

A new divalent organoeuropium(II) fluoride and serendipitous discovery of an alkoxide complex from pentaphenylcyclopentadiene precursors†
Angus C. G. Shephard A , Aymeric Delon A B , Rory P. Kelly C , Zhifang Guo A , Sylviane Chevreux B D , Gilles Lemercier B , Glen B. Deacon C , Galina A. Dushenko E , Florian Jaroschik F * and Peter C. Junk A *
A *
A College of Science & Engineering, James Cook University, Townsville, Qld 4811, Australia.
B ICMR, UMR CNRS 7312, Université de Reims Champagne-Ardenne, Reims, France.
C School of Chemistry, Monash University, Clayton, Vic. 3800, Australia.
D Institut de Recherche de Chimie Paris, UMR CNRS 8247, Chimie ParisTech, PSL University, 75005 Paris, France.
E Institute of Physical and Organic Chemistry, Southern Federal University, Rostov-on-Don, 344090, Russia.
F ICGM, Univ Montpellier, CNRS, ENSCM, Montpellier, France.
Handling Editor: George Koutsantonis
Australian Journal of Chemistry 75(9) 746-753 https://doi.org/10.1071/CH21324
Submitted: 6 December 2021 Accepted: 17 February 2022 Published: 29 March 2022
© 2022 The Author(s) (or their employer(s)). Published by CSIRO Publishing. This is an open access article distributed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License (CC BY-NC-ND)
Abstract
From the redox-transmetallation protolysis (RTP) reaction of europium metal, Hg(C6F5)2 and pentaphenylcyclopentadiene, we isolated and crystallographically characterised small amounts of the first divalent europium fluoride half-sandwich complex [Eu(C5Ph5)(μ-F)(thf)2]2 (1). Subsequently, a rational synthesis of this complex from in situ formed [EuF2(thf)n] and [Eu(C5Ph5)2] was carried out. In addition, the new divalent Eu alkoxide complex [Eu(OC5Ph5*)2(thf)4] (2) (OC5Ph5* = 2,3,4,5,5 pentaphenylcyclopenta-1,3-dienolate) was identified by X-ray diffraction analysis, in which an intriguing phenyl group migration in the cyclopentadiene ligand occurred. This complex was shown to be derived from small impurities of 1,2,3,4,5-pentaphenylcyclopenta-1,3-dienol (C5Ph5OH) in the C5Ph5H starting material and was then synthesised on a larger scale. Density functional theory calculations provided evidence for the facile phenyl group migration observed in the cyclopentadienolate ring.
Keywords: 2,3,4,5,5-pentaphenylcyclopenta-1,3-dienolate, bis(pentafluorophenyl)mercury, C–F activation, DFT calculations, Europium metal, pentaphenylcyclopentadiene, redox transmetallation/protolysis.
Introduction
Pentaphenylcyclopentadiene and related polyarylcyclopentadiene ligands have long been studied in transition metal chemistry,[1–4] whereas in f-element chemistry, they have drawn some attention only over the last 15 years.[5–13] The groups of Harder and ourselves have discovered two complementary synthetic pathways to divalent decaaryl lanthanoid complexes as shown in Scheme 1.[5–10] These highly bulky planar sandwich complexes display limited redox activity for Sm and Yb,[7,9] and in the case of europium, interesting luminescence properties have been observed.[6,10]

|
For the poorly soluble decaphenyleuropocene complex [Eu(C5Ph5)2], we have effected a redox-transmetallation protolysis (RTP) approach starting from europium metal and C5Ph5H and using either HgPh2 or Hg(C6F5)2 as a redox-transmetallating reagent (Scheme 1b).[10] Initially, in thf, a solvent-separated ion pair (SSIP) is formed, from which the sandwich complex was isolated by precipitation from toluene. As Hg(C6F5)2 often shows higher reactivity in RTP reactions than HgPh2,[14] the synthesis was performed by stirring at room temperature, whereas sonication was required with HgPh2. In this paper, we report the synthesis of the first divalent cyclopentadienyleuropium fluoride, and a complex of the 2,3,4,5,5-pentaphenylcyclopenta-1,3-dienolate ion.
