

Spectroscopic and Modelling Analysis on the Interaction of 3′-Azidodaunorubicin Semicarbazone with ctDNA
Fengling Cui A B , Jianhua Jin A , Xiaoqing Niu A , Qingfeng Liu A and Guisheng Zhang A BA College of Chemistry and Chemical Engineering, Key Laboratory of Green Chemical Media and Reactions, Ministry of Education, Henan Normal University, Xinxiang 453007, China.
B Corresponding authors. Email: fenglingcui@hotmail.com; zgs@htu.cn
Australian Journal of Chemistry 67(2) 234-240 https://doi.org/10.1071/CH13372
Submitted: 16 July 2013 Accepted: 16 September 2013 Published: 17 October 2013
Abstract
The synthesis and characterisation of a new anthracycline, 3′-azidodaunorubicin semicarbazone (ADNRS) is reported. The interaction between ADNRS and calf thymus DNA (ctDNA) was investigated by absorption and fluorescence spectroscopy in combination with melting temperature (Tm) curves and molecular modelling in physiological buffer (pH 7.4). Evidence indicates that ADNRS binds in the groove of ctDNA and the fluorescence quenching mechanism is a static quenching type. Calculated thermodynamic parameters show that hydrophobic interactions may play a predominant role in the binding. Furthermore, molecular modelling results corroborate the spectroscopic investigations.
Introduction
In recent years, interactions between small molecules and DNA have attracted increasing interest of many research groups with the aim of understanding the drug–DNA interactions and the design of novel DNA–targeted drugs.[1] Circular dichroism spectroscopy,[2] molecular modelling,[3] isothermal titration calorimetry,[4] electrochemical,[5] and spectroscopic[6] methods have been applied as effective techniques to study these interactions. It is well known that DNA plays a major role in life processes and is quite often the main cellular target.[7] Many anticancer drugs are known to interact with DNA to exert their biological activities, such as anthracycline.
As a clinically useful anthracycline antibiotic for cancer therapy, daunorubicin can rapidly enter the nucleus of cells and bind with high affinity to DNA by non-covalent intercalation between base pairs which leads to inhibition of the synthesis of DNA and RNA.[8] However, several side effects limit its use such as cytotoxicity, myelosuppression, and development of multidrug resistance. Intense efforts in the synthesis of analogues have been made to obtain better therapeutic drugs.[9] In our previous work, a daunorubicin analogue, 3′-azido-3′-deaminodaunorubicin, was synthesised by transforming the 3′-amino group of daunorubicin to an azido group. The bioactivity and the binding to protein/DNA of the compound were investigated.[10–12] This lead compound exhibited potent anticancer activity in both drug-sensitive and drug-resistant leukemia cells, with a 25-fold lower drug resistance index than the parent compound, daunorubicin.
Proper understanding of the mechanism of drugs binding to their targets in cell is a fundamental requirement to develop new drug therapy regimens. In this paper, 3′-azidodaunorubicin semicarbazone (ADNRS, Fig. 1), a hybrid of the lead compound with a semicarbazide group, was synthesised. Its binding behaviours to ctDNA were investigated using ultraviolet (UV) and fluorescence spectroscopy as well as modelling methods under simulated physiological conditions. The binding mode and thermodynamic characteristics were explored. Molecular modelling results further confirmed the spectroscopic results and illustrated the binding mode. This combination of spectroscopic and molecular modelling methods might be widely used in the investigation on the toxic interaction of small molecular pollutants and drugs with biomacromolecules, which might contribute to understanding the molecular mechanism of toxicity or side effects in vivo.
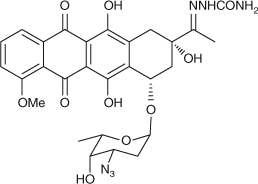
|
Materials and Methods
Materials
UV absorption spectra were recorded on a TU-1810 spectrophotometer (Puxi Analytic Instrument Ltd, Beijing, China) using a 1.0 cm cell. Fluorescence spectra were measured on a Cary Eclipse fluorescence spectrophotometer (Varian, USA) equipped with a thermostatic bath and a 1.0 cm quartz cuvette. pH measurements were carried out with a pFS-80 pF-meter (Shanghai Dazhong Analytical Instrument Plant, Shanghai, China) with a combined glass-calomel electrode.
