

The Benzyne Story
Curt Wentrup AA School of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane, Qld 4072, Australia. Email: wentrup@uq.edu.au
Australian Journal of Chemistry 63(7) 979-986 https://doi.org/10.1071/CH10179
Submitted: 2 May 2010 Accepted: 3 June 2010 Published: 15 July 2010
Abstract
The history of o-benzyne from its early beginnings as an unobservable reactive intermediate until its present status as a very well characterized but still theoretically challenging molecule with important applications in synthesis is reviewed. The m- and p-benzynes, tridehydrobenzenes, and benzdiynes are also known, and p-benzyne is a key intermediate in the action of a potent class of ene-diyne anti-tumour compounds.
Benzyne (C6H4) was first proposed as a reactive intermediate by Bachmann and Clarke at the Eastman Kodak Co., who investigated the reaction of sodium with boiling chlorobenzene (a Wurtz–Fittig reaction), which produces benzene, biphenyl, a trace of p-diphenylbenzene, and significant amounts of o-diphenylbenzene, triphenylene, and o,o’-diphenylbiphenyl.[1] While the formation of the first three products could be explained by means of ionic reactions of the types
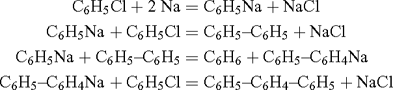
the formation of the last three, in particular triphenylene, was taken as ‘decisive in favour of the free radical explanation’:
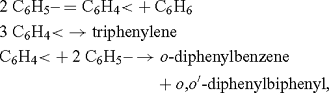
whereby C6H5– is the phenyl radical, and C6H4< was named free phenylene. It was clear from the presentation that the free phenylene was meant to be the biradical 1 (Scheme 1).

|
Compelling experimental evidence was provided by Georg Wittig, then at the University of Freiburg and soon afterwards at Tübingen, in 1942.[2] Wittig and his students discovered that the formation of biphenyl by reaction of the halobenzenes with phenyllithium was the fastest with fluorobenzene, which was contrary to the expected difficulty of displacing fluoride directly in a nucleophilic substitution reaction. A dipolar form 2 of benzyne was proposed as an intermediate in the formation of biphenyl (Scheme 2a) because, in contrast to the corresponding olefinic zwitterions, which would give rise to open-chain acetylenes, ‘the zwitterion [2] would not be able to form dehydrobenzene’, dehydrobenzene being the triple-bonded molecule 3. The trimerization of this zwitterion to triphenylene ‘under certain conditions’ was noted. In the same year, Morton et al. at Massachusetts Institute of Technology (MIT) had also proposed the ‘dipolar phenylene’ intermediate [2], which ‘cannot be stabilized by double bond formation’ in their investigation of the Wurtz reaction of chlorobenzene with pentylsodium (Scheme 2b).[3] Schubert invoked o-phenylene to explain the formation of triphenylene in the reactions of diphenyl ethers with phenylpotassium in 1950 (Scheme 2c).[4]

|
In 1953, John D. Roberts and coworkers, then at MIT and soon afterwards at CalTech, reported a classic 14C labelling experiment, which necessitated the involvement of a symmetrical, electrically neutral intermediate – formulated as benzyne 3 – in the reaction of chlorobenzene with potassium amide in liquid ammonia (Scheme 3).[5] These authors also estimated that the bent acetylene structure would lead to a strain energy comparable to that of cyclopropene.[5b] In fact, the current values for these strain energies are 54 (cyclopropene) and 50 kcal mol–1 (benzyne) (1 cal = 4.184 J).[6,7] The enthalpy of formation of benzyne is ~107 kcal mol–1.[8]