Results and discussion
During an RTP reaction of Eu with Hg(C6F5)2 and C5Ph5H, a small crop of bright yellow crystals formed from the thf solution, and had a colour which was in striking contrast to the bright orange sandwich complex [Eu(C5Ph5)2] (Scheme 2a). X-ray diffraction (XRD) analysis of this new compound revealed the formation of the first divalent europium fluoride half-sandwich complex [Eu(C5Ph5)(μ-F)(thf)2]2 1 (Fig. 1). In the light of previous work on the synthesis of the divalent ytterbium fluoride complex [Yb(C5Ph4H)(µ-F)(thf)2]2,[15] we considered that the in situ formed C6F5H is the fluoride source. Indeed, repeating this reaction and monitoring by 19F NMR spectroscopy revealed the formation of some p-C6F4H2, indicative of C–F activation of the C6F5H by europium metal (used in excess in these reactions). A higher yielding synthesis of the europium fluoride complex 1 was then carried out by treatment of the isolated sandwich complex [Eu(C5Ph5)2] with [EuF2(thf)n] (Scheme 2c), formed from the C–F activation of C6F5H with Eu metal in thf (Scheme 2b). NMR spectroscopic characterisation of this complex was excluded by the paramagnetic nature of Eu2+.

|
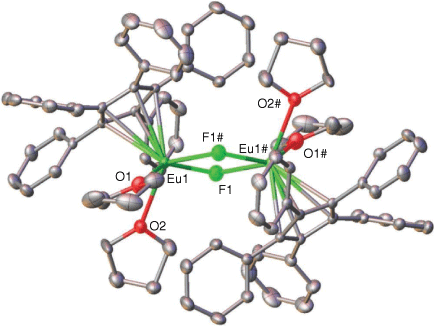
|
Complex 1 (Fig. 1) crystallises in the orthorhombic space group Pbca as a symmetrical dimer. The two Eu atoms are seven-coordinate, ligated by one C5Ph5 ring, two thf molecules and two bridging fluoride ions. The Eu–F bond lengths (Eu(1)–F(1) = 2.406(2) Å, Eu(1)–F(1)# = 2.391(2) Å) are comparable with the Yb–F bond lengths of the previously reported [Yb(C5Ph4H)(µ-F)(thf)2]2 (Yb(1)–F(1) = 2.2515(17) Å and Yb(1)–F(1)# = 2.2546(18) Å),[15] after consideration of the larger ionic radius of Eu2+,[16] alongside the increase in steric bulk from C5Ph4H− to C5Ph5− (steric coordination numbers[17] 3.3 and 3.8 respectively[10]). The Eu–C and Eu–O bond lengths are longer than those in the samarium bromide half-sandwich complex [Sm(C5Ph5)(µ-Br)(thf)2]2,[10] e.g. Eu–C(centroid) = 2.7023 Å vs Sm–C(centroid) = 2.636 Å) despite the larger ionic radius of Sm2+.[16] This might be explained by the shorter fluoride bridge in 1 leading to a more crowded environment around the metal centres as shown by the much shorter Eu–Eu distance (3.878 Å) than the longer bromide-bridged Sm–Sm distance (4.662 Å) in [Sm(C5Ph5)(µ-Br)(thf)2]2.
