

Personal Accounts of Australian Drug Discovery at the Public–Private Interface*
Jonathan B. Baell A
A
A Medicinal Chemistry Theme, Monash Institute of Pharmaceutical Sciences (Parkville Campus), Monash University, Parkville, Vic. 3052, Australia. Email: jonathan.baell@monash.edu
Australian Journal of Chemistry 74(1) 16-27 https://doi.org/10.1071/CH20244
Submitted: 5 August 2020 Accepted: 8 September 2020 Published: 6 October 2020
Journal Compilation © CSIRO 2021 Open Access CC BY-NC-ND
Abstract
The public–private interface is a vibrant and invigorating stage for drug discovery and can allow for relatively higher risk but more rewarding research. Although adequate resourcing is a perennial challenge, persistence, optimism, and flexibility will pay dividends and can allow for a thoroughly rewarding career. In this account of chronological research experiences, selected examples are used to support this contention.
Introduction
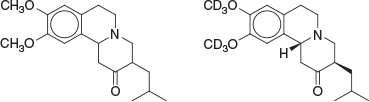
Perhaps the most fitting way to introduce what became a career in medicinal chemistry is to outline that which might be said to underpin it, this being those all-important formative postgraduate studies. This began in the late 1980s at the University of Tasmania, the chemistry department of which operated at a very high standard, with names such as Frank Larkins (physical, Head of Department), John Bremner (organic), Elaine Browne (organic), Alan Arnold (spectroscopy), Michael Hitchman (inorganic), Kingsley Cavell (inorganic), Rudi Thomas (inorganic), Alan Canty (inorganic), Peter Smith (inorganic and industrial), Barry O’Grady (physical), Lawrie Dunn (physical), Noel Roberts (physical), and Adrian Blackman (natural products) being prominent. In 1986, I had the pleasure of undertaking Honours here, under the supervision of John Bremner and Elaine Browne. The Honours year was challenging. Coursework examinations included being able to calculate proton NMR spectra from first principles, and all-night laboratory efforts were commonplace in order to achieve the practical outcomes deemed necessary by us students. For my project, outcomes were satisfying and these included successful identification of a new heterocyclic skeleton, this being indolo[1′,2′:3,4]pyrimido[6,1-a]isoquinoline.[1] Another focus of the project was on the chemistry of tetrabenazine (Fig. 1), an alkaloid-like tetrahydroisoquinoline that was initially synthesised in the 1950s at the research laboratory of Hoffmann–La Roche in Basel.[2]
|
Observed to have reserpine-like effects in mice, tetrabenazine went on to become a relatively well known monoamine-depleting agent, now with decades of clinical use in several countries for various psychoses and commonly known as Xenazine, Xentra, or Nitoman. After sustained pharmacological scrutiny, tetrabenazine was approved by the USA FDA in August 2008 (Ovation Pharmaceuticals/Biovail) for the treatment of chorea associated with Huntington’s disease.[3] With improved activity against the elucidated pharmacological target (VMAT2, or vesicular monoamine transporter 2), the deuterated eutomer deutetrabenazine (Fig. 1) was approved by the USA FDA in early 2017 for the same indication and sold by Teva Pharmaceuticals as Austedo. The first deuterated drug approved by the FDA, deutetrabenazine has improved pharmacokinetic properties as a result of rendering the methoxy groups more metabolically robust. Presumably this combination of features had been deemed sufficiently novel for patenting purposes and may impart ideas to the readers of this article for their own particular projects.
This is a good example of one of the most successful routes to the discovery of new drugs, and that is taking a known bioactive compound, interrogating it, and improving it by some measure using medicinal chemistry. Parenthetically I note that bioactive ketones in small synthetic molecules tend to be relatively less a focus of medicinal chemistry efforts. Possibly this is because they are occasionally associated with toxicity, but I believe this is anecdotal and may be unsubstantiated. It is a functional group that I have observed quite frequently in the context of central nervous system (CNS)-active compounds. Although I am not aware of any published analyses, in my mind I consider that a ketone could be privileged for CNS activity and this influences our approach today in CNS-relevant medicinal chemistry projects.
From 1987, I undertook a PhD studying conformational constraints in peptide-based drug design, under the supervision of Peter Andrews and Paul Alewood, at the Victorian College of Pharmacy (VCP), now called Monash Institute of Pharmaceutical Sciences. I suspect I was a relatively intolerable PhD student and like many such students, only realised many years later the significance of the various roles that supervisors play in the process of a student’s development. Several publications arose from my PhD, with perhaps a reclassification of a protein secondary structure known as the β-turn generating the most interest,[4] now numbering in total some 260 citations. Years later when using Google Scholar metrics in various applications I belatedly noticed that these early publications were absent. The reason was that these publications were listed under my family surname Ball, and did not recognise my subsequent attempt to change the societal tradition through husband (Ball) and wife (Bell) adopting an amalgamated name (Baell) upon marriage. Over the years it has taken some effort to secure formal acknowledgement by various database operators that these were indeed authored by myself!
These were the days when there was considerable freedom for a student without today’s carefully managed approach and its common insistence on back-up plans ‘just in case things do not pan out as expected’: in my experience, things rarely pan out as expected when the best research is undertaken. In any case, I pretty much had a free rein, to the extent that I could see my thesis title in danger of needing alteration to ‘The Medicinal Chemistry of Carbon’ if I did not moderate my explorations. During this period, I recall reading a powerful commentary ‘Why doesn’t Australia have a research-based pharmaceutical industry?’, which was an impassioned plea published in the brave new world of 1984. That article, which embodied the keen interest of Peter Andrews in seeing Australia do a better job of discovering Australian drugs, left me with an indelible impression. Unfortunately, in the intervening years, as a nation we have not done so well in countering this implied criticism. In those halcyon days, Peter (Andrews), Paul (Alewood), and a handful of other disciples made the journey north to Queensland to establish a biomolecular research precinct at Bond University. I chose to finish my PhD in Melbourne, but enjoyed several visits to the site. On the last occasion, Ron Warrener showed me the impressive reagent store which at first glance contained every possible compound one could imagine in alphabetical order. I asked why these stopped at butyrylthiocholine iodide, to be told that unexpected financial restrictions – attributable to the business dealings scrutiny being applied to Alan Bond at the time – had quickly come into force, such that continued ordering from the Sigma–Aldrich catalogue page was not possible, and the page was never able to be turned to reveal those chemicals beginning with the letter ‘c’. Once I had processed this information, I figured that a silver lining could be the encouragement of considerable creativity in retrosynthetic planning. While I make light of the situation, it was clear to me that those early years up north became pretty tough to all those concerned. In any case, whether correct or not, I attribute the current status of Queensland as a biomolecular research and biotech powerhouse largely to the drive and tenacity exemplified by Peter Andrews and colleagues.