Reagents
Preparation procedure for ADNRS: a drop of acetic acid was added to a mixture of ADNR (27.7 mg, 0.05 mmol) and hydrazinecarboxamide monohydrochloride (16.7 mg, 0.15 mmol) in methanol (3 mL) and the mixture was stirred overnight. TLC monitoring indicated the reaction was complete. The solvent was removed, and the resulting residue was diluted with saturated aqueous Na2CO3 solution and extracted with CH2Cl2 (3 × 5 mL). The combined extracts were dried with Na2SO4 and concentrated. The crude product was purified on a column of silica gel to give the pure product ADNRS as a red solid (23.8 mg, 78 %). δH (400 MHz, [D6]DMSO) 13.99 (1H, s), 13.22 (1H, s), 9.08 (1H, s), 7.85–7.79 (2H, m), 7.56 (1H, d, J 8.0), 5.31 (1H, d, J 2.4), 5.19 (1H, d, J 4.0), 5.12 (1H, s), 4.85 (1H, t, J 7.2), 4.07 (1H, d, J 4.0), 3.93 (1H, s), 3.61 (1H, d, J 4.0 Hz), 3.50–3.46 (1H, m), 3.13 (1H, d, J 16.0), 2.77 (1H, d, J 16.0), 2.32 (1H, dd, J 12.0, 6.0 Hz), 3.73 (1H, s), 3.64–3.62 (1H, m), 3.40 (1H, d, J 13.6), 2.14–2.04 (2H, m), 1.86 (3H, s), 1.69 (1H, dd, J 11.6, 3.6 Hz ), 1.15 (3H, d, J 6.0); δC(100 MHz, [D6]DMSO) 186.2, 186.1, 160.6, 157.3, 156.3, 154.2, 150.7, 136.0, 135.9, 135.7, 134.5, 119.8, 119.4, 118.8, 110.4, 99.6, 72.1, 71.2, 68.9, 67.0, 56.5, 55.7, 33.3, 28.1, 16.9, 11.1. m/z (HR-MS ESI) 611.2101; [M+H]+ requires 611.2096.
A stock solution of ADNRS was prepared to a concentration of 8.7 × 10–5 mol L–1. ctDNA (Sigma Chemical Co., St Louis, MO) was used without further purification. Stock solutions of ctDNA were prepared by dissolving an appropriate amount of DNA in doubly distilled water to a final concentration of 5.1 × 10–4 mol L–1 and stored at 4°C. The concentration of DNA was determined spectrophotometrically using the extinction coefficient value of 6600 L mol–1 cm–1 at 260 nm.[12] The ratio of the absorbance at 260/280 nm was found to be >1.8, which indicated that DNA was sufficiently free from any contamination.[13] A pH value of the media of 7.4 was controlled with a Tris-HCl (Tris = tris(hydroxymethyl)aminomethane) buffer solution. All reagents were of analytical reagent grade and doubly distilled water was used throughout.
Methods
Fluorescence Spectra
All fluorescence measurements were recorded on a Varian Cary Eclipse spectrofluorimeter coupled a 1.0 cm path length fluorescence cuvette. ADNRS, variable concentrations of ctDNA, and 2.0 mL Tris-HCl buffer (pH 7.4) were added to a 10 mL volumetric flask, and diluted with doubly distilled water. Both excitation and emission slit widths were set at 5 nm. An excitation wavelength of 254 nm was chosen and the emission spectra were recorded from 520 to 750 nm.
Electronic Absorption Spectra
Absorption spectra were recorded from 200 to 260 nm, using 1 cm path length cuvettes. Absorbance measurements were performed at a fixed concentration of ADNRS while varying the DNA concentration. In order to eliminate the absorbance of DNA, an equal amount of DNA was added to both sample solution and reference cuvette, and the absorbance was recorded after each successive addition of DNA.
Denaturation of DNA
Thermally denatured ctDNA was obtained by incubating double-stranded ctDNA (dsDNA) in a boiling water bath for 30 min and then rapidly cooling in an ice-water bath for 10 min.[14] Comparison of the quenching effects of single stranded DNA (ssDNA) and dsDNA was performed by adding small aliquots of a concentrated ssDNA or dsDNA solution respectively to a solution of ADNRS at a constant concentration.
Fluorescence Quenching Studies
Potassium iodide was used as the quencher for iodide quenching experiments to determine the relative accessibilities of the free and bound ADNRS.
Effect of Ionic Strength
A series of assay solutions containing various amounts of NaCl and a fixed amount of ADNRS–ctDNA and ADNRS were prepared to measure the fluorescence intensity.