From another RTP reaction of Eu, Hg(C6F5)2 and C5Ph5H, we observed the formation of yellow crystals after filtration and leaving the reaction mixture to stand at room temperature for several days. Single crystal XRD analysis revealed the formation of the new divalent Eu–alkoxide complex [Eu(OC5Ph5*)2(thf)4] 2 (OC5Ph5* = 2,3,4,5,5 pentaphenylcyclopenta-1,3-dienolate) (Fig. 2). Complex 2 crystallised in the monoclinic space group P21/c. The Eu atom is six-coordinate with a trigonal prismatic donor array, ligated by two phenyl-migrated 2,3,4,5,5 pentaphenylcyclopenta-1,3-dienolate moieties, and four thf molecules. A near linear O–Eu–O arrangement is observed (O(1)–Eu(1)–O(2) = 166.49(6)°). This coordination environment is rare, with very few examples reported in the literature.[18] Two noteworthy examples include [Eu{P(H)Mes*}2(thf)4] (Mes* = 2,4,6-tBu3C6H2), with P–Eu–P = 160.5(1)°,[19] and [Eu(pz)2(thf)4] (pz = 3,5-diphenylpyrazolate), with C–Eu–C = 152.815°.[20] The more common arrays for alkoxides/aryloxides are alkoxy bridged dimers where the alkoxide has low steric bulk and five coordinate monomers of variable arrangements (both cis and trans OR) with bulky aryloxides.[18] The Eu–O alkoxide bond lengths in 2 (Eu(1)–O(1) = 2.4096(16) Å and Eu(1)–O(2) = 2.3723(16) Å) are longer than those reported for Eu phenolate complexes such as [Eu(OPh(2,6-tBu)2(4-Me))2(thf)3] (Eu–O(phenolate) = 2.315(6) Å and 2.322(5) Å).[21] Despite this difference, the O(phenolate)–Eu–O(phenolate) angle (151.2(3)°) is similar to that of 2.
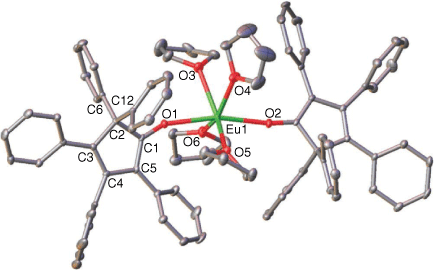
|
The formation of 2 could either be explained by oxygen activation of the divalent europium sandwich complex or more likely by the presence of some impurities of 1,2,3,4,5-pentaphenylcyclopenta-1,3-dienol (C5Ph5OH), a precursor of C5Ph5H in the starting material. In order to gain further insights on the synthesis of this complex, an RTP reaction between Eu metal, Hg(C6F5)2 and C5Ph5OH was conducted in thf at room temperature (Scheme 3). The reaction went readily to completion as indicated by 19F NMR spectroscopy and 2 was isolated in 40% yield. The compound was analysed by infrared spectroscopy (see Supplementary Material) and elemental analysis but, due to the paramagnetism of Eu2+, NMR spectroscopic studies could not be conducted.
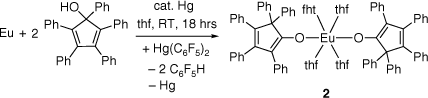
|
Quenching the reaction mixture with water provided the corresponding ketone, 2,2,3,4,5-pentaphenylcyclopent-3-enone 3, as shown by 1H and 13C NMR and IR spectroscopy in agreement with literature reports (Scheme 4).[22] This outcome further confirms the identity of 2, and the phenyl group migration during the RTP reaction. XRD characterisation was undertaken on 3, confirming the connectivity of the ketone (Scheme 4).
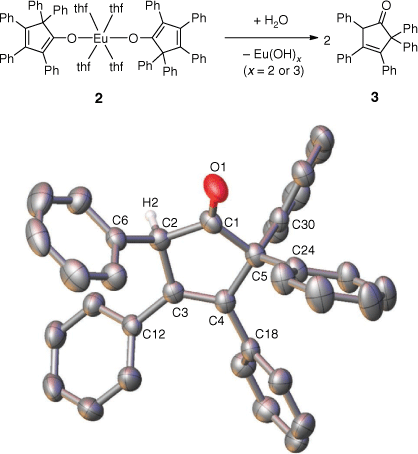
|
In order to exclude that the phenyl ring migration had already occurred in the C5Ph5OH starting material, NMR spectroscopy and XRD characterisations were performed on the starting material, verifying the structure[23] and showing high stability of this ligand under ambient conditions (Fig. 3).