|
Huisgen and Rist (University of Munich) provided further evidence for the benzyne structure (3-methoxybenzyne 4) by demonstrating that identical mixtures of carboxylic acids were obtained on treatment of ortho- and meta-fluoroanisole with phenyllithium followed by carboxylation with CO2 (Scheme 4). The existence of 1,2-naphthyne 5 was demonstrated analogously (Scheme 4).[9] Nevertheless, Huisgen expressed uncertainty as to whether the symmetrical intermediates implied by the results were really free arynes, and, at any rate, ‘an isolation is out of the question’.[9a]

|
Further advances were made when it was discovered that benzyne could be generated from o-dihalobenzenes (e.g. o-bromofluorobenzene) with lithium amalgam or by forming the Grignard reagent with magnesium.[10–12] The first Diels–Alder reaction of benzyne, with furan, was performed in this manner, giving a 76% yield of the adduct 6, which was converted to 1-naphthol with acid (Scheme 5).[10] In the absence of the furan trapping agent, biphenylene and triphenylene were obtained in yields of 24 and 3%, respectively.

|
The concept of mesomerism (resonance) had been developed by Slater, Ingold, Hückel, Pauling, and Wheland in 1931–33[13] and was slowly being adopted by organic chemists. Qualitative valence bond theory was accepted fairly readily, but molecular orbital (MO) theory only took hold in the 1950s. Huisgen and Rist[9] formulated benzyne 3 with a (weakened) in-plane π bond. Lüttringhaus and Schubert were the first to explicitly formulate mesomerism between the three limiting valence structures of benzyne, i.e. the zwitterion 2, the biradical o-phenylene 1, and the ‘acetylene’ 3 (Eqn 1).[4b] Wittig used the two equivalent zwitterionic structures 2 in mesomerism with the ‘cyclohexadienyne’ 3 (Eqn 2) at this time (1955)[10] and continued to do so for many years (the term cyclohexadienyne is incorrect, as it implies that benzyne lacks the resonance stabilization of benzene). Eugen Müller[14] considered resonance between the singlet biradical and the zwitterion, 1 ↔ 2, and Heaney, Mann, and Millar[11] formulated a resonance between the zwitterion and the aryne, 2 ↔ 3.
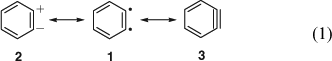
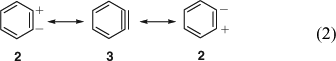
In an early application of MO theory, Coulson[15] estimated the structure of o-benzyne with a pronounced and localized but rather too short C1C2 triple bond (122 pm) and a too long C2C3 bond (144 pm) (the modern experimental values are 125.5 and 138.3 pm, respectively; see below). Clearly, in 1958 MO theory was not yet capable of providing a satisfactory description of benzyne. At the same time, Roberts admitted to not knowing the state of the two electrons formally forming the triple bond.[16] The first detailed MO study of benzyne by Hoffmann, Imamura, and Hehre was published in 1968.[17] An account by Radom et al. in 1986 stressed that very little was still known about the level of theory needed for a proper description of benzynes but concluded from two-configurational calculations that while the triple-bonded valence structure 3 dominates, o-benzyne possesses non-negligible biradical character, i.e. 3 ↔ 1; accordingly, single-configuration descriptions are inadequate.[18] This theme is developed thoroughly in this issue’s Research Front.[8,19,20]
An important experimental advance was made in 1960, when Stiles and Miller discovered that o-benzenediazonium carboxylate 7 is an excellent precursor of benzyne in organic solvents at 40–60°C. This precursor, easily obtained by diazotization of anthranilic acid, allowed benzyne chemistry to move out of the organometallic domain for the first time (Scheme 6).[21] A 30% yield of biphenylene can be obtained by thermal decomposition of 7 in boiling 1,2-dichloroethane (~84°C).[22] The first UV spectrum of gaseous benzyne (λmax = 242 ± 3 nm) was obtained by photolysis of 7.[23] Further details of the UV spectrum of benzyne are reviewed herein.[24] A mass spectrum of benzyne (or most likely a ring-opened[25] C6H4•+ ion) was obtained in the same manner.[26] Compound 7[22] continues to be an important preparative source of benzyne.[27]