DNA Melting Studies
DNA melting experiments were conducted by monitoring the absorption intensity of ctDNA at 260 nm in the temperature range of 30 to 100°C both in the presence and absence of ADNRS. The melting temperature (Tm) was taken as the midpoint of the hyperchromic transition.
Molecular Modelling Studies
A binding model between ADNRS and ctDNA was generated by Sybyl 6.9. The FlexX module of the Sybyl 6.9 suite was applied to calculate the interaction mode of ADNRS binding to DNA. With the help of the Tripos force field, the geometry of ADNRS was subsequently optimised to minimal energy using Gasteiger–Huckel charges. The crystal structure of B-DNA used for docking was extracted from the Protein Data Bank, identifier 453D. Before the docking run, water was removed from the DNA PDB file and essential polar hydrogen atoms and Gasteiger charges were added. A maximum of ten conformers was considered for the ligand. The conformer with the lowest binding free energy was used for further analysis. During the docking process, the ligand atoms were allowed to move within a specified region to achieve the energy conformation and the radius around the solvent molecules was set to 12 Å. DNA was enclosed in a grid having 0.375 Å spacing. Other miscellaneous parameters were set as default. The output from Sybyl was rendered with PyMol.
Results and Discussion
Fluorescence Studies
Fluorescence Quenching Studies
The fluorescence emission spectra of ADNRS in the absence and presence of DNA are shown in Fig. 2. It was obvious that free ADNRS displayed an excitation maximum at 254 nm and an emission maximum at 585 nm. When increasing amounts of DNA was added, a regular decrease resulted in the fluorescence intensity of ADNRS but no shift of fluorescence maximum emission took place, which indicated that the binding of ADNRS to DNA indeed took place.
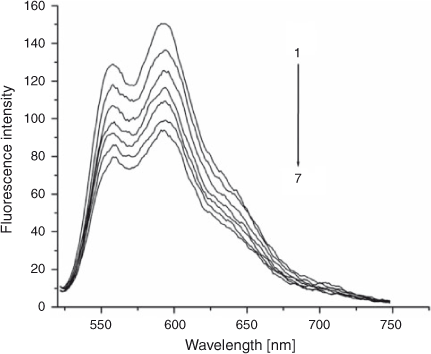
|
Fluorescence quenching includes dynamic quenching and static quenching. If the quenching mechanism is dynamic, it should follow the well known Stern–Volmer equation (Eqn 1):
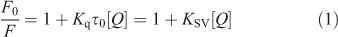
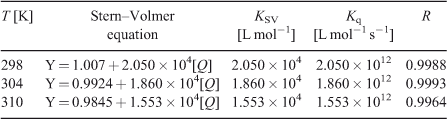
where τ0 is the average life expectancy of the fluorescent molecule; F and F0 are the fluorescence intensities of ADNRS with and without DNA, respectively, [Q] is the concentration of ctDNA, and Kq and KSV are the quenching rate constants and the Stern–Volmer dynamic quenching constant, respectively. Fig. 3 shows the Stern–Volmer plots of F0/F versus [Q] at the three temperatures (298, 304, and 310 K) and the values of KSV and Kq are listed in Table 1. The values of Kq were much greater than 2.0 × 1010 L mol–1 s–1, the maximum diffusion collision quenching rate constant of various quenchers with biopolymers,[15] which indicated that the quenching mechanism of ADNRS by DNA was most likely a static quenching procedure.
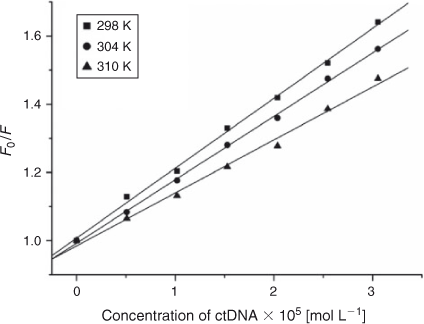
|
|
For static quenching it is often assumed that small molecules bind independently to a set of equivalent sites on a macromolecule, namely, the binding capability of DNA at each binding site is equal. The binding constant K and the binding sites n can be calculated using Eqn 2:[16]
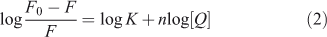
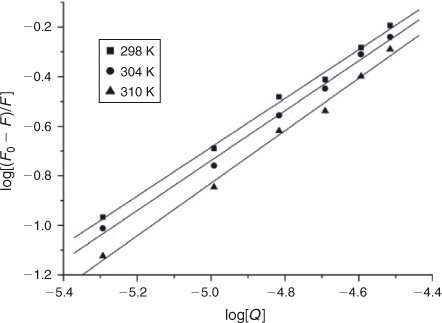
where K and n are the intrinsic binding constant and the number of binding sites, [Q] is the concentration of quenching reagent, F0 is the fluorescence intensity of the complex alone, and F is the fluorescence intensity of the complex in the presence of DNA. Thus, a plot of log(F0 – F)/F versus log[Q] (Fig. 4) yielded the K and n values which are summarised in Table 2.