Given the context of this article, it is particularly germane to note that in the early days of 1987 a cohort of we VCP students had the honour of witnessing an elderly Adrien Albert in person giving a seminar at the Australian National University (ANU). Afterwards some of us had the opportunity to chat with him, and knowing he was a keen astronomer, a fellow student (now known as Professor Phil Thompson), eager to impress, commented on his sighting of Halley’s Comet the year before. Quick as a flash Adrien retorted, ‘I’ve seen it twice’. As we were all under the mistaken impression that Halley’s Comet orbited only once every century or so (rather than its 76 years) we were all suitably impressed with the apparent age of the great man standing before us.
Postdoctoral Experiences at CSIRO
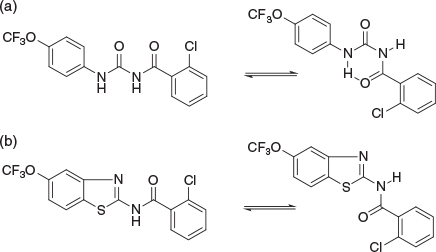
After my PhD there followed an enjoyable period of several years working at CSIRO with Ern Lacey, at the McMaster Laboratories of the Animal Health Division located on the University of Sydney grounds. Here we endeavoured to discover anthelmintics for the treatment of sheep endoparasitic nematode infections. When field trials were run, all hands were on deck to help out, which to my horror I found out included scraping out the abomasum of freshly euthanased sheep in order to determine parasite burden. After my first experience of this, on subsequent occasions I managed to orchestrate laboratory experiments that simply ‘could not be left unattended’, even for a moment, on those particular days. During this period, we focussed significant attention on benzoylphenylureas, which were well known to have potent anthelmintic activity. Over a period of a year or so I made a large number of novel heterocyclic isosteres, the design of which focussed on the hypothesis that this type of core might exist in both an extended (higher calculated dipole moment) and closed conformation (lower calculated dipole moment) as shown in Fig. 2.
|
With the help of the highly collegial Trevor Hambley nearby in the Chemistry Department, X-ray crystallography was applied to crystals that I grew from solvents with high dielectric constants. However, in all cases we determined that the intramolecularly hydrogen-bonded form – the closed conformation – was preferred, and we assumed that this was the bioactive conformation. Had I known at the time about ethylene carbonate, a liquid (above 37°C) with a surprisingly extreme dielectric constant of 90 (twice that of DMSO),[5] and which allowed us to later make novel hindered quaternary amines under conditions mild enough to prevent fragmentation,[6] I would have tried this ‘solvent’ to see if this might stabilise the open form with high dipole moment, albeit that its melting point might have proven challenging for such a use. Readers of this article might consider the use of ethylene carbonate to encourage recalcitrant reactions where the mechanism suggests a dipolar aprotic solvent of the highest dielectric constant would be favourable. Along the way we discovered that benzothiazoles were suitable bioisosteres of closed benzoylphenylureas, and exhibited potent anthelmintic activity. This mimicry was attributed to the sulfur atom, being highly polarisable and hence positively charged in this particular context, emulating features of the anilide NH. Such polarisability is a property generally not accounted for by conventional molecular mechanics forcefields, even today, which consequently can incorrectly predict conformational behaviour. At one point there was confusion when assay data became inconsistent and we determined this was due to an impurity introduced in an altered synthetic route for some compounds. It was during this period that I introduced a self-imposed numbering scheme ABC-XX-YY-ZZ that was diagnostic for every compound sample, ABC being the chemist initials, XX being the book number, YY being the page number, and ZZ being entry number on that page. This resolved the issue of batch-to-batch variations in bioassay data. We still adopt this nomenclature system today, albeit that electronic laboratory notebooks are to some extent obviating the need for such precision.
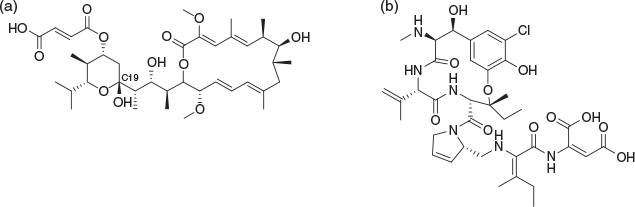
Some time at CSIRO in Sydney was also spent on natural products and a memorable period involved optimisation to find conditions which most efficiently and cleanly gave acetal derivatives from the C19 hemiacetal of bafilomycin C1 (Fig. 3). I proudly announced to my then boss, Ern Lacey, that I had developed the perfect conditions but that I only had 100 mg remaining of the 1 g supply initially kindly provided by the late Rod Rickards at ANU, and that I needed substantial resupply to make the intended focussed analogue library. Ern Lacey delicately ‘made it understood’ that I had just consumed a year’s supply of material. Thereafter I rapidly learned both how to undertake reactions on a single digit milligram scale, while also learning that supplies of complex natural products were generally to be treated with respect. Troubleshooting reactions on a milligram scale is a valuable skill but is not necessarily innate and this is something I often encourage my current students to explore.
|
The early 1990s saw a relocation to CSIRO Animal Health in Parkville, under John Edgar. Here my initial brief was to scale up production of the natural product, phomopsin A. In a very rewarding few months, we worked out how to obtain gram quantities of phomopsin A from lupin seed infected with strains of the fungus, Phomopsis leptostromiformis, which was allowed to grow over many weeks as a solid culture. A large-scale extraction from an aqueous alcoholic solution recycled through hydrophobic exchange resin, followed by large scale silica column purification with 7 parts isopropanol, 1 part water, 2 parts concentrated aqueous ammonia solution, then exhaustive evaporation to remove all traces of water and finally recrystallisation from glacial acetic acid, proved successful. A livestock poison, this compound was conjugated to fetuin to become the first reported vaccine against natural product toxicants.[7] The National Cancer Institute (NCI) also took considerable interest in phomopsin A as a potential anticancer β-tubulin binding agent, taking it all the way to animal models. The key to successful scale up was use of solid phase culture as opposed to liquid phase culture. When I recount this purification procedure, I am often asked about the amount of silica that must have contaminated the product, from being dissolved in this highly basic, aqueous eluent, there continuing to be a widespread belief that silica dissolves in methanol and anything more polar. This is simply not true – at least not to any problematic degree.[8] I think this urban myth arose when university laboratories used to re-use their silica, and, just as I once found when I did so at the University of Tasmania, preparative TLC silica contains binder that really does cause substantial product contamination from silica column elution with highly polar solvents.
Research Scientist – Biomolecular Research Institute
During the period of 1992–1996 CSIRO did not particularly value peer-reviewed publications so these were few and far between. With CSIRO undergoing another of its major restructurings, in the mid-1990s I opted not to follow a secure future at the Australian Animal Health Laboratory (AAHL) in Geelong in order to pursue my passion for drug discovery. With the support of Ray Norton and Peter Colman, very fortunately almost immediately I found myself at the Biomolecular Research Institute (BRI) in Melbourne, an entity that, in a moment of wisdom and vision between CSIRO and the Victorian State Government, had been formed for the purposes of drug discovery, and was directed by Peter Colman.
