossref | GoogleScholarGoogle Scholar | 33144696PubMed |
[16] W. H. D. Bowles, T. M. Gloster, Front. Mol. Biosci. 2021, 8, 705133.
| Crossref | GoogleScholarGoogle Scholar |
[17] A. F. Swindall, S. L. Bellis, J. Biol. Chem. 2011, 286, 22982.
| Crossref | GoogleScholarGoogle Scholar | 21550977PubMed |
[18] A. T. Holdbrooks, C. M. Britain, S. L. Bellis, J. Biol. Chem. 2018, 293, 1610.
| Crossref | GoogleScholarGoogle Scholar | 29233887PubMed |
[19] Y. Liu, D. Pan, S. L. Bellis, Y. Song, Proteins 2008, 73, 989.
| Crossref | GoogleScholarGoogle Scholar | 18536010PubMed |
[20] R. T. Almaraz, Y. Tian, R. Bhattarcharya, E. Tan, S.-H. Chen, M. R. Dallas, et al. Mol. Cell. Proteomics 2012, 11, M112.017558-1.
| Crossref | GoogleScholarGoogle Scholar |
[21] E. C. Seales, G. A. Jurado, B. A. Brunson, J. K. Wakefield, A. R. Frost, S. L. Bellis, Cancer Res. 2005, 65, 4645.
| Crossref | GoogleScholarGoogle Scholar | 15930282PubMed |
[22] J. G. Rodrigues, M. Balmaña, J. A. Macedo, J. Poças, Â. Fernandes, J. C. M. de-Freitas-Junior, et al. Cell. Immunol. 2018, 333, 46.
| Crossref | GoogleScholarGoogle Scholar | 29576316PubMed |
[23] P. M. Drake, W. Cho, B. Li, A. Prakobphol, E. Johansen, N. L. Anderson, et al. Clin. Chem. 2010, 56, 223.
| Crossref | GoogleScholarGoogle Scholar | 19959616PubMed |
[24] W. Huang, L. Sun, B. Wang, Y. Ma, D. Yao, W. Han, et al. Z. Natforsch. C: J. Biosci. 2020, 75, 41.
| Crossref | GoogleScholarGoogle Scholar |
[25] K.-H. Chang, L. Lee, J. Chen, W.-S. Li, Chem. Commun. 2006, 629.
[26] C.-H. Chiang, C.-H. Wang, H.-C. Chang, S. V. More, W.-S. Li, W.-C. Hung, J. Cell. Physiol. 2010, 223, 492.
| 20112294PubMed |
[27] C. Büll, T. J. Boltje, M. Wassink, A. M. A. de Graaf, F. L. van Delft, M. H. den Brok, et al. Mol. Cancer Ther. 2013, 12, 1935.
| Crossref | GoogleScholarGoogle Scholar | 23974695PubMed |
[28] C. D. Rillahan, A. Antonopoulos, C. T. Lefort, R. Sonon, P. Azadi, K. Ley, et al. Nat. Chem. Biol. 2012, 8, 661.
| Crossref | GoogleScholarGoogle Scholar | 22683610PubMed |
[29] M. S. Macauley, B. M. Arlian, C. D. Rillahan, P.-C. Pang, N. Bortell, M. C. G. Marcondes, et al. J. Biol. Chem. 2014, 289, 35149.
| Crossref | GoogleScholarGoogle Scholar | 25368325PubMed |
[30] F. Amann, C. Schaub, B. Müller, R. R. Schmidt, Chem. – Eur. J. 1998, 4, 1106.
| Crossref | GoogleScholarGoogle Scholar |
[31] B. Muller, C. Schaub, R. R. Schmidt, Angew. Chem. Int. Ed. 1998, 37, 2893.
| Crossref | GoogleScholarGoogle Scholar |
[32] G. Dufner, R. Schworer, B. Muller, R. R. Schmidt, Eur. J. Org. Chem. 2000, 1467.
| Crossref | GoogleScholarGoogle Scholar |
[33] D. Skropeta, R. Schworer, T. Haag, R. R. Schmidt, Glycoconj. J. 2004, 21, 205.
| Crossref | GoogleScholarGoogle Scholar | 15486453PubMed |
[34] W. Li, Y. Niu, D.-C. Xiong, X. Cao, X.-S. Ye, J. Med. Chem. 2015, 58, 7972.
| Crossref | GoogleScholarGoogle Scholar | 26406919PubMed |
[35] D. Skropeta, R. Schworer, R. R. Schmidt, Bioorg. Med. Chem. Lett. 2003, 13, 3351.
| Crossref | GoogleScholarGoogle Scholar | 12951124PubMed |
[36] J. Guo, W. Li, W. Xue, X.-S. Ye, J. Med. Chem. 2017, 60, 2135.
| Crossref | GoogleScholarGoogle Scholar | 28165727PubMed |
[37] R. Kumar, R. Nasi, M. Bhasin, N. Huan Khieu, M. Hsieh, M. Gilbert, et al. Carbohydr. Res. 2013, 378, 45.
| Crossref | GoogleScholarGoogle Scholar | 23374752PubMed |
[38] C. Dobie, A. P. Montgomery, R. Szabo, D. Skropeta, H. Yu, J. Mol. Recognit. 2018, 31, e2684.
| Crossref | GoogleScholarGoogle Scholar |
[39] A. Montgomery, R. Szabo, D. Skropeta, H. Yu, J. Mol. Recognit. 2016, 29, 210.
| Crossref | GoogleScholarGoogle Scholar | 26669681PubMed |
[40] A. P. Montgomery, D. Skropeta, H. Yu, Sci. Rep. 2017, 7, 14428.