|

|
The binding constant K increased with temperature, which indicated that the binding was an endothermic reaction, and the capacity of ADNRS binding to DNA was enhanced with the rising temperature.[17] The binding constant of the DNA–ADNRS complex was calculated to be 1.724 × 104 L mol–1 at 298 K, which was smaller than the values of typical intercalators (105 L mol–1).[18] Therefore, it could be concluded that the binding of ADNRS to DNA was not a classical intercalation.
Thermodynamic Parameters and the Nature of Binding Forces
The thermodynamic parameters dependent on temperature were further investigated for characterising the interaction forces between ADNRS and ctDNA. There are several forces acting between small molecules and biomacromolecules including hydrophobic forces, hydrogen bonding, van der Waals forces, and electrostatic interactions.[19] The enthalpy change ΔH, entropy change ΔS, and free energy change ΔG for a binding reaction were calculated from the van’t Hoff equation (Eqn 3) and Gibbs–Helmholtz equation (Eqn 4):
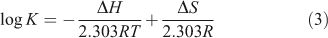
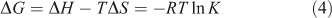
where K is the binding constant at corresponding temperature and R is the gas constant. The values of ΔH and ΔS were obtained from the slope and intercept of the linear van’t Hoff plot by assuming that ΔH is independent of temperature over the range of employed temperatures. The free energy change ΔG was estimated from Eqn 4. The values of thermodynamic parameters are also listed in Table 2. Generally, when ΔH < 0 and ΔS > 0, the main binding force is electrostatic; when ΔH < 0 and ΔS < 0, van der Waals or hydrogen bonding are the main forces; and when ΔH > 0 and ΔS > 0, the main force is hydrophobic.[20] The negative value of ΔG revealed that the interaction process was spontaneous. Meanwhile the positive ΔH and ΔS values indicated that hydrophobic interactions were the main mode of binding of ADNRS to DNA; other non-covalent interactions also cannot be excluded.
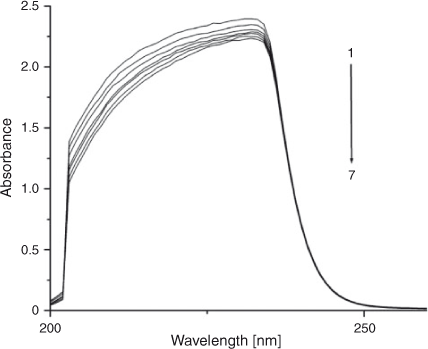
Absorption Spectra Studies
Electronic absorption spectroscopy is a common means to study the interaction of complexes with DNA. Binding of DNA via the intercalative mode usually results in significant hypochromicity (≤40 %) and red shift (>10 nm),[21,22] because the intercalation involves a strong stacking interaction between an aromatic chromophore and the DNA base pairs.[23] However, external groove binding causes a small (or no) spectral shift and an occasional hyperchromicity. The absorption spectra of ADNRS in the absence and presence of ctDNA are shown in Fig. 5. As the concentration of ctDNA increased, the absorption band of ADNRS showed slight hypochromism (~10 %) without any band shift. The phenomena are in agreement with the experimental results of the known groove binder distamycin.[24] As a consequence, the groove binding was most likely the binding mode of ADNRS with ctDNA. The intrinsic binding constant K for ADNRS was calculated from the Wolfe–Shimer equation, (Eqn 5):
|
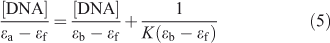
where [DNA] represents the concentration of DNA, ϵa, ϵf, and ϵb are the apparent extinction coefficient for the complex (in the presence of DNA), the free complex, and the fully DNA-bound complex, respectively. The value of K was calculated to be 4.16 × 104 L mol–1 from the ratio of the slope to the y-intercept. It was lower than typical intercalators such as ruthenium(ii) mixed-ligand complexes,[25] which was consistent with the fluorescence studies confirming that the interaction of ADNRS to ctDNA was through non-intercalation.
