

Iodocyclisation of Electronically Resistant Alkynes: Synthesis of 2-Carboxy (and sulfoxy)-3-iodobenzo[b]thiophenes*
Shuqi Chen A and Bernard L. Flynn A B
A and Bernard L. Flynn A B
A Monash Institute of Pharmaceutical Sciences, Monash University, 381 Royal Parade, Parkville, Vic. 3052, Australia.
B Corresponding author. Email: bernard.flynn@monash.edu
Australian Journal of Chemistry 74(1) 65-76 https://doi.org/10.1071/CH20218
Submitted: 13 July 2020 Accepted: 23 September 2020 Published: 23 October 2020
Abstract
The iodocyclisation of alkynes bearing tethered nucleophiles is a highly effective method for the construction and diversification of heterocycles. A key limitation to this methodology is the 5-endo-dig iodocyclisation of alkynes that have an unfavourable electronic bias for electrophilic cyclisation. These tend to direct electrophilic attack of the iodonium atom to the wrong carbon for cyclisation, thus favouring competing addition reactions. Using our previously determined reaction conditions for the 5-endo-dig iodocyclisations of electronically resistant alkynes, we have achieved efficient synthetic access to 2-carboxy (and sulfoxy)-3-iodobenzo[b]thiophenes. The corresponding benzo[b]furans and indoles were not accessible under these conditions. This difference may arise due to the availability of a radical mechanism in the case of iodobenzo[b]thiophenes. The 2-carboxy functionality of the iodocyclised products can be further employed in iterative alkyne-coupling iodocyclisation reactions, where the carboxy group or an imine (Schiff base) partakes in a second iodocyclisation to generate a lactone or pyridine ring.
Introduction
The 5-endo-dig electrophilic cyclisation of phenylalkynes bearing ortho-nucleophiles (Nu), 1, to give benzo[b]furans, benzo[b]thiophenes, benzo[b]selenophenes, and indoles has emerged as one of the most effective methods for the diversification of these ring systems, in particular for drug discovery (Scheme 1a).[1,2] The C2 substituent in 2 is readily diversified through the use of different alkynes in the formation of 1, most often by Sonogashira coupling. Highly effective diversification of the C3 substituent can be achieved through the use of iodonium electrophiles (EX = IX) to give 2 (E = I).[1] The resultant C3-iodo group affords access to a diverse array of C3-substituents through Pd-mediated couplings, metal-for-iodide exchange, and other methods. Broader scaffold diversification can be achieved through the use of other carbocyclic and heterocyclic substrates (replacing benzene) and the optional incorporation of single-atom spacers A in 1′, which on electrophilic cyclisation gives 2′, 2′′ or 2′′′, the latter two via 6-endo-dig cyclisation.[1,2b,e,3]
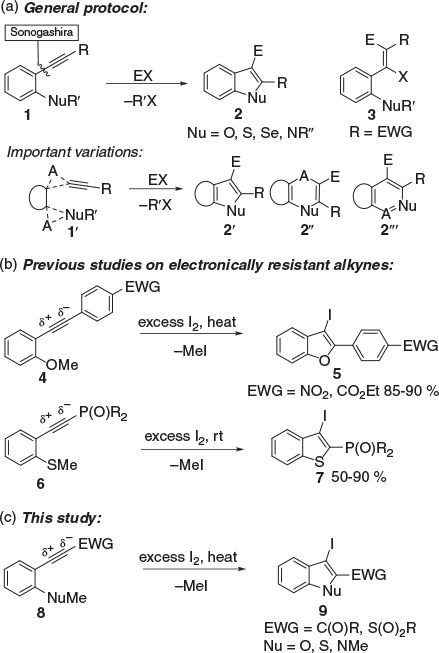
|
A key limitation of the 5-endo-dig electrophilic cyclisation is the incorporation of electron-withdrawing groups (EWGs) into the substrate 1 (R = EWG).[4] This incorporation gives rise to an unfavourable electronic bias in 1 that favours EX addition to give 3 over cyclisation to give 2. In earlier studies, we identified that high I2 concentration (excess I2) and modestly elevated temperatures (60–80°C) could be used to achieve efficient iodocyclisation of such electronically resistant (ER) alkynes (Scheme 1b).[5] These reaction conditions have been rationalised as favouring cyclisation through I2-alkyne complex 10 that takes place at elevated temperatures (Scheme 2).[5] At lower temperatures (room temperature, rt), the I2-addition product 3 or 3′ is favoured and relatively stable (major product). This I2 addition most usually proceeds through a stereospecific termolecular radical addition mechanism, involving either homolysis of 10 to give a bridged radical 14 that then reacts with I2 to give 3, or direct attack of 10 by I• to give 3; the energetics of these two pathways are similar.[5] The rapidly reversible interconversion between 1, 10–14, and 3, achieved at elevated temperatures and high I2 concentrations, is critical to the successful iodocyclisation of ER alkynes. This interconversion provides sustained access to 10 as elevated temperature overcomes the energy barrier for the cyclisation to 2. For neutral alkynes and those with a favourable electronic bias, vinyl cation 12 is predicted to play a major role in the cyclisation, which usually proceeds at rt. Interestingly, our and other recent studies indicate that the often-presented bridged iodonium cation 15 is unlikely to be an intermediate in iodocyclisation or I2 addition as it is higher in energy than the vinyl cations 12 and 13.[5,6]
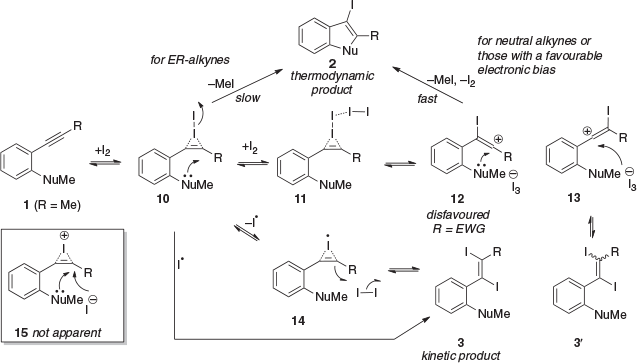
|
To date, our application of the preferred reaction conditions (high [I2] and elevated temperatures) for the iodocyclisation of ER alkynes 1 (R = EWG) has been limited to those bearing electron-deficient benzene rings 4 → 5 and P(O)R2 groups 6 → 7 (Scheme 1b).[5,7] From a diversification standpoint, carbonyls (aldehydes, esters, amides, and ketones) and sulfonyls are exceptionally useful functionalities in synthesis and pharmacophoric units in medicinal chemistry. Accordingly, in the present work, we deemed it of great interest to examine if these reaction conditions could be used to prepare 2-carboxy (and sulfoxy)-3-iodo-benzo[b]thiophenes, benzo[b]furans, and indoles: iodocyclisation of 8 to 9 (Scheme 1c).
Results and Discussion
For the purpose of this study, we prepared a series of phenylalkynylcarbonyl compounds (aldehydes, esters, amides, and ketones) 8aa–cd (Scheme 3), bearing an ortho nucleophile (NuMe, Nu = S, O, NMe). These were readily prepared by lithiation of the corresponding terminal alkyne 16a–c and reaction with suitable electrophiles 17a–e. Iodocyclisation of these using our previously established conditions for ER alkynes, high [I2] (0.6–1 M) heated (50–80°C) in 1,2-dichloroethane (DCE), was only effective for Nu = S, giving 2-carboxy-3-iodobenzo[b]thiophenes 9aa–ae in good to excellent yields. The corresponding benzo[b]furans and indoles 9bb–cd were not obtained and prolonged heating of these led to the formation of complex reaction mixtures.