|
Diphenyliodonium carboxylate 8 requires a higher temperature, 160°C in diglyme, to form benzyne, iodobenzene, and CO2. The benzyne was trapped with anthracene, giving triptycene (Scheme 6). Flash vacuum pyrolysis (FVP) of 8 at 325°C yielded biphenylene and iodobenzene.[28] Much later, the three-phase test was used to demonstrate the occurrence of free o-benzyne in the thermolysis of 8 in solution.[29]
As more and more solid evidence for the existence of benzyne accumulated, further efforts were made to demonstrate the existence of the free molecule in the gas phase. Schüler and coworkers generated benzyne from the monohalobenzenes and even from benzene and phenylacetylene in an electrical glow discharge, the evidence being the observation of biphenylene by means of its gas-phase UV spectrum. They assumed that benzyne existed in a biradical state (1) in the plasma.[30] This method of benzyne generation languished for many years, but a modern variation has risen to prominence recently: high quality microwave spectra of benzyne have been produced by pulsed electrical discharges (0.8–1.4 kV) in benzene vapour seeded in neon, which had been cooled to a few degrees K in a free jet expansion.[31]
Wittig and Ebel found that iodophenylmercuric iodide 9 and bis(2-iodophenyl)mercury 10 yielded biphenylene on gas phase pyrolysis.[32] Ebel and Hoffmann[33] reported detailed experiments on the gas-phase flow pyrolysis of 10 (Ar carrier gas, 60 hPa, 700°C; residence time in the hot zone ~8.5 × 10–3 s), where the benzyne so produced was trapped downstream with furan vapour. Although the furan adduct 6 was isolated in only 0.1% yield, these experiments demonstrated the existence of free benzyne with a lifetime of at least 20 ms under the reaction conditions. FVP of compound 10 was used later by Grützmacher[34] to obtain the mass spectrum and an approximate ionization potential of benzyne (or most likely a ring-opened C6H4•+ ion[25]), whereas Fisher and Lossing[35] used FVP of o-diiodobenzene for the same purpose. Günther demonstrated that FVP of o-diiodobenzene over Zn at 500–550°C (2 hPa N2 carrier gas at a rate of 120 cm s–1) afforded a 21% yield of biphenylene and 10% of triphenylene.[36] The relatively high yields of triphenylene in some of these reactions indicate that it is not (only) formed by free benzyne trimerizing in the gas phase. With H2 as carrier gas a 3–5% yield of benzene and 7% of biphenylene were obtained. Co-pyrolysis with anthracene afforded the Diels–Alder trapping product triptycene in 8% and biphenylene in 10% yields.[36] Monitoring of the dimerization of gaseous benzyne produced by flash photolysis of solid benzene diazonium carboxylate 5 in an evacuated tube by both UV and mass spectrometry permitted the determination of a preliminary rate of dimerization to biphenylene as equal to or greater than 8 ± 108 l mol–1 s–1.[37] This value was considered to be within a factor of 3 of the true bimolecular rate constant, which is just a little short of a rate measured a few years later by flash photolysis of phthalic anhydride vapour in Ne and Ar, 4.6 ± 1.2 × 109 l mol–1 s–1.[38]
The photolysis of either 9, 10, or phthaloyl peroxide 11 in the presence of tetraphenylcyclopentadienone (tetracyclone) yielded 1,2,3,4-tetraphenylnaphthalene, formed via Diels–Alder addition of benzyne followed by cheletropic extrusion of CO (Scheme 7).[32] The fact that the reactions took place photochemically caused the authors to propose a biradical state (1; singlet or triplet) of dehydrobenzene.[32] Benzyne is now recognized to be a ground state singlet molecule, and the currently accepted singlet-triplet splitting is 37.5 kcal mol–1.[8]