From 1996–2000 I worked on a variety of projects at BRI. One of these was the development of FcγRIIA modulators that were licenced to Arthron Pty Ltd (Fig. 4a).[9] In hindsight, the high concentrations required for efficacy and my anecdotal observations of the association of carboxylic acids with non-specific anti-inflammatory activity makes me wonder if targets other than FcγRIIA were responsible. For this reason, academics hoping to licence their drug discovery intellectual property (IP) will these days face a barrage of questions from candidate licensees demanding evidence of target engagement, and a relationship between pharmacokinetics (PK – ‘what the body does to the compound’) and pharmacodynamics (PD – ‘what the compound does to the body’) that makes sense. At BRI we also discovered khellinone-based blockers of the voltage-gated Kv1.3 potassium channel as potential treatments of multiple sclerosis (Fig. 4b).[10] These were licensed initially to Pharmaxis in 2001, then increasingly more advanced compounds and associated patent suites to Iliad (2004). Illiad was then acquired by Bionomics in 2005 in order to develop a chemistry capacity. Bionomics, with its more advanced compounds, and with increasing contributions by Andrew Harvey and Bernard Flynn, then on-licenced to Merck–Serono in 2008. I was humbled that this work was recognised by the 2004 Biota Award, now most fittingly called the Peter Andrews Award for Innovation in Medicinal Chemistry.

|
A quite different area that emerged while I was at BRI was the design and synthesis of peptidomimetics of pharmacologically active toxins such as ShK (Fig. 5a) and omega-conotoxin GVIA. Success required development of a new approach we termed ‘De Novo Type III Peptidomimetics’ to generate topographical mimetics (Fig. 5b) of hot-spots (Fig. 5a), since limited available chemical diversity space (Fig. 5c) did not yield binding epitope pharmacophores.[11] This was my first appreciation of the extraordinarily limited representation of chemical diversity by commercially available compounds, even if the latter number in the many millions.

|
De novo design of type III peptidomimetics requires a knowledge of synthesisability and conformational preference such that scaffolds are manually and interactively constructed in silico. Properly undertaken, the process does not fail and is highly publishable, yet no other groups appear to have taken this technique up, perhaps because it is deceptively difficult to start the process – the inertia and psychological barrier is high when faced with a largely blank screen and only a peptide epitope as starting point – and people tend to become distracted by the inherent apparent suitability of this task to automation, which in our hands was not successful. Indeed, it was necessity – the mother of all invention – that broke the inertia to develop this technique as time was running out for this project, which up to that point had been without success. Given the wealth of protein–protein interface structural information in the RCSB protein data bank and the fact that others are not fully utilising this information, one could imagine that whole careers could be built designing, synthesising, and publishing type III peptidomimetics. I was driven more by translation than publication and from that perspective, the road from an early mimetic to clinical candidate appeared challenging with academic-level resourcing.
The WEHI Era of Enlightenment 2001–2012
None the less, for new disease targets where there is structural information but no small molecule starting point, type-III peptidomimetic design remains worthy of investigation, particularly when the scaffold component can be kept relatively minor, thus increasing what today we term ligand efficiency.[12] Such was the position in which I found myself in 2001, at the Walter and Eliza Hall Institute of Medical Research (WEHI), which, directed then by Suzanne Cory, had established a Structural Biology Division, headed by Peter Colman, with medicinal chemistry capacity. The core team comprised a handful of structural biologists led by Ray Norton and medicinal chemists led by myself, having come from what had by then become the recently defunct Biomolecular Research Institute. Our focus was the Bcl-2 family of proteins such as Bcl-2, Bcl-w, Bcl-xL, and Mcl-1, because WEHI biologists had amassed considerable evidence over many years that their inhibition could have anti-cancer therapeutic applications. In silico screening proved problematic and our analysis of emerging small molecule inhibitors raised serious concerns about their utility. This was later convincingly shown by David Huang and his team,[13a] demonstrating that several reported inhibitors of supposed good selectivity actually maintained cytotoxicity even in cells incapable of apoptosis triggered via the Bcl-2 pathway. Even today there is a disturbing lack of evidence that links anticancer drugs in development to their anticipated target.[13b]
In the absence of a genuine inhibitor, we resurrected de novo type-III peptidomimetic design. First, we analysed the published structure of the BH3-only peptide derived from Bad, bound to the Bcl-2 family member known as Bcl-xL. We also looked at alanine scanning data, and together our analyses suggested a binding hotspot involving an α-helical segment of the Bad peptide that projected hydrophobic residues, essential for tight binding, into the binding site of Bcl-xL. We then resurrected the benzoylphenylurea as a scaffold,[14a] recognising that, if appropriately substituted, it might mimic the projection of three important hydrophobic residues into the binding site. Taken up by Guillaume Lessene, we synthesised a set of analogues, optimising based on structure–activity relationship (SAR) and potency, and successfully came up with a low micromolar-level binder and inhibitor of Bcl-xL (Fig. 6a). Our most active compound was later shown[14b] intriguingly to adopt a bound conformation different from initial design, but one that was closely geometrically related. In so doing, our compound induced a hitherto unknown conformational form of Bcl-xL. We felt this was in effect capturing a protein in mid-breath and wondered if this concept and phenomenon might be able to be extrapolated more generally to capture snapshots of mechanistically relevant protein conformational dynamics.

|
At the diametrically opposite end of the spectrum, an excellent alternative approach for ligand discovery is to systematically test large numbers of small molecules to identify one or more that modulate the target of interest. Termed high throughput screening (HTS), sophisticated automation and an optimised and miniaturised assay applied to tens of thousands and even up to millions of compounds, is now regarded as one of the best sources of what later become, after intensive medicinal chemistry, first-in-class drugs.[15] At the time, however, the virtues of HTS were relatively unknown. A case was made around 2001 to establish HTS and the WEHI hierarchy showed great foresight in approving a substantial investment in an HTS facility, located at the La Trobe University R&D Park in Bundoora. Here we had new medicinal chemistry laboratories designed principally by myself and Margaret Brumby, augmenting pre-existing infrastructure that had been previously serving Rio Tinto administration, along with an associated ‘rock warehouse’. The latter was the principal initial attraction to WEHI as it was envisaged that with some smart engineering, a positive-pressure facility suited to housing mice might be possible and indeed, this was subsequently realised. At the time there was no bespoke small molecule libraries available of diverse lead-like compounds, nor any manual about how to establish one, so we spent many months developing chemoinformatics approaches to designing an HTS library from commercially available compounds. We were fortunate to have assay technology experts such as Ian Street and John Parisot to develop HTS assays that were both precise and accurate. HTS against Bcl-w followed by extensive and convoluted triage by Keith Watson and myself led to around 100 screening hits that we felt represented our best compounds (Fig. 7). Bcl-w was chosen in part because it was most familiar to the Institute and accompanied by all required biological tools, and we had assumed that any small molecule inhibitor active against one Bcl-2 family member would be equivalently active against another, since in all cases a hydrophobic groove that bound an amphipathic α-helix with similar sequences was involved. This was later shown to be the exception rather than the rule, but at the time the dataset against Bcl-w was rather uninspiring and compounds were only weakly active, causing us considerable disappointment: we felt therefore no Bcl-2 family member would represent a druggable target, especially in light of the very high screening concentrations applied (50 µM).