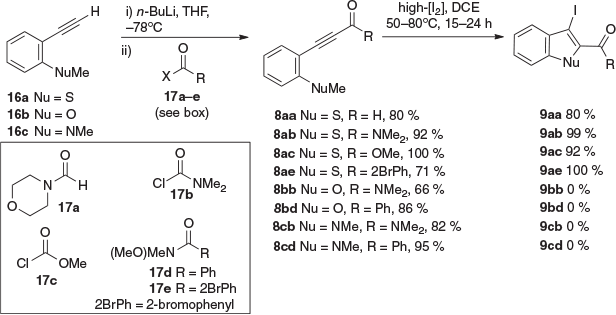
|
We also explored equivalent iodocyclisations where the EWG on the terminal end of the alkyne is a sulfone (Scheme 4). The alkyne substrates were generated from the terminal alkynes 16a–c. Magnesiation of 16a–c (better than lithiation in this case) followed by reaction with methanesulfonic anhydride (Ms2O) gave the alkynyl methyl sulfones 8a–cf in modest yield (33–52 %). Although the overall conversions were generally high, chromatography of these sulfones was associated with some decomposition.
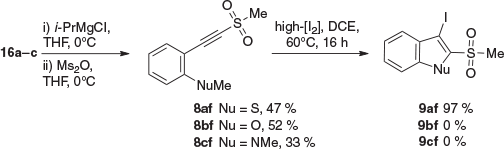
|
Iodocyclisation of 8a–cf under our conditions for ER alkynes gave the 3-iodo-2-(methylsulfonyl)benzo[b]thiophene (9af) in excellent yield of 97 %, but did not give any of the corresponding benzo[b]furan or indole 9b–cf (Scheme 4).
Another type of ER alkyne arises where the aryl ring bearing the nucleophile is very electron-rich; this also directs the electrophile to the wrong (distal) alkyne carbon, favouring I2 addition over 5-endo-dig cyclisation. We had previously encountered this limitation in the iterative use of iodocyclisation for the preparation of poly-[3,2-d]-fused thiophenes, which was overcome using our high-[I2], elevated-temperature conditions.[4] In some ongoing work, we have recently revisited this chemistry to generate a series of different heteroacenes (see also below). For this, we required the iodobromobenzo[b]thieno[3,2-d]thiophene 24 (Scheme 5). Following our earlier approach,[4] a facile iodocyclisation of electronically favoured 18 to 19 (91 %) was followed by Sonogashira coupling to give 21 in 65 % yield. Reaction of 21 with I2 at room temperature rapidly forms 23 through the stabilised cation 22. Under our cyclisation conditions for ER alkynes (refluxing DCE at high [I2] for 20 h), the initially formed 23 is converted to 24 in 85 % yield.

|
To explore the iodocyclisations of these electron-rich ER alkynes further, we prepared three other substituted 2-(methylthio)-3-ethynylbenzo[b]thiophenes, 25, 27, and 29, from 19 or 20 (Scheme 5). The carbomethoxy-substituted system 25 represents an extreme version of an ER alkyne, bearing a push–pull combination of electron-donor and electron-withdrawing groups in a negatively biased orientation for iodocyclisation. Treatment of 25 under our iodocyclisation conditions for ER alkynes successfully afforded the iodocyclised product 26, albeit in modest yield (19 %). In the cases of 27 and 29, there are two possibilities for iodocyclisation. In both these cases, the cyclisation is directed to the electronically favoured outcome. In the case of 27, this involves a reversal of the usual chemoselectivity, where iodocyclisation through SMe is normally favoured over CO2Me, e.g. 31 → 32 (Scheme 6).[8] Additionally, the higher strain associated with the formation of two fused five-membered rings may also play a role in disfavouring the formation of the benzo[b]thieno[3,2-d]thiophene rings.
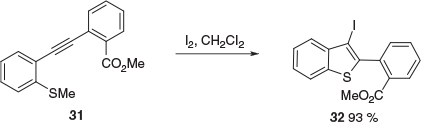
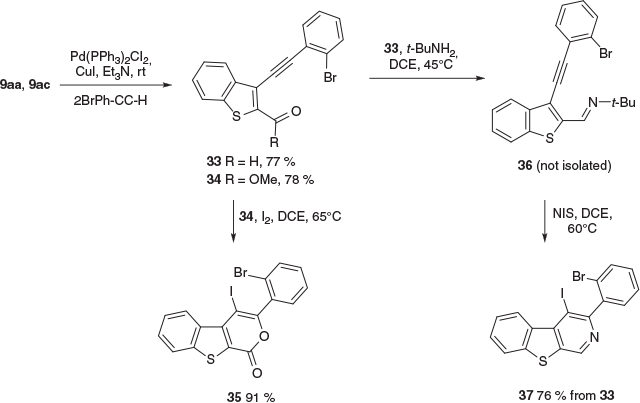
Finally, to illustrate the benefit of inclusion of a C2-carboxy group in 9 for heteroacene synthesis, we explored the use of 2-carboxy-3-iodobenzo[b]thiophenes (9aa and 9ac) in iterative iodocyclisations (Scheme 7). Ester 9ac was coupled to (2-bromophenyl)ethyne to give 34, which was iodocyclised to give the benzothienopyranone 35 in 91 % yield. Schiff base condensation of the aldehyde 33 gave aldimine 36 (not isolated), which was iodocyclised to the corresponding benzothienopyridine 37 (76 % from 33). Interestingly, the iodocyclisation of both 34 and 36 operated under relatively mild conditions, the latter using N-iodosuccinimide (NIS), and did not require our specialised conditions for ER alkyne. Presumably, the C2-carbonyls rebalance the alkyne electronics to that of a neutral or electronically favoured alkyne.
|
The iodobromo-containing compounds 9ae, 24, 35, and 37 generated in this work are to be used in a broader scaffold-diversification effort involving the additional transformation of these halide functionalities into other rings; such work is ongoing and will be described elsewhere.
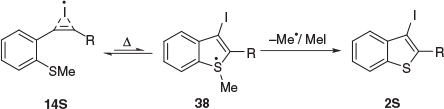
These studies reveal a stark difference in yield between the 5-endo-dig iodocyclisation of ER alkynes 8 where Nu = S (80–100 %) compared with Nu = O or NMe (0 %). This difference can be explained by the greater nucleophilicity of sulfur over oxygen and nitrogen in the key intermediate 10 (Scheme 2). In addition to its lower electronegativity and its more polarisable lone pairs of electrons, sulfur is also larger than oxygen and nitrogen and better able to accommodate stereoelectronic requirements of 5-endo-dig cyclisation.[9] Another possibility for this very stark difference may be the accessibility to a radical pathway for Nu = S that is not available for Nu = O or NMe. Radical attack on alkylsulfides to give S-centred radicals that undergo subsequent homolytic cleavage to release an alkyl radical is well established.[10] The rapidly reversible reaction of 1S (1, Nu = S) with I2 at elevated temperature to give 3S provides sustained access to bridged radical 14S, which may undergo a reversible and energetically disfavoured (uphill) rearrangement to the S-centred radical 38, which undergoes irreversible homolysis to give 2S and MeI (Scheme 8). Similar 5-exo and endo-dig cyclisation reactions involving tandem radical addition and alkyl sulfide homolytic cleavage have been described previously.[11] Further investigation of these mechanistic possibilities is under way and will be reported elsewhere.