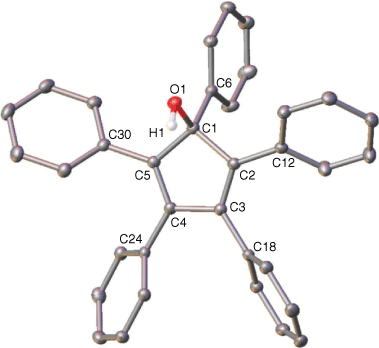
|
The phenyl group migration in pentaphenylcyclopentadienol has previously been studied and requires harsh reaction conditions, e.g. heating C5Ph5OH in tetraethylene glycol at 150–210°C afforded the ketone 3 with an experimentally determined energy barrier of 34.7 kcal/mol.[22,24] On the other hand, reaction of tetraphenylcyclone with pentafluorophenyllithium at −78°C followed by hydrolysis led to modest yield of both 2-pentafluorophenyl-2,3,4,5-tetraphenylcyclopent-3-ene-1-one (the migration product) and 1-pentafluorophenyl-2,3,4,5-tetraphenylcyclopenta-2,4-dienol.[25] In our case phenyl migration occurs at room temperature, and without oxidation of the alkoxide functionality. We therefore carried out some theoretical investigations on the phenyl group migration starting either from the neutral alcohol precursor A or the anionic alkoxide C to obtain some understanding of this process (Fig. 4).

|
Density functional theory quantum chemical calculations were performed at the CAM-B3LYP/Def2TZVP level of theory using the Gaussian-09 program for the gas phase (see Supplementary Material for full information). Supplementary Fig. S1 shows the calculated geometric parameters of the structure A, which agree well with X-ray diffraction data. In compound A a 1,5-sigmatropic shift of a phenyl group around the five-membered ring through transition state TS1 occurs. This shift has an energy barrier of ΔEZPE≠ = 35.0 kcal/mol and results in the formation of isomer B. Isomer A is slightly more stable than B by ΔEZPE 1.9 kcal/mol. In TS1, the distances between the migrating carbon atom of the phenyl group and nearest carbon atoms of the Cp ring are: 1.875 and 1.889 Å. The calculated total charge (Mulliken hereinafter) on the migrating phenyl group is close to 0 (0.003 e), which is typical for sigmatropic shifts. In all the calculated structures hereinafter, phenyl groups at sp2-hybridised carbon atoms occupy the propeller conformation. The result is highly consistent with the experimental data.[22]
In contrast, in the anion C, intramolecular migration of the phenyl group occurs much faster than in A through the transition state TS2 with an energy barrier of ΔEZPE≠ = 14.0 kcal/mol resulting in the formation of isomer D (Fig. 4). In the anionic structure of TS2, the distances between the migrating carbon atom of the phenyl group and nearest carbon atoms of the Cp ring are longer than in TS1: 2.046 and 2.121 Å (Fig. S2). The calculated total charge on the migrating phenyl group is negative (−0.247 e). The value of the calculated energy barrier in C indicates the possibility of rearrangement during the reaction at room temperature almost instantaneously. Isomeric anion D is significantly more stable than C at ΔEZPE 23.5 kcal/mol. Such a difference in energies for C and D, as well as a rather low migration barrier for the phenyl group in comparison with the previously known ones,[26] is most likely associated with the delocalisation of the negative charge between the oxygen atom and the cyclopentadiene ring in TS2 and D. This is indicated by the alignment of the corresponding bonds of the five-membered ring, a decrease in the length of the C–O bond in TS2 and D, and a lower value of the negative charge on the oxygen atom in TS2 (−0.476 e) and in D (−0.436 e) compared to C (−0.626 e).
Isomer B then further transforms to the final ketone product 3, which is more stable than isomer A by ΔEZPE 4.4 kcal/mol, via migration of a hydrogen. The migration of a hydrogen atom in B through transition state TS3 (Fig. S3) by intramolecular 1,3-sigmatropic shift (which is forbidden by the Woodward–Hoffmann rules) requires overcoming a very high energy barrier ΔEZPE≠ = 66.72 kcal/mol according to the calculations. This barrier value indicates the impracticability of the B→TS3→3 reaction mechanism. An alternative mechanism could be the intermolecular transfer of hydrogen with the participation of a solvent or, for example, water, which may be contained in the solvent as an impurity. Since the A→B→3 conversion was carried out in tetraethylene glycol, to simplify the calculations, the complex B × MeOH (B with one methanol molecule) was considered. For the methanol complexes B × MeOH and 3 × MeOH (3 with one methanol molecule) calculations were performed at CAM-B3LYP/Def2SVP level. In complex B × MeOH, the hydrogen atom H1 (at the oxygen bound with the Cp ring) migrates to the methanol oxygen atom, whereas the methanol hydrogen atom H2 migrates to the Cp carbon atom through the formation of the six-membered transition state TS4 (Fig. S4). As a result, the complex 3 × MeOH is formed. Calculated energy barrier for the proccess B × MeOH→TS4→3 × MeOH is ΔEZPE≠ = 28.6 kcal/mol. Сomplex 3 × MeOH is more stable than B × MeOH by ΔEZPE 4.1 kcal/mol. In TS4, the distances between the migrating hydrogen atoms and the oxygen at the Cp ring and the Cp carbon atoms are: 1.489 and 1.555 Å respectively. The charges on the migrating hydrogen atoms in TS4 are positive 0.244 e (H1) and 0.253 e (H2). The calculated energy barrier value for intermolecular mechanism of the hydrogen migrations indicates that the rate determining stage of the A→B→3 conversion is the 1,5-shift of a phenyl group A→B and agrees with experimental data.