|
Nevertheless, we profiled our screening hits against the Bcl-2 family panel and one of these returned interesting activity against Bcl-xL. This compound was identified as WEHI-100308 because it was registered as the 100 308th compound in our purchased library (Table 1). I later enjoyed pointing out to Peter Colman that transgressing his preferred limit of 100 000 compounds had paid unexpected dividends, a witticism that he bore with great humour. Nevertheless, the point is raised how much of HTS is a numbers game, and you get out what you put in.

|
Compound WEHI-100308 was what is called by chemoinformaticians as a singleton, a compound without a close analogue in the screening deck, as judged by a similarity metric known as the Tanimoto coefficient, which for such cases is less than 0.8 (0 being entirely dissimilar and 1.0 being entirely similar). Keith Watson therefore assembled a small analogue set by his own hand, and became convinced the sharp SAR showed the hit to be genuine. This is clearly shown in Table 2 for analogue WEHI-0105785 that lost all activity when the right-hand-side benzoic acid was modified, and analogue WEHI-0105786 that gained activity and selectivity when the hydrazone of an aldehyde was changed to that of a ketone.

|
As was the case for many of our screening hits, judging by the scientific literature for related compounds, the origin of this compound likely lay in Eastern Europe, many institutions of which had fuelled the establishment of early vendor-supplied compound libraries. We were struck by the notion that a substantial body of Eastern European chemistry was largely unknown by the West simply because of the language barrier. In this case, the original synthesis almost certainly involved a Meerwein reaction[16] and we took this route for our early compounds in coupling a phenyldiazonium salt to the appropriate furan derivative, shortly before changing to a Suzuki coupling for our discovery of breakthrough core-changed variants[17,18] as shown schematically in Fig. 8. A key to the success that followed was the ability of WEHI to contribute central resourcing for the early medicinal chemistry that was then not reliant on the boom or bust granting cycle.

|
These rapid advances in valuable small molecule composition of matter sharpened considerably partner appetite for licensing, which duly took place. With Guillaume Lessene largely taking up the WEHI Bcl-xL project helm, and joined by Genentech and Abbott, the program furnished highly successful outcomes. The evolution described graphically in Fig. 8 encodes for several important lessons. First, the representation of possible chemical diversity space is necessarily vanishingly small in any HTS library no matter how big it may be.[19,20] This means that perfectly matched femtomolar-binding ligands as exemplified by the biotin–streptavidin partnership will not be found straight up but weaker, ill-fitting ligands might. Acceptance of this fact – that any screening hit will come with inherited suboptimal features – then requires acknowledgement that optimisation within the screening hit molecular volume is an important initial undertaking. This improves ligand efficiency and avoids the trap of accessing cheap potency through development of molecular obesity, which simply increases the likelihood of future failure. Another lesson to be drawn from the progression illustrated in Fig. 8 is that target-based screening hits are typically inactive in cell-based assays, requiring optimisation, so that premature triage on cell-based activity often selects for off-target compounds. This is therefore to be discouraged. A third lesson I would like to highlight is that medicinal chemistry is far from routine chemistry. While at some stages of medicinal chemistry optimisation there can be repetitive aspects associated with analogue generation, generally later-stage compounds such as A-1155463 can become synthetically interesting and challenging, and is one of the reasons that serious medicinal chemistry groups prefer to hire skilled organic chemists as a priority over those whose primary skills reside in medicinal chemistry knowledge. A fourth and final lesson is that, while late stages of optimisation may have involved – and typically do involve – a dozen or more chemists at any given time, key breakthroughs to get to the value-inflexion compounds WEHI-727 and then WEHI-462 (Fig. 8) were made on public funds. The importance of the subsequently developed WEHI-539 as a unique biochemical probe in teaching us about Bcl-XL pharmacological modulation, and thus transforming the field, cannot be underestimated. The green crystallography thumbs of Peter Czabotar at WEHI need to be acknowledged throughout this project for key driving of the structural biology excellence.
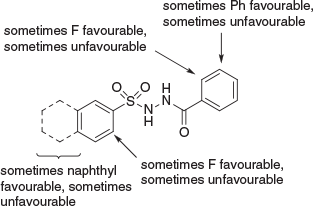
Another illustrative example of medicinal chemistry optimisation starting with HTS comes from our interest in lysine acetyltransferases (KATs). Here, Tim Thomas and Anne Voss at WEHI had been developing expertise over many years in KAT6A, which was becoming increasingly interesting as an anti-cancer target. HTS of ~200000 compounds was undertaken and gave a single hit, that once again could essentially be regarded as a singleton.[20] Chemistry resources being scarce, we managed to scrape together an analogue set of ~30 compounds and have them tested. Initial data were troubling, pictorially described in Fig. 9.
|
The main problem was that the SAR was not rational. For those experienced in medicinal chemistry, it will be understood that clear and logical SAR is the ultimate arbiter for whether it will be possible to optimise a screening hit. Clear, early SAR with sharp negatives and occasional sharp positives is all one needs to ensure that it is largely just a matter of effort for a given HTS hit against a disease-relevant target to progress all the way through to clinical trials, albeit that the entire journey may typically require a dozen (or several dozen) FTE chemistry years of medicinal chemistry. I do not mean to undervalue PK-PD here, which is essential, but it is the SAR which tells us that it will be possible, and even probable. In short, an absence of logical SAR precludes rational optimisation. However, as shown in Fig. 10, we were able to discern certain patterns in the SAR, such that when defined as three SAR streams, all inconsistencies in this early SAR dissolved.

|
We did not know at the time whether these represented quite different binding modes – and the pseudosymmetry had us thinking about horizontally flipped engagement – or broadly the same binding modes with minor positional changes, nuanced perhaps through active site sidechain movement. Either way, our segregation into these three SAR streams allowed for efficient optimisation to very potent and selective compounds.
Absolutely essential for any success in medicinal chemistry optimisation is the bioassay: its relevance, robustness, accuracy, and reproducibility. This point cannot be overemphasised. We were fortunate to have access to the WEHI HTS experts and for both the Bcl-xL and KAT6A programs, this was a key to project success.