|
Wittig and Hoffmann introduced yet another, convenient albeit explosive, benzyne precursor, 1,2,3-benzothiadiazole S,S-dioxide 12, which decomposes to benzyne at 10–20°C in solution. Benzyne was trapped in Diels–Alder cycloaddition reactions (Scheme 8).[39] When 12 was allowed to decompose in substance in an evacuated flask at 60°C, biphenylene was isolated in 38–52% yield together with 2% triphenylene. Köbrich provided evidence for a symmetrical benzyne intermediate in the pyrolysis of the silver salts of o-chlorobenzoic acids 13 in a sealed tube (Scheme 8).[40]

|
Huisgen and Knorr built a solid foundation for mechanistic benzyne chemistry by using competition experiments to demonstrate that the same reactive intermediate – interpreted as benzyne – was formed from three different precursors, namely thermolysis of 7 and 10 and treatment of o-bromofluorobenzene with Li(Hg). The competition constant for the Diels–Alder cycloadditions with furan and 1,3-cyclohexadiene was ~21.5 for all precursors under a variety of conditions.[41]
The oxidation of 1-aminobenzotriazole with lead tetraacetate or nickel peroxide constitutes an extremely mild method for generating benzyne by loss of two molecules of N2. In this way benzyne chemistry can be carried out well below room temperature (Scheme 9).[42] This continues to be an important method in preparative benzyne chemistry.[27] It is interesting to note that benzyne undergoes click chemistry; thus, the cycloaddition of azides to benzynes is a convenient method of synthesis of benzotriazoles.[27]

|
Three groups reported the formation of benzyne by pyrolysis of carbonyl compounds in 1965–66: the gas-phase pyrolysis of indanetrione (FVP over quartz tubings at 600°C/0.2 hPa) yielded up to 40% of biphenylene and ~4% of triphenylene,[43] the solution-phase pyrolysis of phthalic anhydride in benzene afforded 1% biphenylene and ~1% triphenylene,[44] and flow pyrolysis of phthalic anhydride (N2 carrier gas, 50 hPa, ~800°C) yielded 10–15% biphenylene.[45] In the latter work, a 30% yield of octachlorobiphenylene was isolated from a similar pyrolysis of tetrachlorophthalic anhydride.[45] The formation of benzyne by microwave flash pyrolysis of phthalic anhydride is reviewed herein.[7] The mechanism of the deceptively simple formation of benzyne in the thermal reactions (Eqn 3) is still under debate.[19,43c,46]
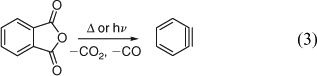
The photolysis of phthalic anhydride in the gas phase and in matrices also affords benzyne. The measurement of the rate of dimerization of gaseous benzyne formed by microsecond flash photolysis was mentioned above.[38] The reaction is reported to take place via a hot, highly vibrationally excited, ground state of phthalic anhydride when using ArF laser photolysis at 193 nm on the nanosecond timescale.[47]
The first matrix-isolation experiments with IR-spectroscopic observation of benzyne were reported in 1973 using phthaloyl proxide and benzocyclobutenedione as precursors. [48] o-Benzyne has now been characterized very thoroughly by IR spectroscopy[49] in noble gas matrices, which is reviewed herein,[19,25] as are the gas-phase microwave,[31,50] photoelectron and UV spectroscopic,[24] and mass spectrometric[25] investigations. Also the 1,2-naphthyne 5 and 2,3-naphthyne have been characterized by IR and UV spectroscopy.[51] Moreover, the 13C dipolar NMR spectrum of benzyne-1,2-13C2 was obtained in an Ar matrix at ~20 K and yielded the important acetylenic C1C2 distance as 124 ± 2 pm,[52] which is in agreement with the more accurate value obtained by microwave spectroscopy, 125.5 pm.[31,50] This bond distance lies between the values for ethyne (120.3 pm) and ethene (133.2 pm), being closer to the ethyne value, and the C2C3 bonds (138.3 pm) are a little shorter than the CC bonds in benzene (139.1 pm).[31] The 1H and 13C NMR spectra of benzyne in solution were obtained after generating the molecule inside a hemi-carcerand, a large molecular container.[53] In this environment benzyne is stable at –100°C for several hours, sufficiently for the recording of its NMR spectra. At higher temperatures it reacts irreversibly with the hemicarcerand host via Diels–Alder addition to benzene moieties.[53] The combined evidence from microwave,[31,50] NMR,[53a] and IR spectroscopy[48,49] as well as theoretical calculations[17,53b] indicates that o-benzyne can be described as a resonance hybrid of the acetylene 3 and the cumulene 14, being closer to the acetylenic structure (Eqn 4).
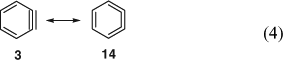
Stable transition metal complexes of benzyne have been prepared and investigated by X-ray crystallography. They have much more normal bond lengths than benzyne itself, e.g. C1C2 is ethene-like (133–136 pm), all other CC bonds are very much benzene-like (138–140 pm) in the Zr and Ni complexes, and they are best described as metallacyclopropenes (e.g. the Ni(0) complex shown in Eqn 5).[54]
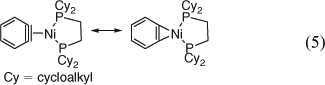
The m- and p-benzynes 15 and 16,[8,19,55] tridehydrobenzenes 17–19,[19] and tetradehydrobenzenes (benzdiynes), e.g. 20[20] (Scheme 10) are described herein. Several resonance structures can be formulated for these species as illustrated for 20 as an example (Scheme 10). Therefore, for a proper quantum-mechanical description, it is preferable to use multireference methods.[8,19,20,56] Sato and Niino present experimental evidence for the multireference character of benzdiynes.[20] Also the highly reactive 1,4-benzdiynes can be stabilized by complexation with transition metals, thus turning them into isolable compounds.[54,57] X-ray crystallography of such complexes indicate that the C1C2 and C4C5 bonds are ethene-like (131–135 pm), and the C2C3 bonds are benzene-like (139–140 pm). The cyclic C6 (hexadehydrobenzene), which according to calculations has D3h symmetry (structure similar to that shown for m-benzyne in Fig. 1), is also known,[20,58] as are linear C6 and other carbon chain clusters.[59]