Effects of Double Stranded DNA and Single Stranded DNA on the Fluorescence of ADNRS
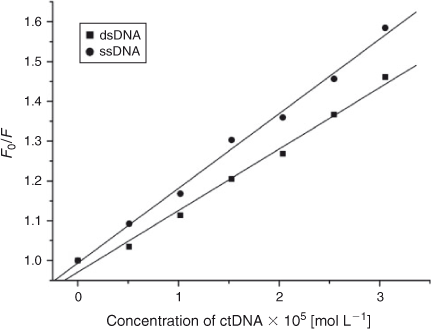
If small molecules interact with the phosphate backbone of DNA through electrostatic interactions, dsDNA and ssDNA would have the same quenching effect on the fluorescence of ADNRS. If the binding mode of small molecules with duplex DNA was intercalation, the quenching effect of ssDNA would be weaker than that of dsDNA.[26] The results of comparison experiments are shown in Fig. 6. It was obvious that both dsDNA and ssDNA could quench the fluorescence of ADNRS but ssDNA had a stronger effect, which confirmed that the interaction between ADNRS and DNA was by groove binding.
|
Iodide Quenching Studies
Further support for the interaction pattern of ADNRS with DNA can be deduced from iodide quenching experiments. Owing to electrostatic repulsion between the negatively charged phosphate backbone of DNA and iodide anions, iodide anions will have difficulty colliding with bound small molecules.[27] So intercalated small molecules should be protected from being quenched by I–, while groove binding molecules can be partly quenched due to being partly protected by DNA.[28] The variation of KSV of ADNRS by I– ion in the absence and presence of ctDNA were obtained by using the Stern–Volmer equation. As shown in Fig. 7, a slight decrease of KSV for ADNRS and the ADNRS–ctDNA system was observed in comparison with a significant change for intercalation binding, which also supported the groove binding mode of ADNRS with ctDNA.
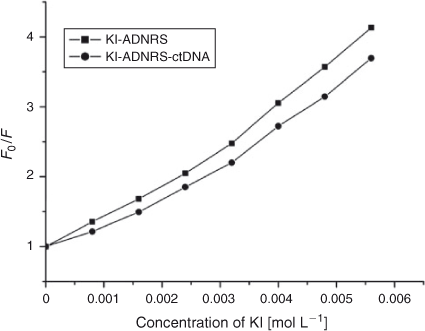
|
Effect of Ionic Strength on the Fluorescence Properties
Monitoring spectral changes with solutions of different ionic strength is an efficient method for distinguishing the binding modes between molecules and DNA.[29] Cations of a strong electrolyte, such as NaCl instead of potassium iodide, can neutralise the negatively charged phosphate. On account of a competition for phosphate anion, the addition of Na+ will weaken the surface-binding interactions which include hydrogen bonding and electrostatic interactions between small molecules and DNA. As shown in Fig. 8, when the concentration of NaCl was from 0 to 0.25 mol L–1, the fluorescence intensities of both free ADNRS and DNA–ADNRS system did not change obviously, showing that the electrostatic interaction between ADNRS and DNA could be excluded.
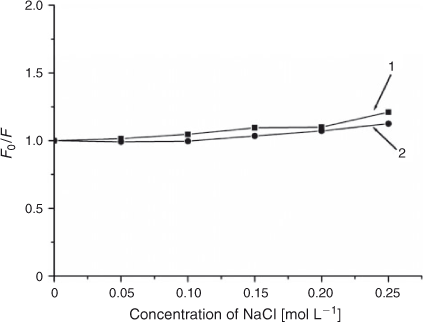
|
DNA Melting Studies
The thermal behaviour of DNA in the presence of small molecules can give insight into their conformational changes and the strength of the interactions. Generally, double-stranded DNA gradually denatures into single strands and generates a hyperchromic effect on the absorption spectra of DNA bases (λ = 260 nm) when the solution temperature increases.[30] In order to identify this transition process, the melting temperature Tm, which is defined as the temperature where half of the total base pairs are unbound, is usually measured. Tm is obtained from the transition midpoint of the curve when fss is plotted against T, where fss = (A – A0)/(Af – A0); A0 is the initial absorbance intensity, A is the absorbance intensity corresponding to its temperature, and Af is the final absorbance intensity.[31] It is well known that intercalation of complexes into DNA base pairs can stabilise the double helix structure and Tm increases by ~5 to 8°C, but that non-intercalative binding causes no obvious increase in Tm.[32] Based on the experimental conditions of this work, thermal denaturation experimental results showed that ctDNA had a Tm of 80 ± 0.5°C, but Tm of 82.5 ± 0.5°C when complexed with ADNRS (Fig. 9). The slight change in Tm value shows that ADNRS provided lower accessibility for ctDNA-binding than the intercalating bound molecules, suggesting the existence of external or groove binding between ADNRS and DNA.