Monash Institute of Pharmaceutical Sciences: 2012–present
In the most invigorating career move to date, I was very fortunate that in 2012, Bill Charman and Peter Scammells appointed me as a Research Professor in Medicinal Chemistry at the Monash Institute of Pharmaceutical Sciences. I took it as a good omen that almost immediately we made breakthroughs in the medicinal chemistry of KAT6A inhibitors. Ultimately, SAR streams B and C converged to furnish WM-1119 (Fig. 11) – where the ‘W’ stands for ‘WEHI’ and the ‘M’ stands for ‘Monash’ – representing the first-in-class AcCoA-competitive inhibitor of this large family of MYST KATs with demonstrably on-target, cell-based activity. Showing efficacy in mouse models of lymphoma, we were fortunate to publish in the journal, Nature, with the detailed chemistry published in the Journal of Medicinal Chemistry.[21–23]

|
Once again, the lesson is schematically given that optimisation within the inherited molecular volume of an HTS hit is the strongly preferred approach to increasing potency without resorting to molecular obesity. With excellent structural biology support from Michael Parker’s group at St Vincent’s Institute of Medical Research through Matthew Chung and Stefan Hermans, it was vividly shown how these AcCoA-competitive compounds bound to the AcCoA binding site in KAT6A (Fig. 12) by mimicking the pyrophosphate interactions of AcCoA with KAT6A, thus representing highly unusual phosphate mimetics that are very cell permeable. Cell-permeable phosphate mimetics are regarded as somewhat of a Holy Grail for medicinal chemists[24] and one might speculate whether the favourable characteristics of this core can be extrapolated to other phosphate-recognition contexts.

|
Selectivity is imparted by features outside the pyrophosphate binding site, raising the prospect that, akin to ATP-competitive kinase inhibitor selectivity, we could fine-tune this chemotype to be more selective for other KATs. In this we were successful, with WM-3835 (Fig. 12) usefully validating KAT7 as a disease-relevant target for acute myeloid leukemia.[25] These discoveries are significant in a broader context, in that until recently, KATs were thought to be undruggable[26] and were associated with only off-target inhibitors.[27] In showing this to not be the case, intense industry interest in this class of enzymes as anti-cancer targets has been generated.
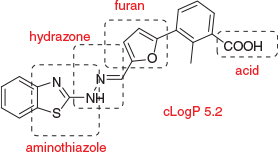
In these efforts directed towards interrogation of KATs, it is important to acknowledge substantial support by the CRC for Cancer Therapeutics in supporting key aspects of biological testing and structural biology. However, it is equally important to note that, just as for the Bcl-XL program, the key small molecule advances were made with medicinal chemistry in an academic funding environment. In my view, this attests to the valuable contribution of free-thinking and risk-taking that may be less obstructed in an academic setting compared with a corporate setting. Pharmaceutical companies may not advance screening hits that are not high affinity, that do not inherit SAR information from the HTS, or that are physicochemically unattractive. In both projects just discussed, we struck out on all three criteria. For example, Bcl-xL inhibitor WEHI-100308 contains several features that vary from mildly unattractive (carboxylic acid), to more strongly unattractive, with potential toxicophores (furan, hydrazone, aminothiazole), and lipophilicity that lies outside Lipinski limits of cLogP 5 (Fig. 13). Too weak (IC50 8 µM) for cell-based activity, this combination in a singleton would render this compound unlikely to progress in a risk averse corporate environment, leaving unanswered the question ‘Is Bcl-xL druggable?’. We did not exclude toxicophores from our initial compound library design because we were more interested in maximising representation of diversity space to capture bioactive compounds for difficult targets.[19] We felt that the combination of Meerwein chemistry and hydrazone linkage presents binding elements that may not be represented by any other means. We saw the need to engineer-out problematic functional groups as a medicinal chemistry task that could be undertaken pursuant to hit identification, and in this we were successful as was shown in Fig. 8.
|
A Perspective on Challenges to Translation of Biomedical Research in Australia
With Australia’s depth of chemical and biomedical research, it is certainly the case that our record of home-grown drug discovery should be much better than is actually the case. A key impediment is ignorance of the processes that result in new drugs, in particular that one of the principal drivers is an intensive and lasting medicinal chemistry effort and significant pharmacokinetic analysis. One of the reasons for this ignorance is that successful drug discovery requires the development of strong, interpretable SAR, but the term ‘structure–activity relationship’ generally cannot manifest as a tangibly understood concept for those with little or no medicinal chemistry training. On the contrary, the concept of structural biology and rational drug design is much more readily imparted to non-experts and I have been in several situations where the major breakthroughs were delivered by SAR-driven medicinal chemistry, with a later association of useful structural biology, yet subsequently the view became that rational drug design drove the project successes. At other times it is the mode of hit discovery that is attributed as the reason for good outcomes. For example, ABT-737 and its subsequently improved versions is often touted as a drug discovery success for SAR by NMR spectroscopy, yet it is the scale and impact of medicinal chemistry that I believe is perhaps most responsible for FDA-approved outcomes such as venetoclax, which is generally not appreciated and remains undervalued.
Neither does it help that successful medicinal chemistry efforts are commonly grossly under-described in outcome-based publications, rendering this science invisible. For example, in the journal, Molecular Cancer Therapeutics, Hirai and co-workers describe the discovery of MK-1775 as follows:[28]
A high-throughput screening was done with a small chemical compound library to find potent inhibitors of Wee1 kinase in enzymatic assay. Modification of the initial hit compounds by leveraging the information on structure-activity relationships led to the identification of a potent and selective small-molecule inhibitor of Wee1 kinase, MK-1775 (Fig. 1A), with an IC50 value of 5.2 nmol/L in in vitro kinase assays.
The underlying patent of some 250 pages describes around 200 final compounds and no doubt many FTE years of medicinal chemistry,[29] but the biomedical researcher reading this is not made aware of this fact. Rather, the impression might be imparted that HTS can pretty much deliver a drug candidate. HTS is without doubt one of the best ways to find the starting point for your future first-in-class drug,[30] but not only does the medicinal chemistry gap need to be bridged, but with the process of HTS itself an alarming number of things can go awry en route.[31] Inexperienced researchers may screen a library that is too few in number for the difficulty of the target under interrogation. The compound library may be of insufficient design quality. The expectations of affinity may be too high, so inappropriate screening concentrations are set. For target-based screens, requirement of cell-based activity may inappropriately be implemented as part of the hit triage, most likely selecting for off-target compounds. The intricacies of the assay technology may not be fully understood and so potential mechanisms for assay interference compounds neither appreciated nor detected by implementation of appropriate positive orthogonal assays and counter screens. For compounds that pass through an appropriate battery of such screens, a lack of awareness of pan-assay interference compounds or PAINS, which can give a signal readout by a variety of mechanisms and which may be selectively but non-progressably protein-reactive,[32–34] is another hindrance to progression. A confounding factor is that attempts at repurposing of FDA-approved drugs – so very popular these days despite a general absence of mechanism to reach the patient for the new use – are done so often without awareness that even these compounds can be non-specific at screening assay concentrations.[35] For all these reasons there are not inconsiderable numbers of researchers in Australia and around the world who, with fingers burnt, are of the view that ‘HTS does not work’. On the contrary, it can work very well, but one needs to understand all the reasons how it may not, if appropriate strategies are not implemented.