Conclusions
The first divalent organoeuropium(II) fluoride, [Eu(C5Ph5)(μ-F)(thf)2]2 1, was prepared initially in low yield by an RTP reaction between Eu metal, Hg(C6F5)2 and C5Ph5H, and then deliberately by reaction of in situ generated [EuF2(thf)n], from C–F activation of C6F5H by Eu metal, with [Eu(C5Ph5)2]. The complex is a symmetrical seven coordinate dimer with two bridging fluoride ions. The alkoxide complex, [Eu(OC5Ph5*)2(thf)4] 2, was serendipitously isolated after a similar RTP reaction, and then deliberately prepared by an RTP reaction between Eu metal, Hg(C6F5)2, and C5Ph5OH in thf. The complex has a six coordinate europium atom with transoid alkoxide ligands and equatorial thf donors. The alkoxide C5Ph5O− was isomerised into C5Ph5O*− as shown by hydrolysis of 2 into the ketone, 2,2,3,4,5-pentaphenylcyclopent-3-enone 3.
Experimental
General remarks
All manipulations were performed under nitrogen, using standard Schlenk techniques. Thf was distilled from sodium benzophenone before use. Pentafluorobenzene was commercially available, and used without further purification. Bis(pentafluorophenyl)mercury,[27] bis(pentaphenylcyclopentadienyl)europium[10] and 1,2,3,4,5-pentaphenylcyclopenta-1,3-dienol[23] were prepared by the literature methods. Infrared spectra (4000–400 cm−1) were obtained as Nujol mulls between NaCl plates, or as neat powders by attenuated total reflectance (ATR) with a Nicolet-Nexus FT-IR spectrometer. 1H and 13C-NMR spectra were recorded on a Bruker 400 MHz spectrometer. The chemical shifts were referenced to residual solvent peaks. Elemental analyses were obtained from the Chemical Analysis Facility, Macquarie University in Sydney. XRD data and refinement details are given in Table S1. CCDC 2126375–2126378 for compound 1–3 and C5Ph5OH respectively, contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
[(Eu(C5Ph5)(μ-F)(thf)2)2] (1)
Method 1
Thf (10 mL) was added to a Schlenk flask charged with freshly filed europium metal (0.39 g, 2.6 mmol), Hg(C6F5)2 (0.19 g, 0.36 mmol) and C5Ph5H (0.32 g, 0.72 mmol) and the suspension was sonicated at 40°C for 3 days giving a dark golden yellow solution. The solution was filtered and concentrated under vacuum. Overnight, a few yellow single crystals of [Eu(C5Ph5)(μ-F)(thf)2]2 deposited that were suitable for X-ray crystallography. No other characterisation could be obtained.