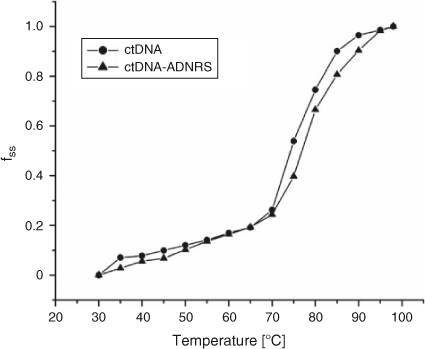
|
Molecular Modelling Studies
As a widely used method for designing new drugs, molecular modelling can provide some insight into the interactions between a macromolecule and a ligand.[33] Many groove binders tend to bind in the region of rich A–T base pairs because it is narrower than the region of rich C–G base pairs, which stabilises the binding.[34] To further confirm the experimental results, complementary application of molecular docking was employed. In the final docked conformation shown in Fig. 10a, ADNRS bound to A–T residues in the minor groove, parallel to the trend of the double helix, which supported our previous results in favour of groove binding. There were hydrogen bonding interactions between ADNRS and ctDNA. The hydroxyl oxygen atom of C(11) was at a distance of 2.1 Å from the O(3′) hydroxyl H atoms of A-17, the carbonyl oxygen atom of the semicarbazone was at a distance of 2.3 Å from the O(3′) hydroxyl H atoms of A-18, the carbonyl O atom of C(10) was at a distance of 2.2 Å from the C(2) azyl H atom of G-10, and the C(14) hydroxy oxygen atom was a distance of 2.0 Å from the C(2) azyl H atom of G-10. Calculation of the binding energy from docking studies revealed the importance of hydrogen bonding in the binding process besides the hydrophobic interaction, which supplemented the binding force results from the thermodynamic data.

|
Conclusion
DNA binding properties of ADNRS were studied in physiological buffer by UV and fluorescence spectroscopy, and molecular modelling techniques. The binding constants and the number of binding sites were calculated based on fluorescence quenching data at different temperatures. Reversible groove binding of ADNRS with ctDNA seems to be at play, according to absorption spectra, iodide quenching studies, the denatured DNA experiment, and the melting temperature determinations. Moreover, the molecular docking results corroborated the experimental results from spectroscopic investigations. Compared with the intercalation mode of ADNR binding to ctDNA,[35] the change in binding mode might be due to the interaction of the semicarbazone group with a phosphate group of ctDNA. The results gained from this study should be a valuable tool for the design and synthesis of new and improved therapeutic agents.
Acknowledgements
We gratefully acknowledge the financial support of National Natural Science Foundation of China (30970696, 21172056) and PCSIRT (IRT1061).
References
[1] M. H. Banitaba, S. S. H. Davarani, A. Mehdinia, Anal. Biochem. 2011, 411, 218.| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjsFOrtb4%3D&md5=5c580c269aba63853542d82f5b30c47bCAS | 21238426PubMed |
[2] G. W. Zhang, P. Fu, L. Wang, M. M. Hu, J. Agric. Food Chem. 2011, 59, 8944.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXpt1Cju74%3D&md5=3ccf51fd27bfbd88a333b564253e370dCAS |
[3] T. Zo1ek, D. Maciejewska, Eur. J. Med. Chem. 2010, 45, 1991.