Conclusion
In conclusion, our previously described method for the iodocyclisation of ER alkynes has been successfully applied to the synthesis of 2-carboxy (and sulfoxy)-3-iodobenzo[b]thiophenes 9aa–af, but was not successful in generating the equivalent benzo[b]furans and indoles 9bb–cf (Schemes 3 and 4). Also, at the limit of this methodology is the efficient iodocyclisation of push–pull ER alkynes, such as 25 → 26, 19 % (Scheme 5). Both polar (nucleophile/electrophile) (Scheme 2) and radical (Scheme 8) mechanisms have been proposed to explain the more efficient cyclisation of sulfides over ethers and amines. An additional aspect of this work is noting the effect of electronic bias on competing iodocyclisation pathways, which can switch the chemoselectivity seen with more neutral alkynes (cf. 27 → 28 with 31 → 32, Schemes 5 and 6). Another is the utilisation of 2-carboxy-3-iodo-benzo[b]thiophenes in the formation of polyfused heteroacenes through iterative iodocyclisation.
Experimental
All reactions were performed under an inert atmosphere of anhydrous N2, unless otherwise stated. Solvents were dried using a commercial solvent purification system. DCE and THF were purchased in an anhydrous form and stored under N2. Solvents used in reaction extractions and chromatography and all other reagents were used as supplied without further purification or drying. All glassware was dried by heating with a heat gun under high vacuum. Hexanes with a boiling point range of 40–60°C were used in chromatography. Flash column chromatography was performed on 40–60-µm silica gel. 1H NMR spectra were recorded at 400 MHz. 13C NMR spectra were recorded at 101 MHz; the number of hydrogens attached to each carbon atom was determined using Distortionless Enhancement by Polarisation Transfer with detection of quaternary carbons (DEPTQ-135), as indicated. All chemical shifts (δ) were calibrated using residual non-deuterated solvent (e.g. chloroform) as an internal reference and are reported in parts per million (ppm) relative to trimethylsilane (0 ppm). Coupling constants (J) are reported in Hz. Thin-layer chromatography (TLC) was performed using 0.25-mm thick plates precoated with Merck Kieselgel 60 F254 silica gel, and visualised using UV light (254 and 365 nm). Liquid chromatography–mass spectrometry (LCMS) was performed using either APCI (atmospheric-pressure chemical ionisation) or ESI (electrospray ionisation) LCMS. Both methods used 254 nm detection and a Poroshell 120 EC-C18 2.7 μm 50 × 3.0 mm column. The column temperature was 35°C. The eluent system used was solvent A (H2O with 0.1 % methanoic acid) and solvent B (MeCN with 0.1 % methanoic acid). LCMS ESI method: the gradient starts from 95 % solvent A/5 % solvent B for 1 min, reaches 100 % solvent B over 1.5 min, maintained for 1.3 min, and then changed to 95 % solvent A/5 % solvent B over 1.2 min. LCMS APCI method: the gradient starts from 95 % solvent A/5 % solvent B for 1 min, reaches 100 % solvent B over 1.9 min, maintained for 2 min, and then changed to 95 % solvent A/5 % solvent B over 1.0 min. Analytical HPLC was performed on an Agilent 1260 Infinity analytical HPLC with a G1312B 1260 binary pump and G4212B 1260 DAD detector. The column used was a Zorbax Eclipse Plus C18 Rapid Resolution 4.6 × 100 mm, 3.5 µm. The eluent system used was: solvent A: H2O with 0.1 % methanoic acid; solvent B: MeCN with 0.1 % methanoic acid. All samples were analysed using the same method: the gradient increases from 95 % solvent A/5 % solvent B to 100 % solvent B over 9 min and maintained at 100 % solvent B for 1 min with flow rate of 1.0 mL min−1. High-resolution mass spectrometry (HRMS) was performed on both a time-of-flight mass spectrometer fitted with either ESI or APCI source – the capillary voltage was 4000 V – or on an Exactive mass spectrometer fitted with an atmospheric solids analysis probe (ASAP) ion source.
The following materials were prepared according to literature procedures: 2-bromo-N-methoxy-N-methylbenzamide (17e),[12] methyl(2-((methylthio)ethynyl)phenyl)sulfane (18),[4] and 3-iodo-2-(methylthio)benzo[b]thiophene (19).[4]
General Procedure 1 for the Synthesis of Terminal Alkynes 16a–16c, 20, and 39 (Sonogashira Coupling–Desilylation)
The respective 2-iodobenzene was dissolved in Et3N (0.2 M) in a dry round-bottom flask (RBF), followed by addition of CuI (4–6 mol-%) and Pd(PPh3)2Cl2 (2–3 mol-%). The RBF was then degassed and backfilled with N2 three times. Finally, trimethylsilylacetylene (1.2 equiv.) was added dropwise under an N2 atmosphere. The reaction mixture was stirred at rt overnight. On completion, the suspension was filtered through Celite® and extracted with Et2O twice, and washed with H2O twice and with brine twice; the combined organic extracts were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by a silica plug (100 % hexanes) to yield the TMS-protected terminal alkyne, which was then dissolved in MeOH/Et2O (2 : 1, 0.2 M), followed by addition of K2CO3 (1.5 equiv.). The reaction mixture was stirred at rt overnight. On completion, the mixture was concentrated to a residue and taken up in Et2O. Washed with H2O twice and with brine twice, the organic extract was dried over anhydrous MgSO4, filtered, and concentrated to yield the desired terminal alkynes, which were directly used in the next step without further purification.
General Iodocyclisation Procedure for the Synthesis of Substrates 9aa–af, 24, 26, 28, 30, and 35
I2 (3 equiv.) was added to a stirred solution of the respective alkyne substrates in dry DCE (0.2 M) under an N2 atmosphere. The reaction mixture was heated at 45–80°C. On completion, the reaction mixture was cooled to rt, quenched with saturated Na2S2O3 solution, and extracted with CH2Cl2 twice. The combined organic extracts were washed with H2O twice and with brine twice, dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure to yield the desired cyclised product.