Adequate funding for medicinal chemistry is a real problem and I view early medicinal chemistry as the single, true valley-of-death for Australian drug discovery, with concomitant great loss of potential value to Australian (and worldwide) patients with unmet needs as well as in a commercial sense. However, I deliberately do not begin this section with this issue, since awareness of the drug discovery process in my view must take precedent. From this understanding, all else follows. For example, currently there is major funding flowing through to medical research via the Medical Research Futures Fund (MRFF). Through this visionary scheme, we are witnessing a once-in-a-generation influx of funds for translation. However, it appears that the definition of translation has been limited to clinical translation, because recipients tend to be those running clinical trials. Rather than add value to multinational pharmaceutical company drugs, I would dearly have liked to have seen expansion and improvement of existing schemes. The NHMRC Development Grant scheme represents one such mechanism. Another is the former NHMRC Industry Fellowship scheme that supported transfer of talented researchers from academia to industry placements, of which recipients included a former post-doctoral fellow of mine, Andrew Harvey, who has gone on to be a leader within QEDDI (Queensland–Emory Drug Discovery Initiative). I can see great utility in the invigoration of this scheme, perhaps modified to include major non-clinical translational fellowships to support leading researchers in either a public or private environment. With a better understanding of the role of medicinal chemistry in drug discovery, perhaps some of the MRFF funds might have been diverted towards schemes that encourage efforts to discover and develop Australian drugs.
In any case, sufficient funding is certainly a problem, and in the University setting this means drug discovery projects may need to be initiated by students to get off the ground, such that good preliminary results may lead to a strong grant proposal, which if successful, may employ a medicinal chemistry postdoc. A strategic patent may then become possible, that might allow access to translational funding schemes such as an NHMRC Development Grant. The challenges in this process are significant, which not only rely on successful grants for schemes in which success rates may be less than 15 % and a time measured in years between them, and the requirement for students to not disclose their good research outcomes – which runs counter to student need – but generate patents that are early and, without 15 years of medicinal chemistry effort embedded within, necessarily weak. The danger of a weak patent is that in licensing discussions, the medicinal chemistry revealed may be subsequently exploited, regardless of what agreements are set in place. This happens more often than one might imagine and is a genuine risk that needs to be taken seriously. The relative invisibility of the underlying medicinal chemistry effort in drug discovery publications (see above) has additional far-reaching ramifications. Biomedical researcher careers depend on publishing in high impact journals and inevitably act to engage in medicinal chemistry translation only as their breakthrough research is being published or long afterwards. The consequence is loss of competitive edge to any faster moving global interest that judges the published research outcomes to be commercially relevant. Such are the challenges faced by resourcing early drug discovery, that university technology transfer offices may be tempted to generate translational metrics by licensing biomedical research directly and by-passing the obstacles to generation of in-house medicinal chemistry, one consequence being a large value loss by licensing long before strong internal small molecule composition-of-matter patents are filed.
There is a danger that mounting challenges – whether perceived or real – manifest in a negative sentiment that permeates through to cohorts of early career researchers and on to PhD students, causing general loss of morale. Such a blanket of despondence is, unfortunately, quite a common phenomenon and can be hard to deconstruct, ultimately relying on individuals who tend towards being naturally positive rather than negative. In any case, it is for such reasons that I try to consciously avoid vocalising grant-writing complaints, which can become a habit among academic staff, as this can negatively influence younger career researchers in the vicinity of such ‘elevator’ conversations. Indeed, in my own case, my perception of academic stress steered me towards CSIRO at the end of my PhD (after some interviews with patent firms) rather than the overseas postdoctoral route, a path which I had judged at the time to be all be too hard, but which now I regret not taking. Many early career researchers reading this article will empathise with these sentiments.
At this point I should note that while I make the point that funding for medicinal chemistry is generally insufficient for a competitive rate of translation on a larger scale, at an individual level it can work. I am most grateful to ARC and NHMRC that over the years have contributed significantly to our medicinal chemistry efforts and without which many patents and publications would not have transpired.
Indeed, albeit hard to see beyond the current COVID-19 crisis, one has reason to be cautiously optimistic about the future. Compounds Australia[36] houses high quality HTS compound libraries, and highly subsidised HTS is possible through the National Drug Discovery Centre.[37] I am Director of the Australian Translational Medicinal Chemistry Facility (ATMCF)[38] that can optimise screening hits, and for meritorious collaborative projects can do so in a subsidised manner enabled with NCRIS-TIA funding. Then there are currently several governmental-led initiatives that make it attractive to invest in Australian drug discovery research and development, and that encourage spin-outs and start-up companies, so that a more cohesive process is beginning to be realised. The bottleneck by a long way is still resourcing for the value-adding early medicinal chemistry (and to a considerable extent pharmacokinetic investigations) that then facilitates access to leveraged schemes, and indeed is made considerably more constricting by this increased upstream capacity and throughput of HTS, but this forms a case so strong that some of us in this arena feel positively that this dire need will soon be much better met.
Conclusions
Research and development at the public–private interface has its challenges for those driven by translational outcomes. These most principally reside in the perennial need to garner adequate resourcing, as well as other demands associated with academia. However, the experience of the endeavour is highly invigorating and can be extremely intellectually rewarding for those with tenacity and a positive mindset. Moreover, the societal metric to gauge ultimate medicinal chemistry success is the manifestation of better treatments for patients with unmet needs. While it is the rare medicinal chemist who reaches this end game, successes en route such as licensing deals contribute significantly to the satisfaction that clinical relevance is possible. Indeed, as medicinal chemists, we are fortunate that of the many skill sets represented within the physical, life science, and biomedical arenas, ours is most strongly linked to inventorship and the associated reward schemes in the university setting that are commonly in place these days as incentives to persist on the translational journey. Such options are typically not available in pharmaceutical companies, where the scientist may have little control over their project that for business reasons may suddenly be axed. In addition, there is a certain freedom of thinking available in an academic setting that is not always the case in a corporate environment. I have found many aspects of the University setting quite liberating and if you are smart, driven, and tenacious so as to weather the early postdoctoral years that I acknowledge as particularly challenging, you will be rewarded. With these words of encouragement, I do hope early career researchers reading this account can feel more informed to better value the positive attributes of a career in medicinal chemistry working at the public–private interface, to follow this path, and in so doing, to help Australia develop Australian drugs of the future.
Conflicts of Interest
The author declares no conflicts of interest.
Acknowledgements
Key mentors and colleagues have been named in this article and I acknowledge their support. Many others could additionally be named and I apologise for any omissions. ARC funding over the years, but especially NHMRC funding in particular in the form of Fellowship support, and other competitive funding schemes, are all acknowledged and have been essential to be able to deliver these accounts. For successes in the most recent translational account of KAT6A inhibitors, the support of the Australian Government’s National Collaborative Research Infrastructure Strategy (NCRIS) program via Therapeutic Innovation Australia (TIA) has been important and is acknowledged.