Method 2
A Schlenk flask was charged with Eu metal (0.300 g, 2.0 mmol), C6F5H (1.1 mL, 10 mmol), anhydrous thf (4 mL) and a piece of iodine for metal activation, which was then stirred for 5 days. The suspension was allowed to settle, and the supernatant solution removed by filter cannula, and the solid dried under reduced pressure, leaving unreacted Eu and EuF2(thf)n. A solution of [Eu(C5Ph5)2] (0.042 g, 0.047 mmol) in anhydrous thf (5 mL) was transferred into the EuF2(thf)n (excess) and the resulting suspension was stirred overnight, yielding a bright yellow solution. The suspension was allowed to settle, and the resulting solution isolated by filter cannula. The solvent was then removed under reduced pressure, yielding 1 as a pale brown solid (0.033 g, 46%). Anal. calc. for C86H82F2O4Eu2 (1521.5 g/mol): C, 67.89; H, 5.43. Found C, 67.85; H, 4.75%. IR (Nujol, cm−1): 1594m, 1500m, 1261w, 1155w, 1071w, 1029m, 908w, 802m, 769m, 737w, 697m.
[Eu(OC5Ph5*)2(thf)4] (2)
Method 1
Thf (10 mL) was added to a Schlenk flask charged with freshly filed europium metal (0.39 g, 2.6 mmol), Hg(C6F5)2 (0.19 g, 0.36 mmol) and C5Ph5H (0.32 g, 0.72 mmol) (contaminated with a small amount of C5Ph5OH) and the suspension was sonicated at 40°C for 3 days giving a dark golden yellow solution. The solution was filtered and concentrated under vacuum. After several days, a few yellow single crystals of [Eu(OC5Ph5*)2(thf)4] deposited that were suitable for X-ray crystallography (yield < 5%). No other characterisation could be obtained.
Method 2
A Schlenk flask was charged with C5Ph5OH (0.230 g, 0.5 mmol), Hg(C6F5)2 (0.133 g, 0.25 mmol) and Eu metal filings (0.152 g, 1.0 mmol). Anhydrous thf (5 mL) and a drop of Hg metal (to form a reactive europium-mercury amalgam) were added, and the reaction mixture stirred overnight (18 h) at room temperature. The resulting suspension was allowed to settle before isolating the supernatant solution by a filtration cannula. The resultant dark yellow filtrate was dried under reduced pressure and washed with anhydrous hexane (2 × 5 mL) yielding a dark orange powder 2 (0.135 g, 40%). Anal. calc. for C86H82O6Eu (1363.53 g/mol): C, 75.75; H, 6.06. Found C, 75.71; H, 6.07%. IR (Nujol, cm−1): 3050m, 3024m, 1945w, 1878w, 1804w, 1593s, 1521s, 1486s, 1459s, 1440s, 1378s, 1342m, 1322w, 1305w, 1278w, 1260m, 1157w, 1069w, 1028s, 916m, 878m, 812w, 750s, 739m, 721w, 699s, 637s, 618w, 549m.
Hydrolysis of 2 to afford 2,2,3,4,5-pentaphenylcyclopent-3-enone (3)
An aliquot (~1 mL) of the reaction mixture of 2 was taken and added directly into distilled water and stirred for 5 min. The organic material was extracted with dichloromethane (2 × 5 mL), and combined before washing with brine, and then stirring over MgSO4. The resulting solution was filtered and solvent removed under reduced pressure, yielding 3 as a pale-yellow powder. Crystals of 3 were grown from the slow evaporation of a 1:1 thf:EtOH solution. 1H-NMR (400 MHz, CDCl3, 25°C): δ = 7.63 (m, 2H, ArH), 7.51 (m, 11H, ArH), 7.32 (m, 10H, ArH), 7.09 (m, 2H, ArH), 5.22 (s, 1H, C(Ph)H ppm. 13C-NMR (101 MHz, CDCl3, 25°C): δ 211.45 (s), 142.74 (s), 139.56 (s), 139.40 (s), 138.48 (s), 135.32 (s), 135.16 (s), 134.30 (s), 129.63 (s), 129.24 (s), 128.29 (s), 128.00 (s), 127.56 (s), 127.30 (s), 127.01 (s), 126.89 (s), 126.72 (s), 126.34 (s), 126.29 (s), 126.12 (s), 72.43 (s), 59.89 (s). IR (ATR, cm−1): 3056w, 3026w, 2961m, 1748s, 1598m, 1574w, 1493s, 1442m, 1410w, 1259s, 1181m, 1093s, 1072s, 1028s, 797s, 741s, 693s, 626m, 551s, 504m. MS (APCI) m/z: calc. for C35H26O (462.2 + 1). Found 463 (M+ + 1). Spectroscopic data were in agreement with those reported.[22]
Crystal and refinement data
