

Discovery and optimisation of conotoxin Vc1.1 and analogues with analgesic properties
Majbrit Frøsig-Jørgensen A , Jing Ji A , Declan M. Gorman A , Meng-Wei Kan A and David J. Craik A *
A , Jing Ji A , Declan M. Gorman A , Meng-Wei Kan A and David J. Craik A *
A
Australian Journal of Chemistry 76(10) 655-670 https://doi.org/10.1071/CH23155
Submitted: 16 August 2023 Accepted: 15 September 2023 Published: 6 October 2023
Abstract
A specimen of the marine cone snail Conus victoriae collected from a beach in Broome, Western Australia, by a group from The University of Melbourne led to the discovery of the α-conotoxin Vc1.1, which was found to have analgesic activity in rodents. The discovery of this venom-derived peptide led to a series of structural, mechanistic and pharmacological studies directed towards the development of a new analgesic for neuropathic pain by groups in Australia and internationally. Solid-phase peptide synthesis played an important role in developing structure–activity relationships. Studies in a rat model of neuropathic pain showed that a cyclic analogue of the peptide, cVc1.1, had comparable analgesic activity with that of gabapentin, one of the foremost clinically used drugs for neuropathic pain, with cVc1.1 delivered orally at a 120-fold lower dose than gabapentin. Originally, Vc1.1 was believed to act primarily through nicotinic acetylcholine receptors, but evidence for a mechanism mediated through γ-aminobutyric acid B (GABAB) receptors later emerged. Efforts to optimise the binding and pharmacological properties of analogues of Vc1.1 revealed that the affinity towards either receptor can be modulated by sequence mutations, disulfide bond modifications and backbone cyclisation. This Account describes the discovery, structure, chemistry and pharmacology of Vc1.1, with a focus on studies carried out in Australian laboratories.
Keywords: conotoxin, drug design, drug development, drug discovery, GABABR, gamma-aminobutyric acid receptor, nAChR, nicotinic acetylcholine receptors, pain, peptide, pharmacology, venom.
Introduction
Neuropathic pain imposes a major burden on society and affects millions of people worldwide. Despite this, current treatments are frequently ineffective and associated with a range of adverse effects, including tolerance and addiction. Compared with conventional small-molecule drugs, peptides offer great potential as drug leads because of their high target specificity and reduced off-target effects. However, they present a challenge in drug development due to their poor membrane permeability and poor bioavailability. Australia has a strong record of research in the field of peptides, particularly in peptides derived from the venoms of its terrestrial and marine fauna. One such peptide is Vc1.1, which was originally discovered by a team of Australian researchers who decoded RNA isolated from the venom duct of a specimen of the marine cone snail Conus victoriae.1 The corresponding 16-amino acid peptide sequence shares high similarity with α-conotoxin peptide family members known to act on nicotinic acetylcholine receptors (nAChRs) in primary and peripheral afferent neurons. A chemically synthesised form of the peptide was found to have analgesic activity in a rodent model of pain.1 Thus began a 20-year journey of exploration of this venom-derived pain-killing peptide. Our laboratory focused on synthesis and structure–activity relationship studies, and in collaboration with Professor David Adams, on the pharmacology of Vc1.1 and Vc1.1 analogues.
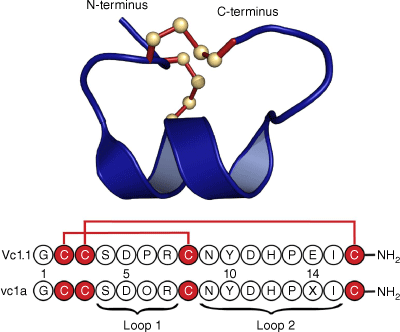
Following initial studies on the synthetically produced Vc1.1 peptide sequence based on the complementary DNA (cDNA) sequence, the native peptide, named vc1a, was isolated from the venom of Conus victoriae and examined by mass spectrometry.1,2 It was found to have post-translational modifications at two residues: a hydroxyproline at position 6 and a γ-carboxyglutamate at position 14, as well as C-terminal amidation. Fig. 1 shows the amino acid sequences of Vc1.1 and vc1a, and the 3-D structure of Vc1.1 as determined in our laboratory by NMR spectroscopy.3 Interestingly, the post-translational modifications in native vc1a are associated with differences in its inhibitory effects compared with Vc1.1.3–5
Sequence and 3-D structure of Vc1.1. The sequence is represented using the one-letter amino acid codes and was derived from a cDNA library associated with the venom duct content of Conus victoriae. Below the sequence of Vc1.1 is that of the corresponding native peptide, vc1a, isolated from the venom duct, which has post-translational modifications at residues 5 and 14 and, like Vc1.1, is amidated at the C-terminus. The 3-D structure was determined in our laboratory by NMR (Protein Data Bank ID: 2H8S). Similarly to other α-conotoxins, Vc1.1 can be considered as comprising two loops in the backbone between successive Cys residues. In the case of Vc1.1, there are four resides in the first loop and seven in the second and it is referred to as a 4/7 α-conotoxin.
Since its discovery 20 years ago, there has been intense interest in Vc1.1, initially because of its potential as a drug lead for the treatment of pain. Synthetically produced Vc1.1 (then named ACV1) entered human clinical trials for sciatica and although it was well tolerated in humans with few side effects, the Phase IIA clinical trial was ultimately discontinued by Metabolic Pharmaceuticals owing to concerns about potentially reduced potency at human nAChRs compared with rat nAChRs.6–8 Nevertheless, interest persisted in analogues of the peptide and in its use as a tool for probing receptor biology. The focus of our studies has been on delineating structure–activity relationships. This Account provides an overview of studies done in our laboratory and of work done in other Australian laboratories, as well as international efforts in the field. Recent reviews have described findings on Vc1.1 as part of broader reviews on α-conotoxins9,10 but here, we focus exclusively on Vc1.1 and its analogues. Nevertheless, we first provide a brief introduction to conotoxins to help place Vc1.1 work in context.
Conotoxins are cysteine-rich peptides from marine Conus species, typically comprising 10–45 amino acids and two or more disulfide bonds.11 They have been widely studied for their potential as drug leads.12–20 α-Conotoxins are a subclass of conotoxins that contain four disulfide-bonded cysteine residues that can potentially form three different disulfide isomers: the bead (Cys I–II and Cys III–IV), the ribbon (Cys I–IV and Cys II–III) and the globular (Cys I–III and Cys II–IV) isomer, with the latter generally being the one found in nature.21 Conotoxins are classified according to their cysteine framework22 and their ‘loop’ structure, the loops being the backbone segments between Cys residues. Vc1.1 displays cysteine framework I (i.e. C–C–CC) and has a four–seven loop structure (i.e. four residues in the first loop and seven residues in the second loop).22 Alterations in the disulfide connectivities can drastically affect the activity of the peptide, as can the sequences of the loops between the Cys residues.23–25 Indeed, much of the published work on Vc1.1 has involved structure–activity relationship (SAR) studies in which individual residues have been substituted or modified to examine effects on receptor binding and activity. In particular, Vc1.1 has been extensively studied by in vitro experiments whereby efforts have been made to understand and improve desired drug-like qualities such as metabolic stability, oral bioavailability and receptor specificity. This has been done by employing techniques such as amino acid substitutions, disulfide bond engineering and backbone cyclisation.
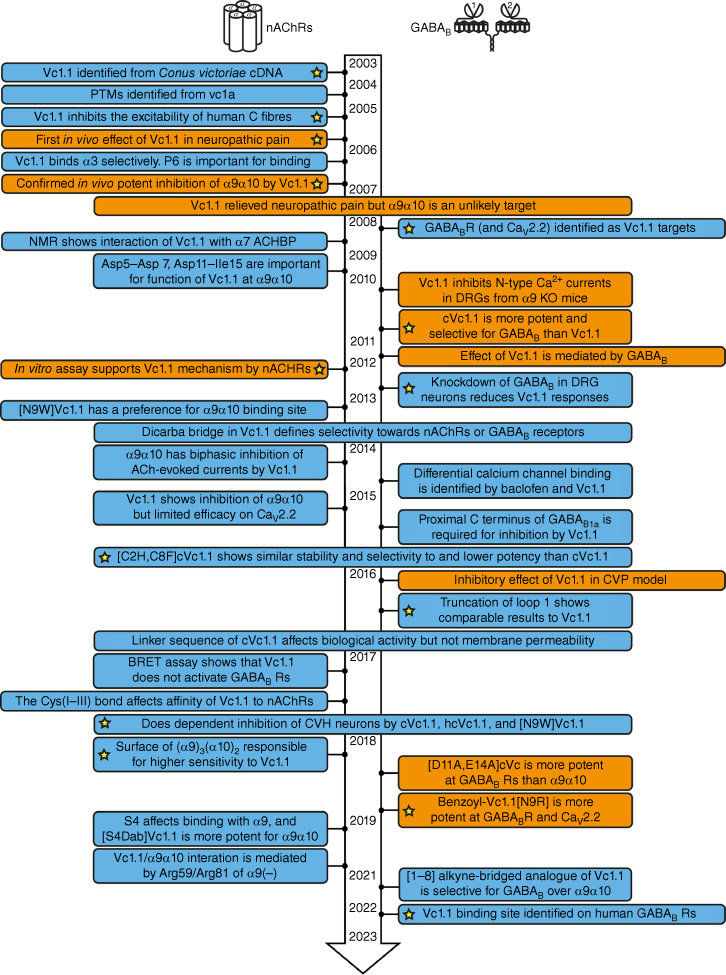
Fig. 2 summarises the timeline of key research papers published on Vc1.1 from 2003 to mid-2023. Because of its α-conotoxin-like structure, Vc1.1 was originally believed to primarily bind at nAChRs; however, evidence for binding to the γ-aminobutyric acid B (GABAB) receptor later emerged.5 Fig. 2 is arranged to highlight the studies focusing on these two main receptor types (nAChR and GABABR). The receptor responsible for the analgesic effects of Vc1.1 has been the subject of debate over a number of years, with evidence now supporting both α9α10 nAChRs26 and GABABRs as being important in mediating its role in the modulation of pain.5
Overview of key messages from published research papers on Vc1.1 from its discovery in 2003 until mid-2023. Boxes shaded blue represent in vitro studies and boxes shaded orange include a combination of in vitro and in vivo studies. The yellow stars highlight papers of particular significance. (PTM, post-translational modification; ACHBP, acetylcholine-binding protein; DRG, dorsal root ganglion; BRET assay, bioluminescence resonance energy transfer assay; KO mice, knockout mice; CVP, chronic visceral pain.)