| Crossref | GoogleScholarGoogle Scholar |
[4] Y. Lu, M. H. Xu, G. K. Wang, Y. Zheng, J. Lumin. 2011, 131, 926.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjtVCqtLo%3D&md5=540a5412860a003237676655e8d66677CAS |
[5] H. K. Choi, J. H. Chang, I. I. Hwan Ko, J. H. Lee, B. Y. Jeong, J. H. Kim, J. B. Kim, J. Solid State Chem. 2011, 184, 805.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXktFKmsb8%3D&md5=bbf1e69eba97e4f45368d5d387e041abCAS |
[6] Q. X. Wang, K. Jiao, F. Q. Liu, X. L. Yuan, W. Sun, J. Biochem. Biophys. Methods 2007, 70, 427.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXislKjtrk%3D&md5=1af8c9a30f21cd2d452953b2166c78e6CAS | 17222457PubMed |
[7] M. B. Prouse, M. M. Campbell, Biochim. Biophys. Acta 2012, 1819, 67.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhsVelug%3D%3D&md5=019c1b4fbc1964d543a5c15545e8c51cCAS | 22067744PubMed |
[8] J. Wang, M. Ozsoz, X. H. Cai, G. Rivas, H. Shiraishi, D. H. Grant, M. Chicharro, J. Fernandes, E. Palecek, Bioelectrochem. Bioenerg. 1998, 45, 33.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXjvFCls7o%3D&md5=50628b1830dc367ec042b2aec83b1012CAS |
[9] C. Temperini, M. Cirilli, M. Aschi, G. Ughetto, Bioorg. Med. Chem. 2005, 13, 1673.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXhtVKjsrw%3D&md5=ea91bb425ceaf09cd25fa5dc6cef06f9CAS | 15698785PubMed |
[10] L. Fang, G. Zhang, C. Li, X. Zheng, L. Zhu, J. J. Xiao, G. Szakacs, J. Nadas, K. K. Chan, P. G. Wang, D. Sun, J. Med. Chem. 2006, 49, 932.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XitVCrtw%3D%3D&md5=8dde20dcba18ce2700b6a700c7439a4aCAS | 16451059PubMed |
[11] Y. Lu, Q. Q. Feng, F. L. Cui, W. W. Xing, G. S. Zhang, X. J. Yao, Bioorg. Med. Chem. Lett. 2010, 20, 6899.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtl2gtLrK&md5=725b0bc82b60746a0691c82f60b7d56dCAS | 21035338PubMed |
[12] F. L. Cui, G. Q. Hui, X. Y. Jiang, G. S. Zhang, Int. J. Biol. Macromol. 2012, 50, 1121.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XjtF2isrY%3D&md5=785305cc98d6bfb2a09da8f32b15607eCAS |
[13] R. Vijayabharathi, P. Sathyadevi, P. Krishnamoorthy, D. Senthilraja, P. Brunthadevi, S. Sathyabama, V. B. Priyadarisini, Spectrochim. Acta A Mol. Biomol. Spectrosc. 2012, 89, 294.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XitFOntbc%3D&md5=2f295893fd2b1ca1e0238108bd5defbcCAS | 22285949PubMed |
[14] G. W. Song, Z. X. Cai, Y. He, Z. W. Lou, Sens. Actuators B Chem. 2004, 102, 320.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXmvFOms7s%3D&md5=0d4a5cc9e018723e6cf38752fa0c6f39CAS |
[15] F. L. Cui, R. N. Huo, G. Q. Hui, X. X. Lv, J. H. Jin, G. S. Zhang, W. W. Xing, J. Mol. Struct. 2011, 1001, 104.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXps1aksbg%3D&md5=11d2cd52c306ff53040bc7673878a4dfCAS |
[16] Y. Lu, G. K. Wang, W. Tang, X. X. Hao, M. H. Xu, X. Li, Spectrochim. Acta A Mol. Biomol. Spectrosc. 2011, 82, 247.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtF2ku7%2FN&md5=4ecb1a9de279722801d5bff95f4d4dffCAS | 21820952PubMed |
[17] G. W. Zhang, J. B. Guo, N. Zhao, J. R. Wang, Sens. Actuators B Chem. 2010, 144, 239.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXptlOlug%3D%3D&md5=b7f0b930c82c1e703d8eb351d278ee63CAS |
[18] Y. T. Sun, S. Y. Bi, D. Q. Song, C. Y. Qiao, D. Mu, H. Q. Zhang, Sens. Actuators B Chem. 2008, 129, 799.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXitVKhur4%3D&md5=aa4b22700854265e82e912185b1c9820CAS |