Single crystals of 1 were covered with viscous hydrocarbon oil and were mounted on loops. Data were obtained at 123 K on a Bruker X8 APEX II CCD diffractometer equipped with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). For complex 2, a single crystal covered with oil based cryoprotectant was mounted on a cryoloop. The single crystal XRD measurement was carried out at 100 K on a Bruker D8 Venture equipped with a fine-focus sealed tube with a Triumph graphite monochromator displaying Mo Kα1 wavelength (λ = 0.7103 Å) and a PHOTON100 CMOS detector. Data were collected using Bruker Apex2 software. Single crystals of C5Ph5OH were coated with viscous hydrocarbon oil and mounted on glass loops, and data were collected on a Rigaku SynergyS diffractometer. The SynergyS operated using microsource Cu-Kα radiation (λ = 1.54178 Å) at 123 K. Data processing was conducted using CrysAlisPro.55 software suite.[28] Single crystals of 3 were mounted on loops. Data were obtained at 190 K on an Oxford Diffraction Gemini Ultra S diffractometer, using Cu-Kα radiation (λ = 1.54184 Å). The structures were solved using SHELXS7 and refined by full-matrix least-squares on all F2 data using SHELX2014[29] in conjunction with the X-Seed graphical user interface.[30] All hydrogen atoms were placed in calculated positions using the riding model. Data collection and refinement details are collated in Table S1.
Data availability
The data that support this study are available in the article and accompanying online supplementary material.
Conflicts of interest
Peter Junk is the Guest Editor of the Australian Journal of Chemistry, but was blinded from the peer review process for this paper. The authors declare no other conflicts of interest.
Declaration of funding
ACGS would like to acknowledge the Australian Government’s support of this research through an Australian Government Research Training Program scholarship. AD thanks the Région Grand Est (France) and the JCU for a PhD scholarship (project EURO2LUM). GAD acknowledges financial support by the Ministry of Science and Higher Education of the Russian Federation (grant No. 0852-2020-0031). The CNRS and the University of Reims Champagne-Ardennes are acknowledged for financial support. The authors acknowledge support by the Australian Research Council (DP190100798). The authors acknowledge crystallographic assistance by Professor P. V. Bernhardt and Dr. C. M. Forsyth.
Supplementary material
Supplementary material containing IR and NMR spectra, crystallographic data and computational results is available online.
References
[1] LD Field, CM Lindall, AF Masters, GKB Clentsmith, Coord Chem Rev 2011, 255, 1733.| Crossref | GoogleScholarGoogle Scholar |
[2] R Stefak, AM Sirven, S Fukumoto, H Nakagawa, G Rapenne, Coord Chem Rev 2015, 287, 79.
| Crossref | GoogleScholarGoogle Scholar |
[3] MC Warner, J-E Baeckvall, Acc Chem Res 2013, 46, 2545.
| Crossref | GoogleScholarGoogle Scholar | 23721454PubMed |
[4] N Kataoka, Q Shelby, JP Stambuli, JF Hartwig, J Org Chem 2002, 67, 5553.
| Crossref | GoogleScholarGoogle Scholar | 12153253PubMed |
[5] C Ruspic, JR Moss, M Schürmann, S Harder, Angew Chem Int Ed 2008, 47, 2121.
| Crossref | GoogleScholarGoogle Scholar |
[6] S Harder, D Naglav, C Ruspic, C Wickleder, M Adlung, W Hermes, M Eul, R Pöttgen, DB Rego, F Poineau, KR Czerwinski, RH Herber, I Nowik, Chem Eur J 2013, 19, 12272.
| Crossref | GoogleScholarGoogle Scholar | 23907896PubMed |
[7] NJC Van Velzen, S Harder, Organometallics 2018, 37, 2263.
| Crossref | GoogleScholarGoogle Scholar |
[8] CM Forsyth, GB Deacon, LD Field, C Jones, PC Junk, DL Kay, AF Masters, AF Richards, Chem Commun 2006, 1003.