Chronic neuropathic pain, which is usually associated with prolonged damage to or disease of sensory nerves, presents a large unmet medical need, and response rates to current treatment options vary widely. Additionally, current treatments, which include antiepileptics, antidepressants and opioids, are associated with increased tolerance over time and can be highly addictive.27–30 A range of conotoxins target receptors that are involved in neurological pathways, including NaV channels, CaV channels, KV channels, nAChRs and G protein-coupled receptors (GPCRs). These receptors are attractive therapeutic targets, and selective ligands for these receptors are often novel candidates for clinical trials assessing new analgesics. So far, only ω-conotoxin MVIIA (ziconotide) has been FDA approved as a drug and is marketed as Prialt.31 It is structurally quite different to Vc1.1 and comprises three disulfide bonds in a cystine knot topology. With this background on conotoxins and pain, we now discuss some of the key studies summarised in the timeline in Fig. 2 that focus specifically on Vc1.1 and its analogues.
Structural studies
Our first entry into the Vc1.1 field was in 2006 and involved determining the 3-D structure of the peptide, which comprises a short helical region stabilised by two disulfide bonds with the globular (I–III, II–IV) connectivity, as shown in Fig. 1.3 At the time, there were no crystal structures of Vc1.1 bound to either nAChRs or the GABABR, and so information on receptor binding mainly relied on mutagenesis or modelling studies. One early study, in 2008, that did involve a direct structural biology approach to probe the ligand receptor interaction was conducted on the binding of Vc1.1 to a model of an nAChR using saturation transfer difference NMR.32 In that case, the soluble acetylcholine binding protein (AChBP) was used to mimic the extracellular domain of the α7 subunit of nAChRs. The study suggested that Tyr10 of Vc1.1 is important for interaction with the nicotinic binding site of the α7 subunit of nAChRs. In a later 2013 study, molecular modelling was used to predict the binding site on α9α10 nAChRs.23 By that stage, it had been established that the α9α10 nAChR was more relevant than α7 or α3 subunits that had been earlier studied.
A wide range of mutagenesis studies have been conducted on Vc1.1, and a selection of the various mutants made by solid-phase peptide synthesis are shown in Fig. 3. There is an extensive literature on SAR studies of these various analogues, with references here cited in order of appearance in Fig. 3 for vc1a,4,5 Vc1.1,4,5,23–25,33–45 [P6O]Vc1.1,4,5,44 [E14γ]Vc1.1,4,5 [G1A]Vc1.1-[N9D]Vc1.1,33,35 [N9F]Vc1.1,23 [N9K]Vc1.1,33,35 [N9W]Vc1.1,23 [D11A]Vc1.1-[D11K]Vc1.1,33,35 [D11N]Vc1.1,34 [E14A]Vc1.1-[I15D]Vc1.1,33,35 [PeIA]Vc1.1,34 [D11A,E14A]Vc1.1,35 cVc1.1,25,36,45 cVc1.1-L1-cVc1.1-L3,46 c[D11A,E14A]Vc1.1,35 c[C2H,C8F]Vc1.1,25 [Ser3]Vc1.1(1-8),47 cis-[2,8]-dicarba Vc1.1 and trans-[3,16]-dicarba Vc1.1.24 We now describe some key examples in more detail.
Sequences of Vc1.1 and selected analogues used in deriving structure–activity relationship (SAR) data, with (a) native disulfide bonds, (b) deleted disulfide bonds, and (c) dicarba bridge substitutions. Red represents cysteine residues with disulfide bonds; green represents substituted amino acids; blue represents linkers used in cyclisation; blue dotted line represents backbone cyclisation; purple with a pink border represents cis-dicarba bridges; pink with a purple border represents trans-dicarba bridges.

Amino acid substitutions
Several studies interrogating the SARs for Vc1.1 examined how single amino acid residue substitutions change the structural or biological properties (e.g. metabolic stability) of the peptide. The resulting sequence changes were then related to the affinity or efficacy at either α9α10 nAChR or GABAB receptors. For example, in one comprehensive study published in 2009, all of the non-cysteine residues of Vc1.1 were individually substituted with either alanine, aspartic acid or lysine, which are neutral, negatively and positively charged respectively.33 Each mutated peptide was examined for its ability to inhibit α9α10 receptor-mediated currents following induction by acetylcholine. NMR spectroscopy was used to monitor the folding of the mutants, and in all cases, the 3-D fold was preserved, apart from when the Pro residues were mutated. In performing SAR studies, it is important to undertake this structural monitoring as changes in activity can then be attributed to different binding interactions of the specific residue that is changed rather than due to a change in the local conformation or global fold. The extensive stabilisation of the Vc1.1 structure by the two disulfide bonds indeed helps to maintain the global fold after single residue mutations, apart from when Pro is changed as this residue is particularly important in defining the local conformation.
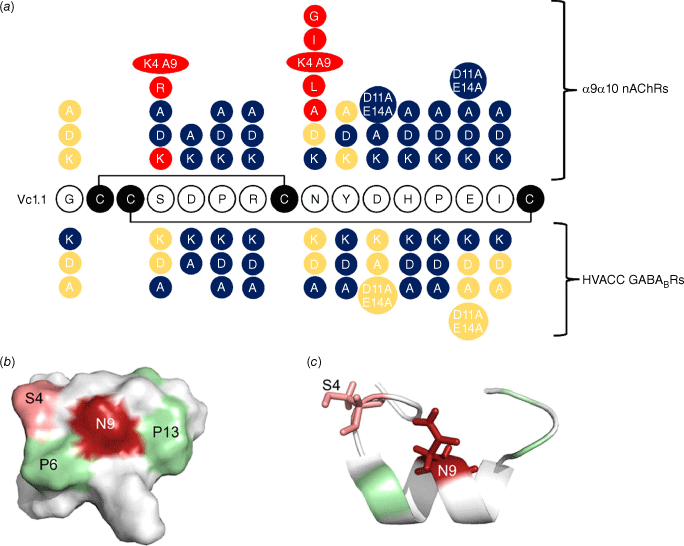
The 2009 study demonstrated that amino acid residues in positions 5–7 and 11–15 were important for the activity of Vc1.1 at the α9α10 nAChR because the peptide lost activity on mutagenesis of these residues, as summarised in Fig. 4a. Furthermore, analogues with certain substitutions at Ser4 and Asn9, separately or simultaneously, exhibited increased biological activity.33 The 3-D structure of Vc1.1 shows that these two residues are located close in space to one another and it was hypothesised that they form important binding interactions with the receptor. Fig. 4b, c shows this proximity in space-filling and ribbon structures of Vc1.1. This hypothesis was subsequently confirmed in 2013 when modelling studies showed that Asn9 has the most extensive interaction with the complementary subunits of nAChRs (i.e. three non-conserved residues at positions 34, 59 and 117).23 Substitution of Asn9 with amino acids that can form energetically more favourable interactions with these three residues on α9α10 or α10α9 nAChRs would significantly improve the potency of the Vc1.1 analogues. Indeed, [N9W]Vc1.1 was found to be more potent than both [N9F]Vc1.1 and Vc1.1 itself.23
Results of mutagenesis studies on Vc1.1 and their effects on activity on α9α10 nAChRs and high-voltage activated (HVA) calcium channels via the GABAB receptor. (a) Red colour highlights changes that improved activity; blue highlights represent substituted amino acids that reduced activity and yellow residues had little effect relative to the parent peptide, Vc1.1. (b) shows a space filling representation showing the location of key positions 4 and 9, and (c) shows the side chains for these residues. Figure adapted from Halai et al.33 and Sadeghi et al.35
A subsequent in silico modelling and amino acid mutagenesis study in 2018 further probed which residues of the peptide and receptor are important for binding and activation and in particular for stoichiometric-dependent activity at the human α9α10 nAChR subtype.34 Energy calculations combined with electrophysiology measurements revealed that residue 154 in the α9 subunit (Asn) and the α10 subunit (Gly) and Asp11 in Vc1.1 were important for the binding interaction. Furthermore, there was a binding preference for the interface α9(+)–α9(−) with a stoichiometry of (α9)3(α10)2 rather than for the interface α10(+)–α9(−), which is present in both (α9)3(α10)2 and (α9)2(α10)3.34 This was followed by another in silico modelling and amino acid mutagenesis study in 2019 that examined the change in α9α10 affinity or potency elicited by the substitution of Ser4 with either diaminobutyric acid (Dab) or diaminopropionic acid (Dap) and led to an increased understanding of the factors that improve potency.48