[19] J. H. Huang, X. M. Wang, J. Mol. Struct. 2012, 1010, 73.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhvVCls74%3D&md5=d3545ed53547dd83ea1da5a3ed623252CAS |
[20] S. Kashanian, M. M. Khodaei, H. Roshanfekr, N. Shahabadi, G. Mansouri, Spectrochim. Acta A Mol. Biomol. Spectrosc. 2012, 86, 351.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhs1CgurfJ&md5=787d1ac1b8ba0d4f19426c8e6d3a7c3cCAS | 22093523PubMed |
[21] B. K. Sahoo, K. S. Ghosh, R. Bera, S. Dasgupta, Chem. Phys. 2008, 351, 163.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXntlSktLg%3D&md5=0bfe7c14bd885ef1ae77eb660d0b6a09CAS |
[22] Y. J. Sun, F. Y. Ji, R. T. Liu, J. Lin, Q. F. Xu, C. Z. Gao, J. Lumin. 2012, 132, 507.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsVWksrjF&md5=ae7fe2fbad18c06d03a6e74af3756b7bCAS |
[23] G. W. Zhang, J. B. Guo, J. H. Pan, X. X. Chen, J. J. Wang, J. Mol. Struct. 2009, 923, 114.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXkt1Snurg%3D&md5=9dd6a82d0322ae5fd94d6a3f7373aa6dCAS |
[24] D. S. Lu, Y. Nonaka, M. Tsuboi, K. Nakamoto, J. Raman Spectrosc. 1990, 21, 321.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3cXltVyrsLc%3D&md5=45f5fe25140f12aefd7ffd743165119dCAS |
[25] L. H. Jin, L. F. Tan, X. Q. Zou, J. Liu, F. Luan, Inorg. Chim. Acta 2012, 387, 253.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xmt1Gmsbo%3D&md5=f45a5f181a410420408ac50f3e39e9e0CAS |
[26] M. B. Gholivand, S. Kashanian, H. Peyman, Spectrochim. Acta A Mol. Biomol. Spectrosc. 2012, 87, 232.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XptlKq&md5=b004d547773533515036ae3a7b078839CAS | 22192418PubMed |
[27] G. W. Zhang, X. Hu, P. Fu, J. Photochem. Photobiol. B 2012, 108, 53.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhvVCnt7s%3D&md5=d3cae5d3eed112087d4656fa514b52d1CAS |
[28] M. Xu, Z. R. Ma, L. Huang, F. J. Chen, Z. Z. Zeng, Spectrochim. Acta A Mol. Biomol. Spectrosc. 2011, 78, 503.
| Crossref | GoogleScholarGoogle Scholar | 21156349PubMed |
[29] Y. T. Sun, H. Q. Zhang, S. Y. Bi, X. F. Zhou, L. Wang, Y. S. Yan, J. Lumin. 2011, 131, 2299.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXosF2rsr8%3D&md5=21a026a07f8cb2d4477072cff0c225f5CAS |
[30] J. H. Li, J. F. Dong, H. Cui, T. Xu, L. Z. Li, Transition Met. Chem. 2012, 37, 175.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xit1Sjur4%3D&md5=7d649bbe7bd3c367b69e2cca01f8d0abCAS |
[31] G. W. Zhang, X. Hu, J. H. Pan, Spectrochim. Acta A Mol. Biomol. Spectrosc. 2011, 78, 687.
| Crossref | GoogleScholarGoogle Scholar |
[32] S. Y. Bi, C. Y. Qiao, D. Q. Song, Y. Tian, D. J. Gao, Y. Sun, H. Q. Zhang, Sens. Actuators B Chem. 2006, 119, 199.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XptlGjsLw%3D&md5=92035a33acc8eb2f912e6daf17b9116dCAS |
[33] R. N. Huo, C. Li, F. L. Cui, G. S. Zhang, Q. F. Liu, X. J. Yao, J. Fluoresc. 2012, 22, 111.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhtVCltrw%3D&md5=1ea3e434d7b27a28014b97fc064008e3CAS |
[34] B. K. Sahoo, K. S. Ghosh, R. Bera, S. Dasgupta, Chem. Phys. 2008, 351, 163.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXntlSktLg%3D&md5=0bfe7c14bd885ef1ae77eb660d0b6a09CAS |
[35] F. L. Cui, G. Q. Hui, X. Y. Jiang, G. S. Zhang, Int. J. Biol. Macromol. 2012, 50, 1121.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XjtF2isrY%3D&md5=785305cc98d6bfb2a09da8f32b15607eCAS |