| Crossref | GoogleScholarGoogle Scholar |
[9] GB Deacon, CM Forsyth, F Jaroschik, PC Junk, DL Kay, T Maschmeyer, AF Masters, J Wang, LD Field, Organometallics 2008, 27, 4772.
| Crossref | GoogleScholarGoogle Scholar |
[10] RP Kelly, TDM Bell, RP Cox, DP Daniels, GB Deacon, F Jaroschik, PC Junk, XF Le Goff, G Lemercier, A Martinez, J Wang, D Werner, Organometallics 2015, 34, 5624.
| Crossref | GoogleScholarGoogle Scholar |
[11] Y Wang, J Cheng, New J Chem 2020, 44, 17333.
| Crossref | GoogleScholarGoogle Scholar |
[12] Y Wang, I Del Rosal, G Qin, L Zhao, L Maron, X Shi, J Cheng, Chem Commun 2021, 57, 7766.
| Crossref | GoogleScholarGoogle Scholar |
[13] G Qin, Y Wang, X Shi, I Del Rosal, L Maron, J Cheng, Chem Commun 2019, 55, 8560.
| Crossref | GoogleScholarGoogle Scholar |
[14] Z Guo, R Huo, YQ Tan, V Blair, GB Deacon, PC Junk, Coord Chem Rev 2020, 415, 213232.
| Crossref | GoogleScholarGoogle Scholar |
[15] GB Deacon, F Jaroschik, PC Junk, RP Kelly, Chem Commun 2014, 50, 10655.
| Crossref | GoogleScholarGoogle Scholar |
[16] RD Shannon, Acta Crystallogr Sect A 1976, 32, 751.
| Crossref | GoogleScholarGoogle Scholar |
[17] J Marçalo, AP De Matos, Polyhedron 1989, 8, 2431.
| Crossref | GoogleScholarGoogle Scholar |
[18] The survey was performed with CCDC: ConQuest 2020.3.0.
[19] GW Rabe, IA Guzei, AL Rheingold, Inorg Chem 1997, 36, 4914.
| Crossref | GoogleScholarGoogle Scholar |
[20] Z Guo, VL Blair, GB Deacon, PC Junk, Chem Eur J 2022, e202103065.
[21] GB Deacon, S Hamidi, PC Junk, RP Kelly, J Wang, Eur J Inorg Chem 2014, 3, 460.
| Crossref | GoogleScholarGoogle Scholar |
[22] GB Oldaker, TA Perfetti, MA Ogliaruso, J Org Chem 1980, 45, 3910.
| Crossref | GoogleScholarGoogle Scholar |
[23] JW Chambers, MD Rausch, AJ Baskar, SG Bott, JL Atwood, Organometallics 1986, 5, 1635.
| Crossref | GoogleScholarGoogle Scholar |
[24] CW Spangler, Chem Rev 1976, 76, 187.
| Crossref | GoogleScholarGoogle Scholar |
[25] HK Gupta, M Stradiotto, DW Hughes, MJ McGlinchey, J Org Chem 2000, 65, 3652.
| Crossref | GoogleScholarGoogle Scholar | 10864748PubMed |
[26] VI Minkin, IE Mikhailov, GA Dushenko, A Zschunke, Russ Chem Rev 2003, 72, 867.
[27] Deacon GB, Cosgriff JE, Lawrenz ET, Forsyth CM, Wilkinson DL. Section 2.3: Organolanthanide(II) Complexes, In Herrmann WA, editor. Herrmann-Brauer, Synthetic Methods of Organometallic and Inorganic Chemistry. Stuttgart: Thieme; 1997, vol. 6, p. 48.
[28] CrysAlisPRO v.39. Yarnton, Oxfordshire, England: Agilent Technologies Ltd.
[29] GM Sheldrick, Acta Crystallogr Sect C Struct Chem 2015, 71, 3.
| Crossref | GoogleScholarGoogle Scholar |
[30] LJ Barbour, J Supramol Chem 2001, 1, 189.
| Crossref | GoogleScholarGoogle Scholar |
† We dedicate this paper to Professor Glen Deacon, being an icon, not only in his beloved field of organolanthanoid chemistry, but in essentially all fields of chemistry. He has been a mentor, supervisor and good friend of many, many students and colleagues.