Despite the availability of large amounts of binding and in vitro activity data, the mechanism of action through which Vc1.1 exerts its analgesic action was subject to debate for a number of years, with Adams and colleagues4,41 focusing mainly on the GABABR and McIntosh and colleagues26,37 working on the α9α10 nAChR. To further explore the apparent dichotomy, a Vc1.1 residue substitution library including alanine, aspartic acid and lysine scanning mutants was screened against the GABABR and both receptor affinity and activity were assessed.35 The results are summarised in Fig. 4a, lower panel. Throughout the three different mutagenesis scans, it was found that substitutions at positions 1, 4, 9, 11, 14, 15 did not significantly change the affinity and activity at the GABABR for Vc1.1. On the basis of the study, a linear and a cyclic version of a double mutant analogue (D11A,E14A) with a 3700- and 8800-fold increased affinity for the GABABR compared with α9α10 receptors was designed and synthesised. Overall, mainly loop 2 of Vc1.1 was found to be important for selectivity towards the GABABR whereas both loop 1 and loop 2 are important for defining the affinity towards α9α10 nAChRs.35
Modification to the N-terminus of Vc1.1 has also been reported to bias the selectivity towards the GABABR over nAChRs when combined with specific residue substitutions.49 Benzoylation of the N-terminus led to increased potency, but neither acetylation nor deletion of glycine was effective in increasing the analgesic effects of Vc1.1.49
Backbone cyclisation
Backbone cyclisation of Vc1.1 proved to be particularly effective in improving potency and pharmacological properties of the peptide. A cyclised version of Vc1.1, referred to as cVc1.1, was synthesised in 2010 in an attempt to improve biological stability and reduce proteolytic degradation of the peptide. In addition, backbone cyclisation improved its oral bioavailability. The compound was cyclised by a GGAAGG-linker sequence from the N to the C terminus, which did not affect the overall structure or activity.36 Three other linker sequences were subsequently designed to study the contribution of charged residues, hydrophobicity and hydrogen bond capacity towards the biological activity. The linker sequences all shared structural similarity to cVc1.1 and had an increased hydrophobicity through N-methylation of either basic (L1: GKGKGK), hydrophobic (L2: GVGVGV), or acidic residues (L3: GEGEGE). However, none of these modifications improved the properties of cVc1.1, and only cVc1.1-L1 showed capacity to inhibit high-voltage activated (HVA) currents in rat dorsal root ganglions (DRGs).46
Disulfide bond modification
Disulfide bonds generally confer a high degree of stability to the structure of a peptide. They also present an attractive target for further modulation and substitution as alternative bond types may result in improved stability or activity. Regioselective replacement of cysteine residues with a cis- or trans-dicarba bridge can shift receptor selectivity. For example, inserting a [2,8]-dicarba bridge in Vc1.1 was found to increase affinity at the GABABR, whereas a [3,16]-dicarba bridge shifted the affinity to the α9α10 receptor compared with the native Vc1.1.24 Conversely, a [1–8] alkyne-bridged analogue of Vc1.1 (alkyne ɑ-Vc1.1) was recently reported to be selective for GABAB over α9α10.50
c[C2H,C8F]Vc1.1 (Fig. 3) is a disulfide-deleted analogue of cVc1.1, designed to have a larger hydrophobic core but with similar structure and stability to the parent double-disufide-bridged peptide.25 In a 2015 study, the potency of c[C2H,C8F]Vc1.1 at both the α9α10 nAChR and GABABR was found to be reduced compared with cVc1.1 but higher than when the corresponding disulfide bond was replaced with a dicarba bridge. Notably, c[C2H,C8F]Vc1.1 has the advantage of avoiding disulfide bond shuffling and thus the formation of unwanted isomers during synthesis.25 In a 2017 study examining the direct contribution of the disulfide bond between CysI and CysIII, the linear version of [C2H,C8F]Vc1.1 was synthesised and it was determined that the inhibitory effect on the α9α10 receptor was almost completely abolished.51 Interestingly, a truncated version of Vc1.1 with a deleted disulfide bond and a truncated peptide sequence, [Ser3]Vc1.1(1–8) (Fig. 3b), maintains similar potency at GABAB to that of the full-length peptide.47 Truncation offers structural simplicity, as well as a faster and more efficient synthesis. Conversely, replacement of Cys3 with a linear alkynyl-derived residue resulted in significantly reduced activity at nAChRs.52
In summary, Vc1.1 has been subjected to a wide range of SAR studies that have identified residues that are important for binding to the α9α10 and GABAB receptors. The types of chemical modifications include single and multiple residue substitutions, disulfide bond engineering, sequence truncation, N-terminal functionalisation and cyclisation, as summarised in Fig. 5. In the following section, we delve more deeply into the pharmacological aspects of these various SAR studies.
Pharmacology of VC1.1 and analogues
Pharmacology at nAChRs
Vc1.1 was first reported as an antagonist for nAChRs, as demonstrated in both binding and functional studies using bovine chromaffin cell cultures.1 The initial [3H]epibatidine displacement studies found half-maximal inhibitory concentration (IC50) values in the range 1–3 µM.1 A later study confirmed these values, reporting a IC50 for Vc1.1 of 1.54 ± 0.14 µM at nicotine-evoked nAChRs in bovine adrenal chromaffin cells.3 Another study specifically looking at axonal excitability found that Vc1.1 reduced the excitability of human C-fibres. After activation of the fibres with 10 µM of the nAChR agonist nicotine, excitability was reduced in a dose-dependent manner by 26.7 ± 11.1, 61.0 ± 8.4, and 92.7 ± 4.9% following applications of 0.2, 2.0 and 10.0 µM Vc1.1 respectively. In that study, the authors detected colocalisation of α3 and α5 subunits of the receptor with a marker for unmyelinated axons, further supporting that the inhibitory effect of Vc1.1 is mediated through nAChRs.53
At around the same time, as well as demonstrating the first analgesic effect of Vc1.1 in neuropathic pain, Satkunanathan et al. described its effect in facilitating an enhanced ability to recover nerve function following injury to the sciatic nerve induced by a blister.54 At doses of 0.036, 0.36 and 3.6 µg, Vc1.1 caused functional recovery of the nerve by 74, 83 and 77% respectively, compared with only 47% recovery for the nerves treated with saline.54 The specific mechanism of action underlying the analgesic activity of Vc1.1 was investigated by examining nAChR subtype specificity. A dose of 30 µM Vc1.1 failed to inhibit the central and skeletal subtypes α3α5β4, α4β2, α4β4, α7, αβγδ, displaying a maximum inhibition of <20% and a IC50 of >30 000 nM.3 However, 30 µM Vc1.1 had a maximum inhibition at the peripheral subtypes α3α5β2, α3β2 and α3β4 of 94, 90 and 70% respectively, with corresponding IC50 values of 7200 ± 200, 7300 ± 700 and 4200 ± 1600 nM respectively.3 The post-translationally modified peptides vc1a and [P6O]Vc1.1 were inactive against any of the subtypes.3