Synthetic Methods
(2-Ethynylphenyl)(methyl)sulfane (16a)
Compound 16a was synthesised according to General Procedure 1. A yellow oil (4.13 g, 94 % from 2-iodothioanisole) was obtained. δH (400 MHz, CDCl3) 7.46 (dd, J 7.6, 1.4, 1H), 7.32 (td, J 7.7, 1.4, 1H), 7.18 (d, J 7.3, 1H), 7.09 (td, J 7.5, 1.1, 1H), 3.47 (s, 1H), 2.50 (s, 3H). The spectroscopic data are consistent with those previously reported for this compound in the literature.[13]
1-Ethynyl-2-methoxybenzene (16b)
Compound 16b was synthesised according to General Procedure 1. A rosy oil (1.54 g, 76 % from 2-iodoanisole) was obtained. δH (400 MHz, CDCl3) 7.46 (dd, J 7.5, 1.7, 1H), 7.32 (ddd, J 8.4, 7.6, 1.7, 1H), 6.91 (td, J 7.5, 1.0, 1H), 6.89 (d, J 8.4, 1H), 3.90 (s, 3H), 3.31 (s, 1H). The spectroscopic data are consistent with those previously reported for this compound in the literature.[14]
2-Ethynyl-N,N-dimethylaniline (16c)
Compound 16c was synthesised according to General Procedure 1. A light yellow oil (1.28 g, 87 % from 2-iodo-N,N-dimethylaniline) was obtained. δH (400 MHz, CDCl3) 7.47 (dd, J 7.6, 1.7, 1H), 7.27 (ddd, J 8.4, 7.4, 1.7, 1H), 6.93 (dd, J 8.3, 1.1, 1H), 6.89 (td, J 7.5, 1.1, 2H), 3.43 (s, 1H), 2.94 (s, 6H). m/z (LCMS APCI) (%) tR 2.97 min, 146.1 (100, [M + H]+). The spectroscopic data are consistent with those previously reported for this compound in the literature.[15]
3-(2-(Methylthio)phenyl)propiolaldehyde (8aa)
n-BuLi (2.38 mL, 5.77 mmol, 2.42 M in hexanes) was added dropwise to a stirred solution of 16a (900 mg, 6.07 mmol) in dry THF (35 mL) at −78°C under an N2 atmosphere. The solution was stirred at 0°C for 0.5 h, followed by slow addition of 17a (4.19 g, 36.4 mmol). The reaction mixture was stirred at 0°C for 1 h, quenched with saturated NH4Cl solution (50 mL), and extracted with Et2O (2 × 45 mL). The combined organic extracts were washed with H2O (2 × 45 mL) and brine (2 × 45 mL), dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography (19 : 1 hexanes/EtOAc, Rf 0.25) to yield the title compound (816 mg, 80 %) as a yellow oil. δH (400 MHz, CDCl3) 9.49 (s, 1H), 7.55 (dd, J 7.7, 1.5, 1H), 7.43 (ddd, J 8.1, 7.6, 1.5, 1H), 7.22 (d, J 8.0, 1H), 7.15 (td, J 7.6, 1.1, 1H), 2.53 (s, 3H). δC (101 MHz, CDCl3) 174.5 (C), 163.5 (C), 160.9 (C), 144.5 (C), 135.6 (CH), 135.6 (CH), 134.8 (CH), 132.9 (CH), 131.4 (CH), 125.8 (C), 125.7 (C), 124.7 (CH), 124.7 (CH), 124.3 (CH), 124.3 (CH), 118.3 (C), 117.3 (CH), 117.1 (CH), 94.0 (C), 90.22 (C), 90.20 (C), 15.3 (CH3). HPLC: tR 5.64 min, 96.9 % purity at 254 nm. m/z (HRMS ESI) 177.0362; calcd for C10H9OS+ [M + H]+ 177.0369.
3-Iodobenzo[b]thiophene-2-carbaldehyde (9aa)
I2 (1.73 g, 6.81 mmol) was added to a stirred solution of 8aa (400 mg, 2.27 mmol) in dry DCE (10 mL) under an N2 atmosphere. The reaction was stirred at rt overnight. The reaction mixture was quenched with saturated Na2S2O3 solution (20 mL) and extracted with CH2Cl2 (2 × 25 mL). The combined organic extracts were then washed with H2O (2 × 25 mL) and brine (25 mL), dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure yield the title compound (618 mg, 94 %) as a tan solid without further purification. δH (400 MHz, CDCl3) 10.16 (s, 1H), 8.00 (dd, J 8.0, 1.4, 1H), 7.87 (dd, J 8.0, 1.2, 1H), 7.58–7.50 (m, 2H). δC (101 MHz, CDCl3) 187.6 (CH), 141.2 (C), 140.7 (C), 138.8 (C), 129.4 (CH), 127.5 (CH), 126.3 (CH), 123.5 (CH), 92.6 (C). The spectroscopic data are consistent with those previously reported for this compound in the literature.[16] m/z (LCMS ESI) (%) tR 4.33 min, 288.8 (100, [M + H]+). HPLC: tR 6.40 min, 85.1 % purity at 254 nm. m/z (HRMS ESI) 288.9186; calcd for C9H6IOS+ [M + H]+ 288.9179.
N,N-Dimethyl-3-(2-(methylthio)phenyl)propiolamide (8ab)
n-BuLi (0.98 mL, 2.13 mmol, 2.17 M in hexanes) was added dropwise to a stirred solution of 16a (300 mg, 2.02 mmol) in dry THF (6.7 mL) at −78°C under an N2 atmosphere. The solution was stirred at −78°C for 0.5 h, followed by dropwise addition of 17b (0.20 mL, 2.23 mmol). The reaction mixture was raised to rt, quenched with 10 % citric acid solution (15 mL), and extracted with Et2O (2 × 30 mL). Washed with H2O (2 × 30 mL) and brine (2 × 20 mL), the combined organic extracts were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure to yield the title compound (409 mg, 92 %) as a brown oil without further purification. δH (400 MHz, CDCl3) 7.56 (dd, J 7.7, 1.5, 1H), 7.37 (ddd, J 8.0, 7.5, 1.5, 1H), 7.19 (d, J 7.5, 1H), 7.13 (td, J 7.6, 1.1, 1H), 3.38 (s, 3H), 3.04 (s, 3H), 2.51 (s, 3H). δC (101 MHz, CDCl3) 154.8 (C), 143.2 (C), 134.1 (CH), 130.6 (CH), 124.6 (CH), 124.4 (CH), 119.0 (C), 87.7 (C), 87.6 (C), 38.7 (CH3), 34.4 (CH3), 15.3 (CH3). m/z (LCMS ESI) (%) tR 3.22 min, 219.9 (100, [M + H]+). HPLC: tR 5.57 min, 97.9 % purity at 254 nm. m/z (HRMS ESI) 220.0794; calcd for C12H14NOS+ [M + H]+ 220.0791.
3-Iodo-N,N-dimethylbenzo[b]thiophene-2-carboxamide (9ab)
I2 (1.42 g, 5.59 mmol) was added slowly to a stirred solution of 8ab (409 mg, 1.86 mmol) in dry DCE (9.3 mL) under an N2 atmosphere. The reaction was heated at reflux for 17 h. On completion, the reaction mixture was cooled to rt, quenched with saturated Na2S2O3 solution (30 mL), and extracted with CH2Cl2 (2 × 30 mL). The combined organic extracts were washed with H2O (2 × 30 mL) and brine (2 × 30 mL), dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure to yield the title compound (614 mg, 99 %) as a brown oil without further purification. δH (400 MHz, CDCl3) 7.81 (ddd, J 7.9, 1.2, 0.7, 1H), 7.76 (ddd, J 8.0, 1.3, 0.7, 1H), 7.48 (ddd, J 8.1, 7.0, 1.1, 1H), 7.43 (ddd, J 7.9, 7.1, 1.3, 1H), 3.19 (br s, 3H), 3.03 (br s, 3H). δC (101 MHz, CDCl3) 164.9 (C), 140.3 (C), 138.4 (C), 136.7 (C), 126.4 (CH), 126.1 (CH), 125.9 (CH), 122.6 (CH), 79.7 (C), 39.0 (CH3), 35.4 (CH3). m/z (LCMS ESI) (%) tR 3.35 min, 331.7 (100, [M + H]+). HPLC: tR 5.80 min, 96.4 % purity at 254 nm. m/z (HRMS ESI) 330.9606; calcd for C11H11INOS+ [M + H]+ 330.9601.