References
[1] J. B. Ball, J. B. Bremner, E. J. Browne, Heterocycles 1987, 26, 1573.| Crossref | GoogleScholarGoogle Scholar |
[2] N. Kaur, P. Kumar, S. Jamwal, R. Deshmukh, V. Gauttamb, Ann. Neurosci. 2016, 23, 176.
| Crossref | GoogleScholarGoogle Scholar | 27721587PubMed |
[3] M. R. Hayden, B. R. Leavitt, U. Yasothan, P. Kirkpatrick, Nat. Rev. Drug Discov. 2009, 8, 17.
| Crossref | GoogleScholarGoogle Scholar | 19116624PubMed |
[4] (a) J. B. Ball, T. R. Hughes, P. F. Alewood, P. R. Andrews, Tetrahedron 1993, 49, 3467.
| Crossref | GoogleScholarGoogle Scholar |
(b) J. B. Ball, P. F. Alewood, J. Mol. Recognit. 1990, 3, 55.
| Crossref | GoogleScholarGoogle Scholar |
[5] R. Naejus, D. Lemordant, R. Coudert, P. Willmann, J. Chem. Thermodyn. 1997, 29, 1503.
| Crossref | GoogleScholarGoogle Scholar |
[6] R. O. Dror, H. F. Green, C. Valant, D. W. Borhani, J. R. Valcourt, A. C. Pan, D. H. Arlow, M. Canals, R. Lane, R. Rahmani, J. B. Baell, P. M. Sexton, A. Christopoulos, D. E. Shaw, Nature 2013, 503, 295.
| Crossref | GoogleScholarGoogle Scholar | 24121438PubMed |
[7] J. A. Edgar, K. A. Than, A. L. Payne, N. Anderton, J. Baell, Y. Cao, P. A. Cockrum, A. Michalewicz, P. L. Stewart, J. G. Allen, in Proceedings of the 5th International Symposium on Poisonous Plants 1998, San Angelo, Texas, 18–23 May 1997, pp. 196–200.
[8] https://selekt.biotage.com/blog/does-methanol-really-dissolve-silica-during-flash-column-chromatography.
[9] G. A. Pietersz, P. L. Mottram, N. C. van de Velde, C. T. Sardjono, S. Esparon, P. A. Ramsland, G. Moloney, J. B. Baell, T. D. McCarthy, B. R. Matthews, M. S. Powell, P. M. Hogarth, Immunol. Cell Biol. 2009, 87, 3.
| Crossref | GoogleScholarGoogle Scholar | 19030019PubMed |
[10] J. B. Baell, R. W. Gable, A. J. Harvey, N. T. Toovey, T. Herzog, W. Haensel, H. Wulff, J. Med. Chem. 2004, 47, 2326.
| Crossref | GoogleScholarGoogle Scholar | 15084131PubMed |
[11] J. B. Baell, A. J. Harvey, R. S. Norton, J. Comput. Aided Mol. Des. 2002, 16, 245.
| Crossref | GoogleScholarGoogle Scholar | 12400855PubMed |
[12] C. H. Reynolds, Future Med. Chem. 2015, 7, 1363.
| Crossref | GoogleScholarGoogle Scholar | 26230875PubMed |
[13] (a) M. F. van Delft, A. H. Wei, K. D. Mason, C. J. Vandenberg, L. Chen, P. E. Czabotar, S. N. Willis, C. L. Scott, C. L. Day, S. Cory, J. M. Adams, A. W. Roberts, D. C. S. Huang, Cancer Cell 2006, 10, 389.
| Crossref | GoogleScholarGoogle Scholar | 17097561PubMed |
(b) A. Lin, C. J. Giuliano, A. Palladino, K. M. John, C. Abramowicz, M. L. Yuan, E. L. Sausville, D. A. Lukow, L. Liu, A. R. Chait, Z. C. Galluzzo, C. Tucker, J. M. Sheltzer, Sci. Transl. Med. 2019, 11, eaaw8412.
| Crossref | GoogleScholarGoogle Scholar |
[14] (a) G. Lessene, B. J. Smith, R. W. Gable, J. B. Baell, J. Org. Chem. 2009, 74, 6511.
| Crossref | GoogleScholarGoogle Scholar | 19711992PubMed |
(b) R. M. Brady, A. Vom, M. J. Roy, N. Toovey, B. J. Smith, R. M. Moss, E. Hatzis, J. P. Parisot, H. Yang, I. P. Street, P. M. Colman, P. E. Czabotar, J. B. Baell, G. Lessene, J. Med. Chem. 2014, 57, 1323.
| Crossref | GoogleScholarGoogle Scholar |
[15] J. Eder, R. Sedrani, C. Wiesmann, Nat. Rev. Drug Discov. 2014, 13, 577.
| Crossref | GoogleScholarGoogle Scholar | 25033734PubMed |
[16] (a) H. Meerwein, E. Buchner, K. van Emster, J. Prakt. Chem. 1939, 152, 237.
| Crossref | GoogleScholarGoogle Scholar |
(b) C. S. Rondestvedt, Org. React. 1960, 11, 189.
[17] B. Sleebs, W. Kersten, S. Kulasegaram, G. Nikolakopoulos, E. Hatzis, R. Moss, J. Parisot, H. Yang, P. Czabotar, W. Fairlie, E. Lee, J. Adams, C. L. Chen, M. van Delft, K. Lowes, A. Wei, D. Huang, P. Colman, I. Street, J. Baell, K. Watson, G. Lessene, J. Med. Chem. 2013, 56, 5514.
| Crossref | GoogleScholarGoogle Scholar | 23767404PubMed |
[18] G. Lessene, P. Czabotar, B. E. Sleebs, K. Zobel, K. L. Lowes, J. M. Adams, J. B. Baell, P. M. Colman, K. Deshayes, W. J. Fairbrother, J. F. Flygare, P. Gibbons, W. J. A. Kersten, S. Kulasegaram, R. M. Moss, J. P. Parisot, B. J. Smith, I. P. Street, H. Yang, D. C. S. Huang, K. G. Watson, Nat. Chem. Biol. 2013, 9, 390.
| Crossref | GoogleScholarGoogle Scholar | 23603658PubMed |
[19] J. B. Baell, J. Chem. Inf. Model. 2013, 53, 39.
| Crossref | GoogleScholarGoogle Scholar | 23198812PubMed |
[20] X. Yi, L. Xue, T. Thomas, J. B. Baell, Future Med. Chem. 2020, 12, 423.