In 2006, Vincler et al. questioned whether the analgesic mechanism of Vc1.1 was mediated as a result of these subtypes, as other antagonists for these subtypes were not analgesics.37 They found that at α6/α3β2β3 and α6/α3β4, the IC50 values were 140 and 980 nM respectively, whereas the affinity for the α9α10 subtype was 19 nM.37 The biased affinity of Vc1.1 towards α9α10 instead of the subtypes α3β2, α3β4 and α7 was confirmed by Halai et al. in 2009, who reported IC50 values of 109, 5532, 3000 and 7123 nM for each subtype respectively.33
Earlier, and at the time the development of Vc1.1 progressed into human clinical studies, the nAChR subtype specificity was unknown and the differences in potencies that existed between the rat and human receptor subunits had not yet been explored. At that time, the receptor subtype specificity had been tested using rat receptors recombinantly expressed in Xenopus laevis oocytes, and not in human nAChRs. Eventually, when the compound reached Phase IIA, it was discontinued as results showed lower potency in humans than was expected based on the in vitro assays.8 In fact, Vc1.1 has nanomolar affinity for the rat receptor subtype α9α10, but the affinity is greatly increased when changing to hybrid human–rat receptors (Table 1). The first indication that the analgesic mechanism was not solely mediated through nAChRs occurred when Nevin et al. reported that the compounds vc1a, [P6O]Vc1.1 and [E14γ]Vc1.1, which are all reportedly inactive in vivo, demonstrated similar in vitro potencies to Vc1.1 at inhibiting ACh-evoked currents at α9α10.4
| Peptide | Receptor | Inhibition (%) | IC50 (nM) | Ref. | |
|---|---|---|---|---|---|
| vc1a | α3β2, α3β4, α4β2, α4β4, α7, α1β1γδ | n.s. | – | 3 | |
| α9α10 | – | 62.9 ± 5.2 | 4 | ||
| [P6O]Vc1.1 | α3β2, α3β4, α4β2, α4β4, α7, α1β1γδ | n.s. | – | 3 | |
| α9α10 | – | 99.1 ± 29.7 | 4 | ||
| [E14γ]Vc1.1 | α9α10 | – | 65.3 ± 14.9 | 4 | |
| Vc1.1 | α3α5β2 | 94 | 7200 ± 200 | 3 | |
| α3β2 | 90 | 7300 ± 700 | |||
| α3β4 | 70 | 4200 ± 1600 | |||
| α3α5β4, α4β2, α4β4, α7, α1β1γδ | <20 | >30 000 | |||
| α6/α3β2β3 | – | 140 | 37 | ||
| α6/α3β4 | – | 980 | |||
| α9α10 | – | 19 | |||
| – | 64.2 ± 15.0 | 4 | |||
| – | 64.2 ± 15.0 | 36 | |||
| – | 70 ± 25 | 23 | |||
| – | 75.0 ± 1.0 | 24 | |||
| – | 109 | 33 | |||
| 85 ± 13 | – | 55 | |||
| α9α10 (100 nM) | 35 ± 5 | – | 41 | ||
| α9α10 (1 µM) | 89 ± 5 | ||||
| α9α10 (1:1) | – | 34 (1) A | 38 | ||
| – | 1100 (2) A | ||||
| α9α10 (1:3) | – | – [1] | |||
| – | 1600 (2) | ||||
| α9α10 (10:1) | – | 34 [1] | |||
| – | 3200 (2) | ||||
| hα9rα10 | – | 549 | 33 | ||
| – | 975.4 ± 314 | 23 | |||
| hα9hα10 | – | 320 | 25 | ||
| hα9hα10 (1:1) | – | 1000 ± 100 | 34 | ||
| hα9hα10 (1:3) | 25.5 ± 1.8 | 3570 ± 310 | |||
| hα9hα10 (3:1) | 65.8 ± 1.8 | 750 ± 90 | |||
| hα9[N154G]hα10 (1:3) | 19.5 ± 1.0 | 11 250 ± 150 | |||
| hα9[N154G]hα10 (3:1) | 21.8 ± 2.2 | 17 020 ± 3170 | |||
| hα9hα10[G154N] (1:3) | 64.57 ± 4.78 | 750 ± 50 | |||
| CaV2.1 | 88.1 ± 8.2 | – | 5 | ||
| CaV2.2 | 91.3 ± 2.2 | – | |||
| [D11N]Vc1.1 | hα9hα10 (1:3) | – | 17 540 ± 2110 | 34 | |
| hα9hα10 (3:1) | – | 10 790 ± 1140 | |||
| hα9[N154G]hα10 (1:3) | – | 10 240 ± 690 | |||
| hα9[N154G]hα10 (3:1) | – | 8590 ± 770 | |||
| [PeIA]Vc1.1 | hα9hα10 (1:3) | – | 13 230 ± 1800 | ||
| hα9hα10 (3:1) | – | 9300 ± 860 | |||
| hα9[N154G]hα10 (1:3) | – | 12 960 ± 1890 | |||
| hα9[N154G]hα10 (3:1) | – | 15 200 ± 1340 | |||
| [N9F]Vc1.1 | α9α10 | 10 | 500 ± 163 | 23 | |
| hα9rα10 | – | 763.3 ± 164 | |||
| [N9W]Vc1.1 | α9α10 | 80 | 43 ± 9 | ||
| hα9rα10 | – | 33 ± 9 | |||
| cis-[2,8]-dicarba Vc1.1 | α9α10 | – | n.s. | 24 | |
| trans-[2,8]-dicarba Vc1.1 | – | n.s. | |||
| cis-[3,16]-dicarba Vc1.1 | – | 12 000 ± 3000 | |||
| trans-[3,16]-dicarba Vc1.1 | – | 2400 ± 3000 | |||
| [D11A,E14A]Vc1.1 | hα9hα10 | 3.0 ± 2.6 | 9200 ± 1300 | 35 | |
| cVc1.1 | α9α10 | – | 765.6 ± 98.2 | 36 | |
| hα9hα10 | – | 6000 | |||
| c[C2H,C8F]Vc1.1 | hα9hα10 | – | 13 000 | 25 | |
| c[D11A,E14A]Vc1.1 | α9α10 | – | 17 900 ± 2600 | 35 | |
| hα9α10 | 6.4 ± 2.9 | 29 300 ± 3000 |
The first study into how the affinity of Vc1.1 changes depending on the origin of the α subunit was conducted in 2009 by Halai et al., who showed that the potency was ~5-fold higher for the rat subunits than the hybrid human–rat subunits, with IC50 values of 109 and 549 nM respectively.33 Substituting amino acids can also shift the affinity towards the different subunits. For example, Yu et al. found that [N9F]Vc1.1 had a lower affinity for the rat subunit compared with Vc1.1, whereas [N9W]Vc1.1 displayed a higher affinity for both the human–rat α9α10 hybrid and rat α9α10 receptor.23 However, the affinity of [N9W]Vc1.1 to the human–rat α9α10 hybrid increased markedly compared with the rat α9α10 receptor, with a 30-fold increase at the human–rat hybrid and only a 1.6-fold increase at the rat receptor. The IC50 values for [N9W]Vc1.1 were reported to be 33 and 43 nM at the human–rat α9α10 hybrid and rat α9α10 receptor respectively.23 Introducing a double mutation, [D11A,E14A]Vc1.1, lowers the potency of the peptide. Vc1.1 has been tested at the hybrid human–rat receptor and exhibited IC50 values ranging between 320 and 975 nM.23,25,33 When [D11A,E14A]Vc1.1 was tested at the human–rat α9α10 receptor, it showed an affinity of 9.2 µM, which is almost 10-fold lower than the parent peptide, Vc1.1.35 Cyclisation further reduces the potency of [D11A,E14A]Vc1.1 to 17.9 and 29.3 µM for the rat α9α10 v. the human–rat α9α10 receptor.35
Removal of one of the two disulfide bonds in Vc1.1 or cVc1.1 simplifies manufacture by reducing the possibility of multiple disulfide isomer formation and is thus potentially advantageous if it can be done without loss of activity. However, deletion of the Cys I–III disulfide bond in cVc1.1 to produce c[C2H,C8F]Vc1.1 (Fig. 3b) results in a 2-fold decrease in potency at the human–rat receptor compared with cVc1.1 (Table 1).25 The potency of cVc1.1 is itself higher at the rat α9α10 receptor than the human–rat α9α10 receptor, with IC50 values of 765 and 6000 nM respectively.25,36
The nAChRs are pentameric receptors and the most common stoichiometry of the α9α10 receptor is (α9)2(α10)3, but varying the stoichiometry of the subunits can influence the potency of a ligand.56 Originally, it was believed that the preferred binding site for Vc1.1 was the interface between α10(+)α9(−) but it was later suggested that there exist two different stoichiometries where Vc1.1 can bind either to the highly sensitive α9(+)α9(−) binding site present in the (α9)3(α10)2 receptor or the less sensitive α10(+)α9(−) and α9(+)α10(−) binding sites that can be found in either receptor subunit arrangements.38 As noted earlier, Yu et al. investigated how varying the stoichiometry, as well as mutations of the subunits, influences the selectivity of the peptides.34 Molecular dynamics simulations found similar salt bridges at the interfaces for α9(+)α9(−) and α10(+)α9(−) involving charged residues of Vc1.1 at positions Arg7, Asp11, His12 and Glu14. Furthermore, Asp11 of Vc1.1 has been found to interact with a hydrogen bond with N154 of the human α9(+)α9(−), which G154 at the corresponding position of the α10 subunit cannot form.