Methyl 3-(2-(Methylthio)phenyl)propiolate (8ac)
n-BuLi (1.42 mL, 3.54 mmol, 2.5 M in hexanes) was added dropwise to a stirred solution of 16a (500 mg, 3.37 mmol) in dry THF (11.2 mL) at −78°C under an N2 atmosphere. The solution was stirred at −78°C for 45 min, followed by dropwise addition of 17c (0.29 mL, 3.71 mmol). The reaction mixture was stirred at −78°C for 1 h, then stirred at rt for 0.5 h. On completion, the mixture was quenched with saturated NH4Cl solution (30 mL) and extracted with Et2O (2 × 30 mL). Washed with H2O (2 × 50 mL) and brine (2 × 50 mL), the combined organic extracts were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure to yield the title compound (746 mg, 100 %) as a brown oil without further purification. δH (400 MHz, CDCl3) 7.53 (ddd, J 7.7, 1.5, 0.4, 1H), 7.40 (ddd, J 8.1, 7.5, 1.5, 1H), 7.20 (d, J 8.1, 1H), 7.12 (td, J 7.6, 1.1, 1H), 3.85 (s, 3H), 2.51 (s, 3H). δC (101 MHz, CDCl3) 154.5 (C), 144.2 (C), 134.3 (CH), 131.2 (CH), 124.7 (CH), 124.6 (CH), 117.8 (C), 86.2 (C), 83.9 (C), 53.0 (CH3), 15.3 (CH3). m/z (LCMS ESI) (%) tR 3.44 min, 207.0 (30, [M + H]+). HPLC: tR 6.22 min, 91.8 % purity at 254 nm. m/z (HRMS ESI) 207.0468; calcd for C11H11O2S+ [M + H]+ 207.0474.
Methyl 3-Iodobenzo[b]thiophene-2-carboxylate (9ac)
I2 (2.45 g, 10.4 mmol) was added to a stirred solution of 8ac (714 mg, 3.46 mmol) in dry DCE (20 mL) under an N2 atmosphere. The reaction was heated at 60°C for 17 h. On completion, the mixture was cooled to rt, quenched with saturated Na2S2O3 solution (100 mL) and extracted with CH2Cl2 (2 × 50 mL). The combined organic extracts were washed with H2O (2 × 80 mL) and brine (2 × 50 mL), dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure to yield the title compound (1.0 g, 92 %) as a brown oil without further purification. δH (400 MHz, CDCl3) 7.97–7.95 (m, 1H), 7.82–7.80 (m, 1H), 7.53 – 7.47 (m, 2H), 3.98 (s, 3H). δC (101 MHz, CDCl3) 162.2 (C), 142.0 (C), 140.4 (C), 130.2 (C), 128.3 (CH), 128.3 (CH), 126.0 (CH), 122.8 (CH), 88.4 (C), 52.7 (CH3). HPLC: tR 7.06 min, 90.4 % purity at 254 nm. m/z (HRMS ESI) 318.9283; calcd for C10H8IO2S+ [M + H]+ 318.9284.
1-(2-Bromophenyl)-3-(2-(methylthio)phenyl)prop-2-yn-1-one (8ae)
n-BuLi (0.89 mL, 2.13 mmol, 2.4 M in hexanes) was added dropwise to a stirred solution of 16a (300 mg, 2.02 mmol) in dry THF (10 mL) at −78°C under an N2 atmosphere. The white suspension was stirred at −78°C for 0.5 h, followed by slow addition of 17e (543 mg, 2.23 mmol). The reaction mixture was stirred at −78°C for 45 min, then stirred at 0°C for 1 h. On completion, the reaction mixture was warmed to rt, quenched with 10 % citric acid solution (50 mL), and extracted with Et2O (2 × 30 mL). The combined organic extracts were washed with H2O (2 × 50 mL) and brine (50 mL), dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography (3 : 2 hexanes/CH2Cl2, Rf 0.3) to yield the title compound (478 mg, 71 %) as a yellow solid; mp 64–66°C. δH (400 MHz, CDCl3) 8.32 (dd, J 7.7, 1.7, 1H), 7.71 (dd, J 8.0, 1.1, 1H), 7.62 (dd, J 7.7, 1.5, 1H), 7.47 (td, J 7.6, 1.2, 1H), 7.45–7.36 (m, 2H), 7.23 (d, J 7.7, 1H), 7.16 (td, J 7.6, 1.1, 1H), 2.53 (s, 3H). δC (101 MHz, CDCl3) 177.2 (C), 144.7 (C), 137.1 (C), 135.2 (CH), 134.7 (CH), 133.9 (CH), 133.5 (CH), 131.5 (CH), 127.5 (CH), 124.70 (CH), 124.67 (CH), 121.5 (C), 118.2 (C), 93.6 (C), 91.2 (C), 15.4 (CH3). HPLC: tR 7.26 min, 93.6 % purity at 254 nm. m/z (HRMS ESI) 330.9789; calcd for C16H12BrOS+ [M + H]+ 330.9787. m/z (HRMS APCI) 330.9791; calcd for C16H12BrOS+ [M + H]+ 330.9787.
(2-Bromophenyl)(3-iodobenzo[b]thiophen-2-yl)methanone (9ae)
I2 (605 mg, 2.38 mmol) was added to a stirred solution of 8ae (137 mg, 630 µmol) in dry DCE (5 mL) under an N2 atmosphere. The reaction was heated at 50°C for 15 h. On completion, the mixture was quenched with saturated Na2S2O3 solution (25 mL) and extracted with CH2Cl2 (2 × 20 mL). The combined organic extracts were washed with H2O (2 × 30 mL) and brine (2 × 30 mL), dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure yield the title compound (366 mg, 100 %) as a yellow oil without further purification. δH (400 MHz, CDCl3) 7.97 (d, J 7.8, 1H), 7.81 (d, J 7.0, 1H), 7.67 (dd, J 7.8, 1.0, 1H), 7.54–7.38 (m, 5H). δC (101 MHz, CDCl3) 189.5 (C), 142.5 (C), 141.1 (C), 140.6 (C), 139.3 (C), 133.5 (CH), 132.1 (CH), 129.5 (CH), 128.8 (CH), 128.7 (CH), 127.7 (CH), 126.3 (CH), 122.9 (CH), 120.3 (C), 88.3 (C). HPLC: tR 7.57 min, 96.7 % purity at 254 nm. m/z (HRMS APCI) 442.8598; calcd for C15H9BrIOS+ [M + H]+ 442.8597.
3-(2-Methoxyphenyl)-N,N-dimethylpropiolamide (8bb)
n-BuLi (1.0 mL, 2.44 mmol, 2.4 M in hexanes) was added dropwise to a stirred solution of 16b (350 mg, 2.65 mmol) in dry THF (12 mL) at −78°C under N2. The solution was stirred at −78°C for 0.5 h, followed by dropwise addition of 17b (0.27 mL, 2.91 mmol). The reaction mixture was stirred at −78°C for 5 min, then the temperature slowly raised to rt. The reaction mixture was quenched with saturated NH4Cl solution (35 mL) and extracted with Et2O (2 × 25 mL). Washed with H2O (2 × 50 mL) and brine (2 × 50 mL), the combined organic extracts dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure to yield the title compound (326 mg, 66 %) as a brown oil without further purification. δH (400 MHz, CDCl3) 7.51 (dd, J 7.6, 1.6, 1H), 7.38 (ddd, J 8.4, 7.5, 1.7, 1H), 6.94 (td, J 7.5, 1.0, 1H), 6.90 (d, J 8.4, 1H), 3.88 (s, 3H), 3.33 (s, 3H), 3.03 (s, 3H). δC (101 MHz, CDCl3) 161.2 (C), 154.9 (C), 134.3 (CH), 131.7 (CH), 120.6 (CH), 110.8 (CH), 110.0 (C), 86.9 (C), 85.8 (C), 55.8 (CH3), 38.4 (CH3), 34.2 (CH3). m/z (LCMS ESI) (%) tR 4.30 min, 204.0 (100, [M + H]+). HPLC: tR 4.96 min, 98.5 % purity at 254 nm. m/z (HRMS ESI) 204.1025; calcd for C12H14NO2+ [M + H]+ 204.1019.