| Crossref | GoogleScholarGoogle Scholar | 32064938PubMed |
[21] J. B. Baell, D. J. Leaver, S. J. Hermans, G. L. Kelly, M. S. Brennan, N. L. Downer, N. Nguyen, J. Wichmann, H. M. McRae, Y. Yang, B. Cleary, H. R. Lagiakos, S. Mieruszynski, G. Pacini, H. K. Vanyai, M. I. Bergamasco, R. E. May, B. K. Davey, K. J. Morgan, A. J. Sealey, B. Wang, N. Zamudio, S. Wilcox, A. L. Garnham, B. N. Sheikh, B. J. Aubrey, K. Doggett, M. C. Chung, M. de Silva, J. Bentley, P. Pilling, M. Hattarki, O. Dolezal, M. L. Dennis, H. Falk, B. Ren, S. A. Charman, K. L. White, J. Rautela, A. Newbold, E. D. Hawkins, R. W. Johnstone, N. D. Huntington, T. S. Peat, J. K. Heath, A. Strasser, M. W. Parker, G. K. Smyth, I. P. Street, B. J. Monahan, A. K. Voss, T. Thomas, Nature 2018, 560, 253.
| Crossref | GoogleScholarGoogle Scholar | 30069049PubMed |
[22] D. J. Leaver, B. Cleary, N. Nguyen, D. L. Priebbenow, H. R. Lagiakos, J. Sanchez, L. Xue, F. Huang, Y. Sun, P. Mujumdar, R. Mudududdla, S. Varghese, S. Teguh, S. A. Charman, K. L. White, K. Katneni, M. Cuellar, J. M. Strasser, J. L. Dahlin, M. A. Walters, I. P. Street, B. J. Monahan, K. E. Jarman, H. J. Sabroux, H. Falk, M. C. Chung, S. J. Hermans, M. W. Parker, T. Thomas, J. B. Baell, J. Med. Chem. 2019, 62, 7146.
| Crossref | GoogleScholarGoogle Scholar | 31256587PubMed |
[23] D. L. Priebbenow, D. J. Leaver, N. Nguyen, B. Cleary, H. R. Lagiakos, J. Sanchez, L. Xue, F. Huang, Y. Sun, P. Mujumdar, R. Mudududdla, S. Varghese, S. Teguh, S. A. Charman, K. L. White, D. M. Shackleford, K. Katneni, M. Cuellar, J. M. Strasser, J. L. Dahlin, M. A. Walters, I. P. Street, B. J. Monahan, K. E. Jarman, H. J. Sabroux, H. Falk, M. C. Chung, S. J. Hermans, N. L. Downer, M. W. Parker, A. K. Voss, T. Thomas, J. B. Baell, J. Med. Chem. 2020, 63, 4655.
| Crossref | GoogleScholarGoogle Scholar | 32118427PubMed |
[24] C. S. Rye, J. B. Baell, Curr. Med. Chem. 2005, 12, 3127.
| Crossref | GoogleScholarGoogle Scholar | 16375706PubMed |
[25] L. MacPherson, J. Anokye, M. M. Yeung, E. Y. N. Lam, Y.-C. Chan, C.-F. Weng, P. Yeh, K. Knezevic, M. S. Butler, A. Hoegl, K.-L. Chan, M. L. Burr, L. J. Gearing, T. Willson, J. Liu, J. Choi, Y. Yang, R. A. Bilardi, H. Falk, N. Nguyen, P. A. Stupple, T. S. Peat, M. Zhang, M. de Silva, C. Carrasco-Pozo, V. M. Avery, P. S. Khoo, O. Dolezal, M. L. Dennis, S. Nuttall, R. Surjadi, J. Newman, B. Ren, D. J. Leaver, Y. Sun, J. B. Baell, O. Dovey, G. S. Vassiliou, F. Grebien, S.-J. Dawson, I. P. Street, B. J. Monahan, C. J. Burns, C. Choudhary, M. E. Blewitt, A. K. Voss, T. Thomas, M. A. Dawson, Nature 2020, 577, 266.
| Crossref | GoogleScholarGoogle Scholar | 31827282PubMed |
[26] J. B. Baell, W. Miao, Future Med. Chem. 2016, 8, 1525.
| Crossref | GoogleScholarGoogle Scholar | 27557114PubMed |
[27] J. L. Dahlin, K. M. Nelson, J. M. Strasser, D. Barsyte-Lovejoy, M. M. Szewczyk, S. Organ, M. Cuellar, G. Singh, J. H. Shrimp, N. Nguyen, J. L. Meier, C. H. Arrowsmith, P. J. Brown, J. B. Baell, M. A. Walters, Nat. Commun. 2017, 8, 1527.
| Crossref | GoogleScholarGoogle Scholar | 29142305PubMed |
[28] H. Hirai, Y. Iwasawa, M. Okada, T. Arai, T. Nishibata, M. Kobayashi, T. Kimura, N. Kaneko, J. Ohtani, K. Yamanaka, H. Itadani, I. Takahashi-Suzuki, K. Fukasawa, H. Oki, T. Nambu, J. Jiang, T. Sakai, H. Arakawa, T. Sakamoto, T. Sagara, T. Yoshizumi, S. Mizuarai, H. Kotani, Mol. Cancer Ther. 2009, 8, 2992.
| Crossref | GoogleScholarGoogle Scholar | 19887545PubMed |
[29] T. Sagara, S. Otsuki, S. Sunami, T. Sakamoto, K. Niiyama, F. Yamamoto, T. Yoshizumi, H. Furuyama, Y. Goto, M. Bamba, K. Takahashi, H. Hirai, T. Nishibata, U.S. Patent 7,834,019 B2 2010.
[30] J. Eder, R. Sedrani, C. Wiesmann, Nat. Rev. Drug Discov. 2014, 13, 577.
| Crossref | GoogleScholarGoogle Scholar | 25033734PubMed |
[31] J. B. Baell, ACS Med. Chem. Lett. 2015, 6, 229.
| Crossref | GoogleScholarGoogle Scholar | 25941544PubMed |
[32] J. B. Baell, G. A. Holloway, J. Med. Chem. 2010, 53, 2719.
| Crossref | GoogleScholarGoogle Scholar | 20131845PubMed |
[33] J. B. Baell, Future Med. Chem. 2010, 2, 1529.
| Crossref | GoogleScholarGoogle Scholar | 21426147PubMed |
[34] J. Baell, M. A. Walters, Nature 2014, 513, 481.
| Crossref | GoogleScholarGoogle Scholar | 25254460PubMed |
[35] J. Baell, J. Nat. Prod. 2016, 79, 616.
| Crossref | GoogleScholarGoogle Scholar | 26900761PubMed |
[36] https://www.griffith.edu.au/griffith-sciences/compounds-australia.
[37] https://nddc.wehi.edu.au/.
[38] https://www.monash.edu/atmcf.
* Jonathan B. Baell is the recipient of the 2018 Adrien Albert Award. Dedicated to my beloved younger brother, Jeremy Ball (10 August 1968 – 15 September 2014) – a blaze of light.