At 1:1 and 1:3 ratios of α9:α10 messenger RNA (mRNA), the resulting stoichiometry of the nAChR expressed in Xenopus laevis oocytes is (α9)3(α10)2/(α9)4(α10)1 and (α9)2(α10)3 respectively. When Vc1.1 was incubated with α9[N154G]α10 at ratios of 3:1 and 1:3, the potency was reduced 23- and 3-fold respectively, compared with potency at wild-type receptors. Conversely, mutating Gly to Asn at position 154 of α9 increased the affinity for Vc1.1 when α9:[G154N]α10 was expressed in a ratio of 1:3, further substantiating the evidence that Asn interacts by a hydrogen bond with Vc1.1. Compared with Vc1.1, [D11N]Vc1.1 had a significantly lower potency at the human α9α10 receptor, with IC50 values of 17.5 µM (equivalent to a 5-fold decrease) and 10.8 µM (equivalent to a 14-fold decrease) when tested at ratios of 1:3 and 3:1 respectively. However, when tested against the [N154G]α9α10, Vc1.1 and [D11N]Vc1.1 show comparable results. An analogue with the grafted residues S9-L15 of PeIA, hereafter named [PeIA]Vc1.1, inhibited wild-type hα9α10 and [N154G]α9α10 to a similar extent regardless of the mRNA injection ratios. However, these results were still 4- and 12-fold reduced compared with Vc1.1.34
Pharmacology at the GABAB receptor
Following the first suggestion that α9α10 was unlikely solely responsible for the analgesic activity elicited by Vc1.1, Callaghan et al. reported that Vc1.1 did not directly inhibit CaV2.1 or CaV2.2 currents when expressed in Xenopus laevis oocytes.5 However, when Vc1.1 was applied to DRG neurons, it had an inhibitory effect on the HVA Ca2+ channel currents that was abolished by 4.6 ± 4.4, 6.8 ± 4.5 and 7.4 ± 2.3% when the GABAB specific antagonists phaclofen (50 µM), CGP55485 (1 µM) and CGP54626 (1 µM) were applied alongside Vc1.1 respectively.5
As can be seen in Table 2, the majority of studies examining the inhibitory effect of Vc1.1 indirectly through the GABABR measured the HVA Ca2+ currents in DRG neurons. Two years after it had first been suggested that Vc1.1 acted indirectly through the GABABR, Callaghan and Adams assessed the ability of Vc1.1 to inhibit Ca2+ currents in DRGs from α9 KO mice.57 Following application of 100 nM Vc1.1, Ca2+ currents were inhibited to 69.0 ± 3.6% in 55% of the wildtype (WT) DRG neurons, whereas the Ca2+ current was decreased to 63.4 ± 7.5% in 71% of the α9 KO DRG neurons. When the DRG neurons were treated with the GABAB antagonist CGP55845, Vc1.1 was not able to reduce Ca2+ currents. This supports the argument that the mechanism by which Vc1.1 inhibits Ca2+ currents is indirectly through GABAB.57 Apart from confirming that the effect of Vc1.1 is mediated through GABABRs by in vitro studies using DRG neurons where GABAB was knocked down, Cuny et al. also showed that successful inhibition of CaV2.2 channels by Vc1.1 in co-transfected HEK293 cells requires both GABAB subunits.39 The cyclic analogue cVc1.1 was also tested on co-transfected HEK293 cells and although it did not have an inhibitory effect on CaV2.1, it did have an inhibitory effect in the picomolar range on CaV2.3 channels.40 Clark et al. showed that IC50 values for cVc1.1 at the α9α10 nAChR were almost 10-fold higher than that of linear Vc1.1 and almost 6-fold higher potency for the GABABR. The affinity for the GABABR was further supported by an extinguishing of cVc1.1’s effect in the presence of CGP55845.36
| Peptide | Receptor | Inhibition (%) | IC50 (nM) | Ref. | |
|---|---|---|---|---|---|
| vc1a | Rat DRG | 100.0 ± 4.8 | – | 5 | |
| [P6O]Vc1.1 | Rat DRG | 103.7 ± 7.2 | – | 5 | |
| Human DRG | n.s. | – | 44 | ||
| [E14γ]Vc1.1 | Rat DRG | 110.1 ± 9.2 | – | 5 | |
| Vc1.1 | Rat DRG | 57.8 ± 3.8 | 1.7 | 5 | |
| – | 1.7 | 36 | |||
| 61 ± 6.0 | – | 41 | |||
| 6.5 ± 12.7 (pre-pulse) | – | 55 | |||
| 6.2 ± 12.5 (post-pulse) | – | ||||
| Human DRG | 40 | – | 44 | ||
| Mouse colonic DRG | 10.5 ± 1.6 | – | 45 | ||
| CaV2.1 A | 0 | – | 40 | ||
| CaV2.3 A | 25.5 ± 4.2 | – | |||
| CaV2.2 A | 41.5 ± 5.3 | 19.1 | 43 | ||
| S246A + CaV2.2 A | 26.6 ± 7.9 | – | |||
| S270A + CaV2.2 A | 47.1 ± 14.4 | – | |||
| S887 + CaV2.2 A | 37.9 ± 5.6 | – | |||
| G863 + CaV2.2 A | 9.5 ± 4.0 | – | |||
| ΔPCT + CaV2.2 A | 5.3 ± 3.9 | – | |||
| Vc1.1 (bead) | Rat DRG | 18.9 B | – | 47 | |
| Vc1.1 (globular) | 26.8 B | – | |||
| Vc1.1 (ribbon) | 23.6 B | – | |||
| [Ser3]Vc1.1(1–8) (100 pM) | Mouse DRG | 20.6 ± 3.3 | – | 47 | |
| [Ser3]Vc1.1(1–8) (30 nM) | 31.6 ± 4 | – | |||
| [Ser3]Vc1.1(1–8) (1–3 µM) | 30.2 ± 3.9 | – | |||
| cis-[2,8]-dicarba Vc1.1 | Rat DRG | 40 | – | 24 | |
| trans-[2,8]-dicarba Vc1.1 | 30 | – | |||
| cis-[3,16]-dicarba Vc1.1 | n.s. | – | |||
| trans-[3,16]-dicarba Vc1.1 | n.s. | – | |||
| [G1A]Vc1.1 | Mouse or rat DRG | 30.1 ± 1.3 | 13.1 | 35 | |
| [G1D]Vc1.1 | 33.1 ± 4.2 | – | |||
| [S4D]Vc1.1 | 23.8 ± 3.7 | – | |||
| [S4K]Vc1.1 | 24.8 ± 3.6 | – | |||
| [N9D]Vc1.1 | 25.7 ± 2.3 | – | |||
| [N9K]Vc1.1 | 23.8 ± 2.7 | – | |||
| [D11A]Vc1.1 | 36.9 ± 3.8 | 6.8 | |||
| [D11K]Vc1.1 | 39.1 ± 4.1 | – | |||
| [E14A]Vc1.1 | 29.9 ± 2.7 | 23.1 | |||
| [E14D]Vc1.1 | 32.7 ± 3.8 | – | |||
| [I15A]Vc1.1 | 26.8 ± 1.4 | 170.7 | |||
| [I15D]Vc1.1 | 25.4 ± 5.4 | – | |||
| [D11A,E14A]Vc1.1 | Mouse DRG | 38.4 ± 2.5 | 2.5 ± 1.1 | ||
| c[D11A,E14A]Vc1.1 | 48.9 ± 4.7 | 3.3 ± 1.1 | |||
| cVc1.1 | Rat DRG | – | 0.3 | 36 | |
| Mouse colonic DRG | 26.7 ± 6.8 | – | 45 | ||
| CaV2.1 A | 0 | – | 40 | ||
| CaV2.3 A | 34.8 ± 2.9 | 0.29 ± 0.08 | |||
| cVc1.1-L1 | Rat DRG | 6.8 ± 1.3 | – | 46 | |
| cVc1.1-L2 | n.s. | – | |||
| cVc1.1–L3 | n.s. | – | |||
| c[C2H,C8F]Vc1.1 | Rat DRG | – | 0.857 ± 0.516 | 25 | |
| hCaV2.3 A | – | 0.961 ± 0.254 |
Klimis et al. found that Vc1.1 reduced the HVA Ca2+ currents in DRG neurons and that this effect was abolished when SCH50911 (a specific GABAB antagonist) was applied.41 They also reported that AuIB, another α-conotoxin, has high selectivity towards the GABABR and a long-lasting anti-allodynic effect and, thus proposed that it is more likely that the anti-allodynic effect of Vc1.1 is mediated through GABAB rather than the α9α10 receptor.41
Although several studies using both transfected cells and DRG neurons had shown data supporting a mechanism through GABABRs, Wright et al. were unable to replicate the data and disputed that the analgesic mechanism of Vc1.1 is through the calcium channels, suggesting that it is more plausible to be by nAChRs.55 Furthermore, Christensen et al. were unable to show that Vc1.1 was capable of displacing CGP54626 in HEK293 cells in a bioluminescence resonance energy transfer (BRET) assay, and thus concluded that the target for Vc1.1 is not GABAB.58 When the ability of Vc1.1 to inhibit evoked excitatory post-synaptic currents (eEPSCs) on spinal cord slices was compared with baclofen, it was found that inhibition amplitudes were 3 and 81% respectively, and it was thought unlikely that the action is mediated through GABAB in primary afferents as opposed to the responses seen for DRG neurons.42
Improving the affinity of Vc1.1 for either nAChR or GABAB receptors through molecular modifications has been an area of interest as biased analogues may help in the interrogation of the analgesic mechanism and may also generate promising drug leads. A thorough study of the effect of substituting the amino acids in Vc1.1 with Ala, Lys or Asp on the ability to inhibit α9α10 and HVA calcium currents was thus undertaken.35 Several alanine substitutions caused a decrease in the HVA inhibition by Vc1.1, but only G1A, D11A, E14A and I15A substitutions showed comparable inhibition with Vc1.1, and only [D11A]Vc1.1 had similar effects to baclofen when compared with Vc1.1. Substitutions of aspartic acid at G1, S4, P6, R7, N9, Y10, H12, P13, E14 and I15 showed that only positions 1, 4, 9, 14 and 15 retained activity to inhibit HVA currents and only [E14D]Vc1.1 had comparable results with baclofen. All of these alterations are in loop 2 and retain activity to inhibition by GABABRs, thus supporting the importance of loop 1 for inhibition, especially as D11A and E14A retained activity at HVA currents but not α9α10. The analogue [D11A,E14A]Vc1.1 was synthesised to characterise its ability to selectively inhibit HVA calcium currents and the α9α10 receptor; it was only able to inhibit by GABABRs.35
Several studies suggest that modifications to the C-terminus of the GABABR is important for the function of Vc1.1. In early studies (not involving Vc1.1), Galvez et al. altered the venus fly trap (VFT) domain of the GABABR with two mutations (S270A and S246A) and, based on the results, postulated a VFT model for receptor activation by ligands.59 Building on this work, Huynh et al.43 looked at binding to the C-terminus of the GABABR and systematically truncated several C-terminal residues. Deleting 21 amino acids within the proximal C-terminal domain (R857-S877) strongly suggested that the proximal C-terminal domain of GABAB1a is involved in the inhibition of Ca2+ currents by Vc1.1.43