|

|
The structure of m-benzyne has been the subject of much debate, partly because there is experimental evidence for the existence of an isomer, bicyclo[3.1.0]hexatriene 22 as a reactive intermediate formed by base treatment of the bicyclohexene 21 (Scheme 11).[60] Similarly, there is good experimental evidence for the existence of butalene 23, an isomer of p-benzyne (Scheme 11).[61] However, both these reactions are complex and not fully understood. Similar experiments on a benzologue of 21 indicated that a m-naphthyne was formed rather than a benzologue of 22.[62] Early Hückel MO calculations indicated, not surprisingly, that 22, being a non-alternant ‘azulenoid’ 6π electron system, retains a major portion of the benzenoid resonance energy.[63] In spite of its high strain energy, it is, therefore, a reasonable intermediate. In contrast, butalene 23 is a cyclobutadiene derivative with antiaromatic character and consequential high energy. MINDO/3 calculations indicated the separate existence of m- and p-benzynes 15 and 16 as well as the bicyclic structures 22 and 23.[64] General valence bond calculations predicted that the lowest energy forms of the 1,3- and 1,4-dehydrobenzenes are not bicyclic but monocyclic structures with considerable biradical character. However, the m-isomer 15 collapses to the bicyclic structure 22 at the single-determinant level.[65] Thus, again, multireference theoretical methods are required in order to handle these systems correctly. Recent calculations reveal a 1,3-bonding interaction resulting in an interradical distance of 200–210 pm for the singlet state of m-benzyne, depending on the computational method used (Fig. 1).[19,66] Due to this bonding interaction, m-benzyne has much less biradical character and a larger singlet-triplet splitting than p-benzyne. The calculated IR spectra of m-benzyne and tetrafluoro-p-benzyne at the CCSD(T) level are in very good agreement with the matrix-isolation IR spectra.[19] To summarize, the species observed in matrix isolation[19] as well as gas phase[8] experiments definitely possess the monocyclic aryne structures 15 and 16, although they are not regular hexagons. The bicyclic structures 22 and 23 do not correspond to the observed species. The natures of the intermediates generated in the reactions shown in Scheme 11 remain to be clarified.