| Crossref | GoogleScholarGoogle Scholar | 29089525PubMed |
[41] A. P. Montgomery, K. Xiao, X. Wang, D. Skropeta, H. Yu, Adv. Protein Chem. Struct. Biol. 2017, 109, 25.
| Crossref | GoogleScholarGoogle Scholar | 28683920PubMed |
[42] A. K. Ghosh, M. Brindisi, J. Med. Chem. 2015, 58, 2895.
| Crossref | GoogleScholarGoogle Scholar | 25565044PubMed |
[43] B. Kuhn, J. Benz, M. Greif, A. M. Engel, H. Sobek, M. G. Rudolph, Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1826.
| Crossref | GoogleScholarGoogle Scholar | 23999306PubMed |
[44] A. P. Montgomery, C. Dobie, R. Szabo, L. Hallam, M. Ranson, H. Yu, et al. Bioorg. Med. Chem. 2020, 28, 115561.
| Crossref | GoogleScholarGoogle Scholar | 32616185PubMed |
[45] C. Dobie, A. P. Montgomery, R. Szabo, H. Yu, D. Skropeta, RSC Med. Chem. Advance article 2021,
[46] M. Noel, P. A. Gilormini, V. Cogez, C. Lion, C. Biot, A. Harduin-Lepers, et al. Bioconjug. Chem. 2018, 29, 3377.
| Crossref | GoogleScholarGoogle Scholar | 30192128PubMed |
[47] D. Das, M. T. C. Walvoort, V. Lukose, B. Imperiali, Sci. Rep. 2016, 6, 33412.
| Crossref | GoogleScholarGoogle Scholar | 27624811PubMed |
[48] S. G. Smith, J. M. Goodman, J. Am. Chem. Soc. 2010, 132, 12946.
| Crossref | GoogleScholarGoogle Scholar | 20795713PubMed |
[49] L. Grauso, R. Teta, G. Esposito, M. Menna, A. Mangoni, Nat. Prod. Rep. 2019, 36, 1005.
| Crossref | GoogleScholarGoogle Scholar | 31166350PubMed |
[50] E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly, N. A. Meanwell, J. Med. Chem. 2015, 58, 8315.
| Crossref | GoogleScholarGoogle Scholar | 26200936PubMed |
[51] C.-W. Fu, H.-E. Tsai, W.-S. Chen, T.-T. Chang, C.-L. Chen, P.-W. Hsiao, et al. J. Med. Chem. 2021, 64, 527.
| Crossref | GoogleScholarGoogle Scholar | 33371679PubMed |
[52] H. Xiao, E. C. Woods, P. Vukojicic, C. R. Bertozzi, Proc. Natl. Acad. Sci. USA 2016, 113, 10304.
| Crossref | GoogleScholarGoogle Scholar | 27551071PubMed |
[53] C. T. Manhardt, P. R. Punch, C. W. L. Dougher, J. T. Y. Lau, J. Biol. Chem. 2017, 292, 13514.
| Crossref | GoogleScholarGoogle Scholar | 28717006PubMed |
[54] Q. Z. Lin, R. X. Yin, T. Guo, J. Wu, J. Q. Sun, S. W. Shen, et al. Lipids Health Dis. 2014, 13, 123.
| Crossref | GoogleScholarGoogle Scholar | 25086711PubMed |
[55] J. Song, C. Xue, J. S. Preisser, D. W. Cramer, K. L. Houck, G. Liu, et al. PLoS One 2016, 11, e0160757.
| Crossref | GoogleScholarGoogle Scholar | 27584569PubMed |
[56] K. K. Leung, G. M. Wilson, L. L. Kirkemo, N. M. Riley, J. J. Coon, J. A. Wells, Proc. Natl. Acad. Sci. USA 2020, 117, 7764.
| Crossref | GoogleScholarGoogle Scholar | 32205440PubMed |
[57] K. W. Moremen, M. Tiemeyer, A. V. Nairn, Nat. Rev. Mol. Cell Biol. 2012, 13, 448.
| Crossref | GoogleScholarGoogle Scholar | 22722607PubMed |
[58] B. Kuhn, J. Benz, M. Greif, A. M. Engel, H. Sobek, M. G. Rudolph, Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1826.
| Crossref | GoogleScholarGoogle Scholar | 23999306PubMed |
[59] G. Volkers, L. J. Worrall, D. H. Kwan, C. C. Yu, L. Baumann, E. Lameignere, et al. Nat. Struct. Mol. Biol. 2015, 22, 627.
| Crossref | GoogleScholarGoogle Scholar | 26192331PubMed |
[60] K. W. Moremen, A. Ramiah, M. Stuart, J. Steel, L. Meng, F. Forouhar, et al. Nat. Chem. Biol. 2018, 14, 156.
| Crossref | GoogleScholarGoogle Scholar | 29251719PubMed |
[61] F. V. Rao, J. R. Rich, B. Rakic, S. Buddai, M. F. Schwartz, K. Johnson, et al. Nat. Struct. Mol. Biol. 2009, 16, 1186.
| Crossref | GoogleScholarGoogle Scholar | 19820709PubMed |
[62] J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, et al. Nature 2021, 596, 583.
| Crossref | GoogleScholarGoogle Scholar | 34265844PubMed |
[63] C. Bull, T. J. Boltje, E. A. W. van Dinther, T. Peters, A. M. A. de Graaf, J. H. W. Leusen, et al. ACS Nano 2015, 9, 733.
| Crossref | GoogleScholarGoogle Scholar | 25575241PubMed |
* Danielle Skropeta was the recipient of the 2020 Margaret Sheil Award.