More recently, the binding of Vc1.1 to the GABABR was investigated by a combination of molecular docking and molecular dynamics (MD) simulations, alongside confirmatory mutagenesis, electrophysiology and immunocytochemistry.60 That study reported that Vc1.1 interacts directly with both B1 and B2 subunits of the GABABR , in particular residues S130 and S131 in B1, and K168 in B2. The study also demonstrated that the binding of Vc1.1 has a previously unreported effect on the VFT domain of GABAB, whereby the subunits have both an ‘antagonist-like’ conformation in the case of B1, and an ‘agonist-like’ conformation in the case of B2. Bony et al. postulated that this may be one structural mechanism that causes Vc1.1 to functionally differ both from other α-conotoxins and traditional GABAB ligands.60
The development of Vc1.1 and its analogues originally primarily focused on systemic delivery through intramuscular administration for neuropathic pain. In 2017, a potentially new application for chronic visceral pain was proposed following the response observed for Vc1.1 in an ex vivo assay for chronic visceral hypersensitivity (CVH) when compared with established GABAB agonists. Castro et al. found a dose-dependent inhibition of nociceptor activity in healthy afferents (32%) and in afferents from CVH mice (44%).44 They proposed that the mechanism was mediated through GABAB, as application of [P6O]Vc1.1 was inactive while baclofen was active, and concomitant application of Vc1.1, CVIID and SNX-482 diminished the effect of Vc1.1 in both human and mouse DRGs.44 cVc1.1 showed dose-dependent inhibition, with a IC50 value of 3.5 nM, an almost 3-fold higher potency than for Vc1.1. Although [N9W]Vc1.1 had elicited higher potency than Vc1.1 in in vitro assays, Vc1.1 was more potent in CVH assays compared with [N9W]Vc1.1, with IC50 values of 23.1 and 10.2 nM respectively.45 The double-mutant analogue [D11A,E14A]Vc1.1 was also tested in the CVH model. It showed a similar dose-dependent antinociceptive effect pattern to cVc1.1, with a significantly greater effect on CVH nociceptors than healthy nociceptors.35
In summary, the accumulated in vitro evidence suggests that both GABAB and α9α10 receptors play a role in the mediation of pain by Vc1.1. This is supported by the fact that receptor specificity can be altered by the rational substitution of key residues in Vc1.1. Furthermore, changing the stoichiometry of the nAChR subunits also changes the IC50 values for Vc1.1, thus alluding to a complex mechanism of action.38
In vivo effects of VC1.1 and analogues
Compared with the large number of in vitro studies available, only a few studies have focused on the in vivo effects of Vc1.1 or analogues. A comparison of these published in vivo studies is provided in Table 3. As already noted, the first in vivo analgesic effect of Vc1.1 was demonstrated in 2005 by Satkunanathan et al. in both a chronic constriction injury (CCI) model and partial nerve ligation (PNL) model.54 Vc1.1 was administered intramuscularly for 7 sequential days. One week after cessation, animals dosed with 0.36 and 3.6 µg Vc1.1 maintained a 50% recovery of the paw withdrawal threshold (PWT) in the CCI model, and similar trends were observed for the PNL model.54 The analgesic effect observed for Vc1.1 was later confirmed by Vincler et al., where the PWT for 0.36 µg Vc1.1 resulted in a significant increase to 61 ± 4% after 7 days of dosing.37
| Time point (hours) | 0.5 | 1.0 | 2.0 | 2.5 | 3.0 | 4.0 | 6.0 | 24.0 | Ref. | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| I | R | I | R | I | R | I | R | I | R | I | R | I | R | I | R | ||||
| CCI, im. | Saline | 5 | 45 | 0 | 42 | −1 | 41 | 54 | |||||||||||
| 0.036 µg Vc1.1 | 59 | 68 | −7 | 39 | 44 | 59 | |||||||||||||
| 0.36 µg Vc1.1 | 29 | 55 | 32 | 56 | 61 | 66 | |||||||||||||
| 3.6 µg Vc1.1 | 84 | 78 | 82 | 77 | 151 | 103 | |||||||||||||
| PNL, im. | Saline | −27 | 32 | −18 | 35 | 0 | 42 | 54 | |||||||||||
| 0.36 µg Vc1.1 | 44 | 62 | 11 | 48 | 74 | 75 | |||||||||||||
| CCI, im. | 0.036 µg Vc1.1 | 34 | 37 | ||||||||||||||||
| 0.36 µg Vc1.1 | 89 | ||||||||||||||||||
| PNL, im. | Saline | −44 | 2 | −29 | 2 | 39 | 4 | 4 | |||||||||||
| 60 µg Vc1.1 | 347 | 24 | 267 | 20 | 135 | 13 | |||||||||||||
| 60 µg vc1a | 13 | 10 | −31 | 6 | 3 | 9 | |||||||||||||
| 60 µg [P6O]Vc1.1 | 12 | 4 | 57 | 5 | 264 | 12 | |||||||||||||
| CCI, p.o. | Saline | −16 | 13 | −16 | 13 | −27 | 11 | 36 | |||||||||||
| 0.1 mg kg–1 cVc1.1 | −16 | 13 | −16 | 13 | 20 | 18 | |||||||||||||
| 0.3 mg kg–1 cVc1.1 | 53 | 24 | 88 | 29 | 75 | 27 | |||||||||||||
| 3.0 mg kg–1 cVc1.1 | 141 | 37 | 208 | 47 | 208 | 47 | |||||||||||||
| 30 mg kg–1 gabapentin | 208 | 47 | 288 | 60 | 251 | 54 | |||||||||||||
| PNL, im. | Saline | −35 | 5 | −35 | 5 | −18 | 6 | −18 | 6 | −59 | 3 | 41 | |||||||
| 0.36 µg Vc1.1 | 221 | 28 | 365 | 40 | 462 | 49 | 371 | 41 | 235 | 29 | |||||||||
| 3.6 µg Vc1.1 | 453 | 56 | 659 | 77 | 629 | 74 | 612 | 72 | 400 | 51 | |||||||||
| 36 µg Vc1.1 | 412 | 34 | 744 | 55 | 953 | 69 | 818 | 60 | 694 | 52 | |||||||||
| PNL, it. | Saline | 0 | 7 | 0 | 7 | 0 | 7 | 0 | 7 | 42 | |||||||||
| 0.36 µg Vc1.1 | 68 | 26 | 100 | 31 | 30 | 20 | 10 | 22 | |||||||||||
| 3.6 µg Vc1.1 | 178 | 14 | 594 | 34 | 644 | 37 | 644 | 37 | |||||||||||
| 36 µg Vc1.1 | 1338 | 73 | 1384 | 75 | 1792 | 96 | 1792 | 96 | |||||||||||
Values are ipsilateral and read from graphs available in the referenced papers and rounded to the nearest whole percentage value. im., intramuscular administration; p.o., per oral administration; it., intrathecal administration; I, increase; R, recovery.
Until 2006, the in vivo studies had mainly been focused on showing an analgesic effect of Vc1.1 rather than exploring the underlying molecular mechanism. However, in 2007, Nevin et al. found evidence for the antinociceptive mechanism of action for Vc1.1 being mediated through the GABABR because only Vc1.1 but not the natural peptide, vc1a, or [P6O]Vc1.1 were able to inhibit allodynia.4 In 2011, Klimis et al. not only confirmed the analgesic effect of Vc1.1 after inducing neuropathic pain by PNL surgery but also showed that Vc1.1 did not have any negative effect on motor performance.41 To elucidate the mechanism of action in vivo, SCH50911, a GABABR antagonist, was administered prior to Vc1.1 treatment. This extinguished the analgesic effect of Vc1.1, strongly indicating the mechanism to be mediated through the GABABR. Importantly, no α9α10 agonists were tested, which could have clarified its role in allodynia.41 Another in vivo study in 2012 provided additional insights into effects in animals and found that changing the route of administration to intrathecal did not affect the antinociceptive effect nor did it cause motor impairment in the animals.42
In summary, even though an analgesic effect of Vc1.1 has been unequivocally demonstrated in various animal models and administration routes, the linear peptide Vc1.1 is susceptible to proteolysis and lacks oral activity. Importantly, the cyclised version circumvented these problems. When cVc1.1 was tested in vivo, it reached maximal effect 1 h after treatment and, despite the dose of gabapentin being 120 times higher, the effect of cVc1.1 was comparable.36
Conclusions and outlook
The current treatment options for neuropathic pain leave many patients with insufficient pain relief. Pharmacological treatment with either antidepressants, anticonvulsants or opioids is associated with many side effects, including addiction, tolerance and constipation, and may still be ineffective against the underlying pathology.61,62 The cone snail-derived peptide Vc1.1 reached Phase IIA clinical trials for neuropathic pain but was discontinued, reportedly owing to its lower potency in humans than expected from in vitro assays in rodents. Despite this setback, optimised analogues of Vc1.1 have remained in the pipeline as new drug leads for the treatment of neuropathic pain. This ongoing interest is a result of a greater understanding of Vc1.1 pharmacology and rationalisation of the species receptor bias.
Peptide chemistry has played a pivotal role in not only producing conotoxin peptides and thereby avoiding the need for depletion of natural resources from the snail venoms (and the cone-snails themselves), but also in being able to devise novel analogues not seen in nature. We hope that this article has provided some insights into the valuable role that peptide chemistry has played in exploring the chemistry and biology of Vc1.1.
Although the main focus on Vc1.1 in the past has been on developing novel treatments for neuropathic pain, there is now promising evidence that Vc1.1 can also be a treatment for chronic visceral pain. There is no clear evidence as to the specific pathway that the analgesic effect of Vc1.1 is mediated by, although it most likely involves a combination of α9α10 and GABAB receptors. The interest in Vc1.1 and its analogues, alongside other α-conotoxins, underpins the promising future of these compounds for the treatment of chronic visceral pain and neuropathic pain and highlights their potential to replace traditional opioid therapeutics.
Acknowledgements
David J. Craik was the recipient of the Royal Australian Chemical Institute’s 2024 David Craig Medal. We thank Mengbai Zhang for valuable comments on the manuscript and Bruce Livett for sharing stories on the original discovery of Vc1.1.