|
The Bergman-cyclization[67] of 3-hexene-1,5-diynes (e.g. 24) to p-benzynes (e.g. 16) (Scheme 12) is biologically important because several natural products with powerful anti-tumour, and/or antibiotic properties such as the calicheamicins, esperamicins, lidamycin, and dynemicins (e.g. 25, Scheme 12) contain such enediyne moieties, and their biological activity can be ascribed to formation of transient p-benzynes. Hydrogen abstraction by the p-benzyne generates a more reactive phenyl radical, which abstract a second H atom from DNA, resulting in DNA cleavage.[68,69] There is a related cyclization of enyne-allenes 26 to α,3-didehydrotoluenes 27 or 1,4-didehydroindenes, known as the Myers–Saito cyclization, which is involved in the mechanism of action of the neocarcinostatin anti-cancer agents. Examples of photochemically triggered Bergman and Myers–Saito rearrangements are reviewed herein.[68d]

|
The possible interconversion of the o-, m-, and p-benzynes has been evaluated theoretically.[56] Experiments using femtosecond gas-phase laser irradiation to generate the o-, m-, and p-benzynes from the corresponding dibromobenzenes, together with femtosecond mass spectrometric product observation, indicate lifetimes of ~400 ps for each benzyne isomer under the reaction conditions used. The experiments also suggested that the o- and m-isomers may rearrange to the p-isomer by H-atom tunnelling; the p-isomer then undergoes the Bergman ring opening.[56a]
Several reviews of benzyne chemistry have been published previously.[6a,16,55,67b,70]
Outlook
In spite of extensive experimentation, many challenges remain in benzyne-related chemistry. A better experimental and theoretical understanding of m- and p-benzynes and their relationships with the bicyclic isomers 22 and 23 is needed. It is an experimental challenge but highly desirable to obtain microwave spectra and hence exact geometrical parameters of m- and p-benzynes. The benzynes have always been a testing ground for computational methods (as have carbenes and nitrenes, for example) and continue to be so;[66] their multireference character puts serious demands on the theoretical methods. The interplay between experiment and theory will undoubtedly continue, and systems such as α,n-didehydrotoluenes 27 and 1,n-didehydroindenes involved in Myers–Saito and related rearrangements,[68] didehydrophenyl cations,[67b] and hetarynes[71] need further characterization by matrix-isolation spectroscopy and computational methods. A deeper insight into the mechanisms of rearrangement and interconversion of arynes[25,43c,46,56a] will require exacting experimental and computational investigations. Benzynes continue to be applied widely in synthesis by using classical procedures as well as modern methods, such as the CsF-induced reactions of o-(trimethylsilyl)aryl triflates and o-(trimethylsilyl)aryliodonium triflates, described in this issue.[72] New chemotherapeutic drugs based on enediyne/p-benzyne chemistry are being developed, and their modes of action are being investigated experimentally and computationally.[73] There is fascinating potential for synthetic applications of aryne and cycloalkyne transition metal complexes either as isolable compounds or reactive intermediates under stoichiometric,[54d,74] or catalytic conditions.[75]
[1]
(a) W. E. Bachmann,
H. T. Clarke,
J. Am. Chem. Soc. 1927, 49, 2089.
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
Another 18 computational papers on benzynes from 1969 to 1984 are cited herein.
[19]
H. Winkler,
W. Sander,
Aust. J. Chem. 2010, 63, 1013.
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