3-(2-Methoxyphenyl)-1-phenylprop-2-yn-1-one (8bd)
n-BuLi (1.45 mL, 2.6 mmol, 1.8 M in hexanes) was added dropwise to a stirred solution of 16b (300 mg, 2.27 mmol) in dry THF (11 mL) at −78°C under an N2 atmosphere. The solution was stirred at −78°C for 75 min, followed by slow addition of 17d (0.41 mL, 2.72 mmol). The reaction mixture was stirred at −78°C for 10 min, then at 0°C for 1 h. On completion, the reaction mixture was brought to rt, quenched with saturated NH4Cl solution (40 mL), and extracted with Et2O (2 × 30 mL). The combined organic extracts were washed with H2O (2 × 60 mL) and brine (60 mL), dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography (97 : 3 hexanes/EtOAc, Rf 0.1) to yield the title compound (459 mg, 86 %) as a clear light yellow oil. δH (400 MHz, CDCl3) 8.34–8.31 (m, 2H), 7.65–7.59 (m, 2H), 7.54–7.49 (m, 2H), 7.45 (ddd, J 8.4, 7.5, 1.7, 1H), 6.99 (td, J 7.6, 1.0, 1H), 6.96 (d, J 8.5, 1H), 3.98 (s, 3H). δC (101 MHz, CDCl3) 178.3 (C), 162.0 (C), 137.3 (C), 135.2 (CH), 134.0 (CH), 132.8 (CH), 129.9 (CH), 128.7 (CH), 120.8 (CH), 111.0 (CH), 109.6 (C), 91.4 (C), 90.7 (C), 56.1 (CH3). The spectroscopic data are consistent with those previously reported for this compound in the literature.[17] m/z (LCMS ESI) (%) tR 3.20 min, 237.1 (100, [M + H]+). HPLC: tR 6.08 min, 98.1 % purity at 254 nm. m/z (HRMS ESI) 237.0915; calcd for C16H13O2+ [M + H]+ 237.0910.
3-(2-(Dimethylamino)phenyl)-N,N-dimethylpropiolamide (8cb)
n-BuLi (1.25 mL, 2.27 mmol, 1.8 M in hexanes) was added dropwise to a stirred solution of 16c (300 mg, 2.07 mmol) in dry THF (10 mL) at −78°C under an N2 atmosphere. The solution was stirred at −78°C for 1 h, followed by dropwise addition of 17b (0.22 mL, 2.38 mmol). The reaction mixture was stirred at −78°C for 5 min, then brought to rt and stirred for 0.5 h. The reaction mixture was quenched with saturated NH4Cl solution (25 mL) and extracted with Et2O (2 × 20 mL). Washed with H2O (2 × 50 mL) and brine (2 × 50 mL), the combined organic extracts were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure to yield the title compound (367 mg, 82 %) as a brown oil without further purification. δH (400 MHz, CDCl3) 7.48 (ddd, J 7.6, 1.7, 0.5, 1H), 7.30 (ddd, J 8.3, 7.3, 1.7, 1H), 6.90 (d, J 8.7, 1H), 6.86 (td, J 7.5, 1.1, 1H), 3.30 (s, 3H), 3.03 (s, 3H), 2.96 (s, 6H). δC (101 MHz, CDCl3) 156.0 (C), 155.2 (C), 135.5 (CH), 131.1 (CH), 120.3 (CH), 117.1 (CH), 112.0 (C), 90.3 (C), 87.0 (C), 43.8 (CH3), 38.4 (CH3), 34.3 (CH3). m/z (LCMS ESI) (%) tR 2.64 min, 217.1 (100, [M + H]+). HPLC: tR 3.46 min, 96.3 % purity at 254 nm. m/z (HRMS ESI) 217.1340; calcd for [M + H]+ 217.1335.
3-(2-(Dimethylamino)phenyl)-1-phenylprop-2-yn-1-one (8cd)
n-BuLi (1.32 mL, 2.38 mmol, 1.8 M in hexanes) was added dropwise to a stirred solution of 16c (300 mg, 2.07 mmol) in dry THF (10 mL) at −78°C under an N2 atmosphere. The solution was stirred at −78°C for 1.3 h, followed by slow addition of 17d (410 mg, 2.48 mmol, 0.38 mL). The reaction mixture was stirred at −78°C for 10 min, then at 0°C for 1 h. On completion, the reaction mixture was brought to rt, quenched with saturated NH4Cl solution (40 mL), and extracted with Et2O (2 × 30 mL). The combined organic extracts were washed with H2O (2 × 60 mL) and brine (60 mL), dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography (97 : 3 hexanes/EtOAc, Rf 0.15) to yield the title compound (490 mg, 95 %) as a bright orange oil. δH (400 MHz, CDCl3) 8.29–8.23 (m, 2H), 7.65–7.58 (m, 2H), 7.54–7.49 (m, 2H), 7.37 (ddd, J 8.4, 7.3, 1.7, 1H), 6.94 (d, J 8.4, 1H), 6.90 (td, J 7.5, 1.1, 1H), 3.06 (s, 6H). δC (101 MHz, CDCl3) 178.2 (C), 156.8 (C), 137.4 (C), 136.3 (CH), 133.9 (CH), 132.1 (CH), 129.7 (CH), 128.7 (CH), 120.1 (CH), 117.1 (CH), 110.8 (C), 94.2 (C), 93.0 (C), 43.8 (CH3). m/z (LCMS ESI) (%) tR 3.37 min, 250.1 (100, [M + H]+). HPLC: tR 5.53 min, 94.9 % purity at 254 nm. m/z (HRMS ESI) 250.1230; calcd for C17H16NO+ [M + H]+ 250.1226.
Methyl (2-((Methylsulfonyl)ethynyl)phenyl)sulfane (8af)
i-PrMgCl (2.0 mL, 4.05 mmol, 2.0 M in THF) was added dropwise to a stirred solution of 16a (300 mg, 2.02 mmol) in dry THF (10 mL) at 0°C under an N2 atmosphere. The solution was stirred at 0°C for 1 h, followed by slow addition of methanesulfonic anhydride (1.41 g, 8.1 mmol) at 0°C. The suspension was stirred at 0°C for 1 h, slowly brought to rt, quenched with saturated NH4Cl solution (50 mL), and extracted with Et2O (2 × 30 mL). The combined organic extracts were washed with H2O (2 × 70 mL) and brine (70 mL), dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography (1 : 1 hexanes/CH2Cl2, Rf 0.25) to yield the title compound (147 mg, 32 %) as a yellow oil. The conversion rate of product is >50 % in the crude 1H NMR spectrum, and low yield resulted from column chromatography. δH (401 MHz, CDCl3) 7.52 (dd, J 7.7, 1.2, 1H), 7.45 (td, J 8.1, 1.5, 1H), 7.23 (d, J 8.0, 1H), 7.15 (td, J 7.6, 1.0, 1H), 3.33 (s, 3H), 2.52 (s, 3H). δC (101 MHz, CDCl3) 144.8 (C), 133.9 (CH), 132.1 (CH), 125.0 (CH), 124.7 (CH), 115.8 (C), 90.1 (C), 89.5 (C), 47.2 (CH3), 15.3 (CH3). m/z (LCMS ESI) (%) tR 2.82 min, 227.0 (100, [M + H]+). HPLC: tR 4.85 min, 96.4 % purity at 254 nm. m/z (HRMS APCI) 227.0196; calcd for C10H11O2S2+ [M + H]+ 227.0195.