References
1 Sandall DW, Satkunanathan N, Keays DA, Polidano MA, Liping X, Pham V, Down JG, Khalil Z, Livett BG, Gayler KR. A novel α-conotoxin identified by gene sequencing is active in suppressing the vascular response to selective stimulation of sensory nerves in vivo. Biochemistry 2003; 42: 6904-6911.
| Crossref | Google Scholar | PubMed |
2 Jakubowski JA, Keays DA, Kelley WP, Sandall DW, Bingham J-P, Livett BG, Gayler KR, Sweedler JV. Determining sequences and post-translational modifications of novel conotoxins in Conus victoriae using cDNA sequencing and mass spectrometry. J Mass Spectrom 2004; 39: 548-557.
| Crossref | Google Scholar | PubMed |
3 Clark RJ, Fischer H, Nevin ST, Adams DJ, Craik DJ. The synthesis, structural characterization, and receptor specificity of the α-conotoxin Vc1.1. J Biol Chem 2006; 281: 23254-23263.
| Crossref | Google Scholar | PubMed |
4 Nevin ST, Clark RJ, Klimis H, Christie MJ, Craik DJ, Adams DJ. Are α9α10 nicotinic acetylcholine receptors a pain target for α-conotoxins? Mol Pharmacol 2007; 72: 1406-1410.
| Crossref | Google Scholar | PubMed |
5 Callaghan B, Haythornthwaite A, Berecki G, Clark RJ, Craik DJ, Adams DJ. Analgesic α-conotoxins Vc1.1 and Rg1A inhibit N-type calcium channels in rat sensory neurons via GABAB receptor activation. J Neurosci 2008; 28: 10943-10951.
| Crossref | Google Scholar | PubMed |
6 Metabolic Pharmaceuticals Limited. Metabolic’s neuropathic pain drug, ACV1 – Clinical trials update. Melbourne, Vic., Australia: ASX; 2006. Available at https://www.asx.com.au/asxpdf/20061129/pdf/3zv2c96tyh1nx.pdf[cited 11 October 2018].
7 Metabolic Pharmaceuticals Limited. Metabolic’s neuropathic pain drug ACV1 – additional preclinical studies reveal greater potential. Melbourne, Vic., Australia: ASX; 2006. Available at https://www.asx.com.au/asxpdf/20061123/pdf/3zqm91n1jhpff.pdf [cited 11 October 2018].
8 Metabolic Pharmaceuticals Limited. Metabolic discontinues clinical trial programme for neuropathic pain drug, ACV1. Melbourne, Vic., Australia: ASX; 2007. Available at https://www.asx.com.au/asxpdf/20070814/pdf/313yjgpf7jl4lg.pdf[cited 11 October 2018].
9 Kennedy AC, Belgi A, Husselbee BW, Spanswick D, Norton RS, Robinson AJ. α-conotoxin peptidomimetics: probing the minimal binding motif for effective analgesia. Toxins 2020; 12: 505.
| Crossref | Google Scholar | PubMed |
10 Li X, Tae H-S, Chu Y, Jiang T, Adams DJ, Yu R. Medicinal chemistry, pharmacology, and therapeutic potential of α-conotoxins antagonizing the α9α10 nicotinic acetylcholine receptor. Pharmacol Ther 2021; 222: 107792.
| Crossref | Google Scholar | PubMed |
11 Jin A-H, Muttenthaler M, Dutertre S, Himaya SWA, Kaas Q, Craik DJ, Lewis RJ, Alewood PF. Conotoxins: chemistry and biology. Chem Rev 2019; 119: 11510-11549.
| Crossref | Google Scholar | PubMed |
12 Olivera BM, Hillyard DR, Marsh M, Yoshikami D. Combinatorial peptide libraries in drug design: lessons from venomous cone snails. Trends Biotechnol 1995; 13: 422-426.
| Crossref | Google Scholar | PubMed |
13 Livett BG, Gayler KR, Khalil Z. Drugs from the sea: conopeptides as potential therapeutics. Curr Med Chem 2004; 11: 1715-1723.
| Crossref | Google Scholar | PubMed |
14 Norton RS, Olivera BM. Conotoxins down under. Toxicon 2006; 48: 780-798.
| Crossref | Google Scholar | PubMed |
15 Halai R, Craik DJ. Conotoxins: natural product drug leads. Nat Prod Rep 2009; 26: 526-536.
| Crossref | Google Scholar | PubMed |
16 Muttenthaler M, Akondi KB, Alewood PF. Structure–activity studies on alpha-conotoxins. Curr Pharm Des 2011; 17: 4226-4241.
| Crossref | Google Scholar | PubMed |
17 Lewis RJ, Dutertre S, Vetter I, Christie MJ. Conus venom peptide pharmacology. Pharmacol Rev 2012; 64: 259-298.
| Crossref | Google Scholar | PubMed |
18 Akondi KB, Muttenthaler M, Dutertre S, Kaas Q, Craik DJ, Lewis RJ, Alewood PF. Discovery, synthesis, and structure–activity relationships of conotoxins. Chem Rev 2014; 114: 5815-5847.
| Crossref | Google Scholar | PubMed |
19 Robinson SD, Norton RS. Conotoxin gene superfamilies. Mar Drugs 2014; 12: 6058-6101.
| Crossref | Google Scholar | PubMed |
20 Gao B, Peng C, Yang J, Yi Y, Zhang J, Shi Q. Cone anails: a big store of conotoxins for novel drug discovery. Toxins 2017; 9: 397.
| Crossref | Google Scholar | PubMed |
21 Zhang R, Snyder GH. Factors governing selective formation of specific disulfides in synthetic variants of alpha-conotoxin. Biochemistry 1991; 30: 11343-11348.
| Crossref | Google Scholar | PubMed |
22 Kaas Q, Westermann J-C, Craik DJ. Conopeptide characterization and classifications: an analysis using ConoServer. Toxicon 2010; 55: 1491-1509.
| Crossref | Google Scholar | PubMed |
23 Yu R, Kompella SN, Adams DJ, Craik DJ, Kaas Q. Determination of the α-conotoxin Vc1.1 binding site on the α9α10 nicotinic acetylcholine receptor. J Med Chem 2013; 56: 3557-3567.
| Crossref | Google Scholar | PubMed |
24 van Lierop BJ, Robinson SD, Kompella SN, Belgi A, McArthur JR, Hung A, MacRaild CA, Adams DJ, Norton RS, Robinson AJ. Dicarba α-conotoxin Vc1.1 analogues with differential selectivity for nicotinic acetylcholine and GABAB receptors. ACS Chem Biol 2013; 8: 1815-1821.
| Crossref | Google Scholar | PubMed |
25 Yu R, Seymour VAL, Berecki G, Jia X, Akcan M, Adams DJ, Kaas Q, Craik DJ. Less is more: design of a highly stable disulfide-deleted mutant of analgesic cyclic α-conotoxin Vc1.1. Sci Rep 2015; 5: 13264.
| Crossref | Google Scholar | PubMed |
26 McIntosh JM, Absalom N, Chebib M, Elgoyhen AB, Vincler M. Alpha9 nicotinic acetylcholine receptors and the treatment of pain. Biochem Pharmacol 2009; 78: 693-702.
| Crossref | Google Scholar | PubMed |
27 Gwak YS, Hulsebosch CE. GABA and central neuropathic pain following spinal cord injury. Neuropharmacology 2011; 60: 799-808.
| Crossref | Google Scholar | PubMed |
28 PainAustralia. Painful Facts. Deakin, ACT, Australia: PainAustralia Limited; 2018. Available at http://www.painaustralia.org.au/about-pain/painful-facts [cited 11 October 2018].
29 Colloca L, Ludman T, Bouhassira D, Baron R, Dickenson AH, Yarnitsky D, Freeman R, Truini A, Attal N, Finnerup NB, Eccleston C, Kalso E, Bennett DL, Dworkin RH, Raja SN. Neuropathic pain. Nat Rev Dis Primers 2017; 3: 17002.
| Crossref | Google Scholar | PubMed |
30 Fornasari D. Pharmacotherapy for neuropathic pain: a review. Pain Ther 2017; 6: 25-33.
| Crossref | Google Scholar | PubMed |
31 Miljanich GP. Ziconotide: neuronal calcium channel blocker for treating severe chronic pain. Curr Med Chem 2004; 11: 3029-3040.
| Crossref | Google Scholar | PubMed |
32 Westermann J-C, Clark RJ, Craik DJ. Binding mode of α-conotoxins to an acetylcholine binding protein determined by saturation transfer difference NMR. Protein Peptide Lett 2008; 15: 910-914.
| Crossref | Google Scholar | PubMed |
33 Halai R, Clark RJ, Nevin ST, Jensen JE, Adams DJ, Craik DJ. Scanning mutagenesis of α-conotoxin Vc1.1 reveals residues crucial for activity at the α9α10 nicotinic acetylcholine receptor. J Biol Chem 2009; 284: 20275-20284.
| Crossref | Google Scholar | PubMed |
34 Yu R, Tae H-S, Tabassum N, Shi J, Jiang T, Adams DJ. Molecular determinants conferring the stoichiometric-dependent activity of α-conotoxins at the human α9α10 nicotinic acetylcholine receptor subtype. J Med Chem 2018; 61: 4628-4634.
| Crossref | Google Scholar | PubMed |
35 Sadeghi M, Carstens BB, Callaghan BP, Daniel JT, Tae H-S, O’Donnell T, Castro J, Brierley SM, Adams DJ, Craik DJ, Clark RJ. Structure–activity studies reveal the molecular basis for GABAB-receptor mediated inhibition of high voltage-activated calcium channels by α-conotoxin Vc1.1. ACS Chem Biol 2018; 13: 1577-1587.