3-Iodo-2-(methylsulfonyl)benzo[b]thiophene (9af)
I2 (81 mg, 318 µmol) was added to a stirred solution of 8af (24 mg, 106 µmol) in dry DCE (0.5 mL) under an N2 atmosphere. The reaction was heated at 60°C for 16 h. On completion, the mixture was quenched with saturated Na2S2O3 solution (10 mL) and extracted with CH2Cl2 (2 × 15 mL). The combined organic extracts were washed with H2O (2 × 30 mL) and brine (2 × 20 mL), dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography (1 : 1 hexanes/CH2Cl2, Rf 0.25) to yield the title compound (35 mg, 97 %) as an off-white solid; mp 158–159°C. δH (400 MHz, CDCl3) 7.96–7.90 (m, 1H), 7.90–7.84 (m, 1H), 7.60–7.54 (m, 2H), 3.34 (s, 3H). δC (101 MHz, CDCl3) 141.9 (C), 140.8 (C), 140.4 (C), 128.8 (CH), 128.1 (CH), 126.7 (CH), 122.9 (CH), 86.6 (C), 43.7 (CH3). m/z (LCMS ESI) (%) tR 2.93 min, 338.9 (50, [M + H]+). HPLC: tR 5.20 min, 99.9 % purity at 254 nm. m/z (HRMS ESI) 338.9001; calcd for C9H8IO2S2+ [M + H]+ 338.9005.
1-Methoxy-2-((methylsulfonyl)ethynyl)benzene (8bf)
i-PrMgCl (2.3 mL, 4.54 mmol, 2.0 M in THF) was added dropwise to a stirred solution of 16b (300 mg, 2.27 mmol) in dry THF (12 mL) at 0°C under an N2 atmosphere. The solution was stirred at 0°C for 1 h, followed by slow addition of methanesulfonic anhydride (791 mg, 4.54 mmol) at 0°C. The suspension was stirred at 0°C for 0.5 h, slowly brought to rt, quenched with H2O (50 mL), and extracted with Et2O (2 × 35 mL). The combined organic extracts were washed with H2O (2 × 75 mL) and brine (75 mL), dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography (7 : 3 hexanes/CH2Cl2, Rf 0.2) to yield the title compound (247 mg, 52 %) as a light yellow oil. The conversion rate of product is >70 % in the crude1H NMR spectrum, but low yield resulted from column chromatography. δH (400 MHz, CDCl3) 7.50 (ddd, J 7.7, 1.8, 0.5, 1H), 7.46 (ddd, J 8.4, 1.8, 0.9, 1H), 6.96 (td, J 7.6, 0.9, 1H), 6.92 (d, J 8.5, 1H), 3.89 (s, 3H), 3.30 (s, 3H). δC (101 MHz, CDCl3) 161.9 (C), 134.8 (CH), 133.6 (CH), 120.8 (CH), 111.1 (CH), 106.9 (C), 89.6 (C), 88.1 (C), 56.0 (CH3), 47.1 (CH3). m/z (LCMS ESI) (%) tR 2.74 min, 211.1 (80, [M + H]+). HPLC: tR 4.44 min, 98.6 % purity at 254 nm. m/z (HRMS ESI) 211.0422; calcd for C10H11O3S+ [M + H]+ 211.0423.
N,N-Dimethyl-2-((methylsulfonyl)ethynyl)aniline (8cf)
i-PrMgCl (2.1 mL, 4.13 mmol, 2.0 M in THF) was added dropwise to a stirred solution of 16c (300 mg, 2.07 mmol) in dry THF (10 mL) at 0°C under an N2 atmosphere. The solution was stirred at 0°C for 1 h, followed by slow addition of methanesulfonic anhydride (720 mg, 4.13 mmol) at 0°C. The suspension was stirred at 0°C for 0.5 h, slowly brought to rt, quenched with H2O (50 mL), and extracted with Et2O (2 × 35 mL). The combined organic extracts were washed with H2O (2 × 75 mL) and brine (75 mL), dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography (100 % CH2Cl2, Rf 0.25) to yield the title compound (151 mg, 33 %) as a bright orange oil. The conversion rate of product is >75 % in the crude 1H NMR spectrum, but low yield resulted from column chromatography. δH (400 MHz, CDCl3) 7.45 (dd, J 7.7, 1.7, 1H), 7.36 (ddd, J 8.8, 7.3, 1.7, 1H), 6.87 (d, J 8.5, 1H), 6.82 (t, J 7.5, 1H), 3.28 (s, 3H), 3.02 (s, 6H). δC (101 MHz, CDCl3) 156.6 (C), 135.9 (CH), 132.9 (CH), 119.4 (CH), 116.7 (CH), 106.6 (C), 93.8 (C), 89.5 (C), 47.0 (CH3), 43.4 (CH3). m/z (LCMS ESI) (%) tR 3.06 min, 224.1 (100, [M + H]+). HPLC: tR 4.14 min, 92.7 % purity at 254 nm. m/z (HRMS ESI) 224.0748; calcd for C11H14NO2S+ [M + H]+ 224.0740.
2-Bromo-N-methoxy-N-methylbenzamide (17e)
Compound 17e was prepared using a literature procedure.[12] The crude product (4.21 g, 87 %) obtained was directly used in the next step without purification. δH (400 MHz, CDCl3) 7.58 (dd, J 8.0, 0.7, 1H), 7.40–7.26 (m, 3H), 3.69 (br app d, 3H), 3.27 (br app d, 3H). m/z (LCMS ESI) (%) tR 4.05 min, 243.9 (100, [M + H]+).
Methyl (2-((Methylthio)ethynyl)phenyl)sulfane (18)
Compound 18 was synthesised according to a literature procedure.[4] The crude product (1.39 g, 96 %) obtained was directly used in the next step without purification. δH (400 MHz, CDCl3) 7.36 (dd, J 7.6, 1.5, 1H), 7.26 (ddd, J 8.1, 7.4, 1.6, 1H), 7.13 (dd, J 8.0, 0.9, 1H), 7.06 (td, J 7.5, 1.2, 1H), 2.52 (s, 3H), 2.48 (s, 3H).
3-Iodo-2-(methylthio)benzo[b]thiophene (19)
Compound 19 was synthesised according to a literature procedure.[4] The crude product (2.0 g, 91 %) obtained was directly used in the next step without purification. δH (400 MHz, CDCl3) 7.71 (ddd, J 8.0, 1.1, 0.7, 1H), 7.65 (ddd, J 8.0, 1.2, 0.7, 1H), 7.41 (ddd, J 8.1, 7.1, 1.1, 1H), 7.33 (ddd, J 8.0, 7.2, 1.2, 1H), 2.63 (s, 3H).
3-Ethynyl-2-(methylthio)benzo[b]thiophene (20)
Compound 20 was synthesised according to General Procedure 1. The crude product of trimethyl ((2-(methylthio)benzo[b]thiophen-3-yl)ethynyl)silane was purified by flash column chromatography (100 % hexanes, Rf 0.3) to yield a transparent yellow oil (1.12 g, 82 % from 19). δH (400 MHz, CDCl3) 7.78 (dd, J 8.0, 1.3, 1H), 7.69 (dd, J 8.0, 1.2, 1H), 7.40 (ddd, J 8.1, 7.2, 1.1, 1H), 7.30 (ddd, J 8.0, 7.2, 1.3, 1H), 2.66 (s, 3H), 0.33 (s, 9H). δC (101 MHz, CDCl3) 145.2 (C), 140.2 (C), 138.2 (C), 125.1 (CH), 124.6 (CH), 122.4 (CH), 121.8 (CH), 117.4 (C), 102.7 (C), 97.3 (C), 18.7 (CH3), 0.3 (CH3). HPLC: tR 8.54 min, 98.4 % purity at 254 nm. m/z (HRMS ESI) 277.0524; calcd for C14H17S2Si [M + H]+ 277.0535.