| Crossref | Google Scholar | PubMed |
36 Clark RJ, Jensen J, Nevin ST, Callaghan BP, Adams DJ, Craik DJ. The engineering of an orally active conotoxin for the treatment of neuropathic pain. Angew Chem Int Ed 2010; 49: 6545-6548.
| Crossref | Google Scholar | PubMed |
37 Vincler M, Wittenauer S, Parker R, Ellison M, Olivera BM, McIntosh JM. Molecular mechanism for analgesia involving specific antagonism of α9α10 nicotinic acetylcholine receptors. Proc Natl Acad Sci 2006; 103: 17880-17884.
| Crossref | Google Scholar | PubMed |
38 Indurthi DC, Pera E, Kim H-L, Chu C, McLeod MD, Michael McIntosh J, Absalom NL, Chebib M. Presence of multiple binding sites on α9α10 nAChR receptors alludes to stoichiometric-dependent action of the α-conotoxin, Vc1.1. Biochem Pharmacol 2014; 89: 131-140.
| Crossref | Google Scholar | PubMed |
39 Cuny H, de Faoite A, Huynh TG, Yasuda T, Berecki G, Adams DJ. γ-Aminobutyric acid type B (GABAB) receptor expression is needed for inhibition of N-type (Cav2.2) calcium channels by analgesic α-conotoxins. J Biol Chem 2012; 287: 23948-23957.
| Crossref | Google Scholar | PubMed |
40 Berecki G, McArthur JR, Cuny H, Clark RJ, Adams DJ. Differential Cav2.1 and Cav2.3 channel inhibition by baclofen and α-conotoxin Vc1.1 via GABAB receptor activation. J Gen Physiol 2014; 143: 465-479.
| Crossref | Google Scholar | PubMed |
41 Klimis H, Adams DJ, Callaghan B, Nevin S, Alewood PF, Vaughan CW, Mozar CA, Christie MJ. A novel mechanism of inhibition of high-voltage activated calcium channels by α-conotoxins contributes to relief of nerve injury-induced neuropathic pain. Pain 2011; 152: 259-266.
| Crossref | Google Scholar | PubMed |
42 Napier IA, Klimis H, Rycroft BK, Jin AH, Alewood PF, Motin L, Adams DJ, Christie MJ. Intrathecal α-conotoxins Vc1.1, AuIB and MII acting on distinct nicotinic receptor subtypes reverse signs of neuropathic pain. Neuropharmacology 2012; 62: 2202-2207.
| Crossref | Google Scholar | PubMed |
43 Huynh TG, Cuny H, Slesinger PA, Adams DJ. Novel mechanism of voltage-gated N-type (Cav) calcium channel inhibition revealed through α-conotoxin Vc1.1 activation of the GABAB receptor. Mol Pharmacol 2015; 87: 240-250.
| Crossref | Google Scholar | PubMed |
44 Castro J, Harrington AM, Garcia-Caraballo S, Maddern J, Grundy L, Zhang J, Page G, Miller PE, Craik DJ, Adams DJ, Brierley SM. α-Conotoxin Vc1.1 inhibits human dorsal root ganglion neuroexcitability and mouse colonic nociception via GABAB receptors. Gut 2017; 66: 1083-1094.
| Crossref | Google Scholar | PubMed |
45 Castro J, Grundy L, Deiteren A, Harrington AM, O’Donnell T, Maddern J, Moore J, Garcia-Caraballo S, Rychkov GY, Yu R, Kaas Q, Craik DJ, Adams DJ, Brierley SM. Cyclic analogues of α-conotoxin Vc1.1 inhibit colonic nociceptors and provide analgesia in a mouse model of chronic abdominal pain. Br J Pharmacol 2018; 175: 2384-2398.
| Crossref | Google Scholar | PubMed |
46 Carstens BB, Swedberg J, Berecki G, Adams DJ, Craik DJ, Clark RJ. Effects of linker sequence modifications on the structure, stability, and biological activity of a cyclic α-conotoxin. Biopolymers 2016; 106: 864-875.
| Crossref | Google Scholar | PubMed |
47 Carstens BB, Berecki G, Daniel JT, Lee HS, Jackson KAV, Tae H-S, Sadeghi M, Castro J, O’Donnell T, Deiteren A, Brierley SM, Craik DJ, Adams DJ, Clark RJ. Structure–activity studies of cysteine-rich α-conotoxins that inhibit high-voltage-activated calcium channels via GABAB receptor activation reveal a minimal functional motif. Angew Chem Int Ed 2016; 55: 4692-4696.
| Crossref | Google Scholar | PubMed |
48 Chu X, Tae H-S, Xu Q, Jiang T, Adams DJ, Yu R. α‑Conotoxin Vc1.1 structure–activity relationship at the human α9α10 nicotinic acetylcholine receptor investigated by minimal side chain replacement. ACS Chem Neurosci 2019; 10: 4328-4336.
| Crossref | Google Scholar | PubMed |
49 Cai F, Xu N, Liu Z, Ding R, Yu S, Dong M, Wang S, Shen J, Tae H-S, Adams DJ, Zhang X, Dai Q. Targeting of N-type calcium channels via GABAB-receptor activation by α-conotoxin Vc1.1 variants displaying improved analgesic activity. J Med Chem 2018; 61: 10198-10205.
| Crossref | Google Scholar | PubMed |
50 Belgi A, Burnley JV, MacRaild CA, Chhabra S, Elnahriry KA, Robinson SD, Gooding SG, Tae H-S, Bartels P, Sadeghi M, Zhao F-Y, Wei H, Spanswick D, Adams DJ, Norton RS, Robinson AJ. Alkyne-bridged α‑conotoxin Vc1.1 potently reverses mechanical allodynia in neuropathic pain models. J Med Chem 2021; 64: 3222-3233.
| Crossref | Google Scholar | PubMed |
51 Tabassum N, Tae H-S, Jia X, Kaas Q, Jiang T, Adams DJ, Yu R. Role of CysI–CysIII disulfide bond on the structure and activity of α-conotoxins at human neuronal nicotinic acetylcholine receptors. ACS Omega 2017; 2: 4621-4631.
| Crossref | Google Scholar | PubMed |
52 Knuhtsen A, Whitmore C, McWhinnie FS, McDougall L, Whiting R, Smith BO, Timperley CM, Green AC, Kinnear KI, Jamieson AG. α-Conotoxin GI triazole-peptidomimetics: potent and stable blockers of a human acetylcholine receptor. Chem Sci 2019; 10: 1671-1676.
| Crossref | Google Scholar |
53 Lang PM, Burgstahler R, Haberberger RV, Sippel W, Grafe P. A conus peptide blocks nicotinic receptors of unmyelinated axons in human nerves. Neuroreport 2005; 16: 479-483.
| Crossref | Google Scholar | PubMed |
54 Satkunanathan N, Livett B, Gayler K, Sandall D, Down J, Khalil Z. Alpha-conotoxin Vc1.1 alleviates neuropathic pain and accelerates functional recovery of injured neurones. Brain Res 2005; 1059: 149-158.
| Crossref | Google Scholar | PubMed |
55 Wright AB, Norimatsu Y, McIntosh JM, Elmslie KS. Limited efficacy of α-conopeptides, Vc1.1 and RgIA, to inhibit sensory neuron CaV current. eNeuro 2015; 2(1): e0057-0014.2015.
| Crossref | Google Scholar | PubMed |
56 Plazas PV, Katz E, Gomez-Casati ME, Bouzat C, Elgoyhen AB. Stoichiometry of the α9α10 nicotinic cholinergic receptor. J Neurosci 2005; 25: 10905-10912.
| Crossref | Google Scholar | PubMed |
57 Callaghan B, Adams DJ. Analgesic α-conotoxins Vc1.1 and Rg1A inhibit N-type calcium channels in sensory neurons of α9 nicotinic receptor knockout mice. Channels 2010; 4: 51-54.
| Crossref | Google Scholar | PubMed |
58 Christensen SB, Hone AJ, Roux I, Kniazeff J, Pin JP, Upert G, Servent D, Glowatzki E, McIntosh JM. RgIA4 potently blocks mouse α9α10 nAChRs and provides long-lasting protection against oxaliplatin-induced cold allodynia. Front Cell Neurosci 2017; 11: 219.
| Crossref | Google Scholar | PubMed |
59 Galvez T, Parmentier M-L, Joly C, Malitschek B, Kaupmann K, Kuhn R, Bittiger H, Froestl W, Bettler B, Pin J-P. Mutagenesis and modeling of the GABAB receptor extracellular domain support a venus flytrap mechanism for ligand binding. J Biol Chem 1999; 274: 13362-13369.
| Crossref | Google Scholar | PubMed |
60 Bony AR, McArthur JR, Komori A, Wong AR, Hung A, Adams DJ. Analgesic α-conotoxin binding site on the human GABAB receptor. Mol Pharmacol 2022; 102: 196-208.
| Crossref | Google Scholar | PubMed |
61 Finnerup NB, Sindrup SH, Jensen TS. The evidence for pharmacological treatment of neuropathic pain. Pain 2010; 150: 573-581.
| Crossref | Google Scholar | PubMed |
62 Woolf CJ, Mannion RJ. Neuropathic pain: aetiology, symptoms, mechanisms, and management. Lancet 1999; 353(9168): 1959-1964.
| Crossref | Google Scholar | PubMed |