From trimethyl ((2-(methylthio)benzo[b]thiophen-3-yl)ethynyl)silane, 20 (766 mg, 92 %) was obtained as a purple solid; mp 57–60°C. δH (400 MHz, CDCl3) 7.81 (dd, J 8.0, 1.3, 1H), 7.71 (dt, J 8.0, 0.9, 1H), 7.41 (ddd, J 8.1, 7.2, 1.1, 1H), 7.32 (ddd, J 8.3, 7.2, 1.3, 1H), 3.63 (s, 1H), 2.66 (s, 3H). δC (101 MHz, CDCl3) 145.6 (C), 140.2 (C), 138.4 (C), 125.3 (CH), 124.8 (CH), 122.3 (CH), 121.8 (CH), 116.6 (C), 84.7 (C), 76.6 (CH), 19.11 (CH3). The spectroscopic data of 20 are consistent with those previously reported for this compound in the literature.[4] m/z (LCMS ESI) (%) tR 3.80 min, 205.0 (100, [M + H]+). HPLC: tR 6.90 min, 91.2 % purity at 254 nm. m/z (HRMS ESI) 205.0134; calcd for C11H9S2+ [M + H]+ 205.0140.
3-((2-Bromophenyl)ethynyl)-2-(methylthio)benzo[b]thiophene (21)
19 (373 mg, 1.22 mmol) was dissolved in Et3N (6 mL), followed by addition of CuI (14 mg, 70 µmol) and Pd(PPh3)4 (42 mg, 37 µmol). The RBF was then degassed and backfilled with N2 three times. Finally, 39 (1.70 mL, 1.34 mmol) was added dropwise under an N2 atmosphere. The reaction was stirred at rt for 20 h. CuI (14 mg, 70 µmol) and Pd(PPh3)4 (42 mg, 37 µmol) were added again, and the RBF was degassed and backfilled with N2 three times. On completion, the suspension was filtered through Celite® and extracted with Et2O (3 × 20 mL). Washed with H2O (2 × 50 mL) and brine (2 × 50 mL), the combined organic extracts were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified in three flash column chromatographic attempts (5 : 1 hexanes/EtOAc, Rf 0.6; 9 : 1 hexanes/CH2Cl2, Rf 0.35; 100 % hexanes) to yield the title compound (286 mg, 65 %) as a yellow oil. δH (400 MHz, CDCl3) 7.99 (ddd, J 8.0, 1.2, 0.7, 1H), 7.73 (dt, J 8.0, 0.9, 1H), 7.67–7.64 (m, 2H), 7.43 (ddd, J 8.0, 7.1, 1.0, 1H), 7.36–7.31 (m, 2H), 7.21 (ddd, J 8.1, 7.5, 1.7, 1H), 2.71 (s, 3H). δC (101 MHz, CDCl3) 145.2 (C), 140.1 (C), 138.2 (C), 133.4 (CH), 132.5 (CH), 129.4 (CH), 127.1 (CH), 125.5 (C), 125.2 (CH), 125.2 (C), 124.6 (CH), 122.4 (CH), 121.7 (CH), 116.9 (C), 95.4 (C), 86.8 (C), 19.0 (CH3). HPLC: tR 9.05 min, 98.2 % purity at 254 nm. m/z (HRMS ESI) 358.9556; calcd for C17H12BrS2+ [M + H]+ 358.9558.
2-(2-Bromophenyl)-3-iodobenzo[b]thieno[3,2-d]thiophene (24)
I2 (51 mg, 201 µmol) was added to a stirred solution of 21 (65.8 mg, 183 µmol) in dry DCE (0.92 mL) under an N2 atmosphere. The reaction was heated at 80°C for 20 h. On completion, the mixture was cooled to rt, quenched with saturated Na2S2O3 solution (10 mL), and extracted with CH2Cl2 (2 × 10 mL). The combined organic extracts were washed with H2O (2 × 15 mL) and brine (2 × 15 mL), dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography (3 : 1 hexanes/CH2Cl2, Rf 0.48) to yield the title compound (64 mg, 74 %) as an off-white solid; mp 121–124°C. δH (400 MHz, CDCl3) 8.83 (ddd, J 8.0, 1.3, 0.7, 1H), 7.85 (ddd, J 8.0, 1.2, 0.7, 1H), 7.74 (ddd, J 8.0, 1.1, 0.5, 1H), 7.50 (ddd, J 8.1, 7.1, 1.1, 1H), 7.46–7.41 (m, 3H), 7.35 (ddd, J 8.0, 7.2, 1.3, 1H). δC (101 MHz, CDCl3) 144.0 (C), 142.6 (C), 140.0 (C), 139.1 (C), 135.8 (C), 133.4 (CH), 133.2 (CH), 133.1 (C), 131.0 (CH), 127.5 (CH), 125.7 (C), 125.1 (CH), 124.2 (CH), 123.2 (CH), 121.5 (CH), 75.7 (C). HPLC: tR 9.01 min, 97.1 % purity at 254 nm. m/z (HRMS APCI) 470.8382; calcd for C16H9BrIS2+ [M + H]+ 470.8368.
Methyl 3-(2-(Methylthio)benzo[b]thiophen-3-yl)propiolate (25)
n-BuLi (0.22 mL, 538 µmol, 2.45 M in hexanes) was added dropwise to a stirred solution of 20 (100 mg, 489 µmol) in dry THF (3 mL) at −78°C under an N2 atmosphere. The solution was stirred at −78°C for 1 h, followed by dropwise addition of 17c (44 µL, 563 µmol). The reaction mixture was stirred at −78°C for 0.5 h, then brought to rt and stirred for another 0.5 h. On completion, the reaction mixture was quenched with saturated NH4Cl solution (20 mL) and extracted with Et2O (2 × 15 mL). Washed with H2O (2 × 20 mL) and brine (2 × 20 mL), the combined organic extracts were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography (100 % toluene, Rf 0.5) to yield the title compound (103 mg, 81 %) as a tan solid; mp 84–85°C. δH (400 MHz, CDCl3) 7.82 (dd, J 8.0, 1.3, 1H), 7.72 (dd, J 8.0, 1.1, 1H), 7.43 (ddd, J 8.1, 7.2, 1.1, 1H), 7.34 (ddd, J 8.0, 7.2, 1.3, 1H), 3.88 (s, 3H), 2.70 (s, 3H). δC (101 MHz, CDCl3) 154.7 (C), 152.3 (C), 139.9 (C), 137.8 (C), 125.7 (CH), 125.0 (CH), 122.2 (CH), 121.9 (CH), 112.8 (C), 88.2 (C), 79.7 (C), 53.0 (CH3), 18.8 (CH3). m/z (LCMS ESI) (%) tR 3.39 min, 263.0 (30, M + H+), 285.0 (100, [M + Na]+). HPLC: tR 6.92 min, 97.3 % purity at 254 nm. m/z (HRMS ESI) 263.0191; calcd for C13H11O2S2+ [M + H]+ 263.0195.
Methyl 3-Iodobenzo[b]thieno[3,2-d]thiophene-2-carboxylate (26)
I2 (264 mg, 1.04 mmol) was added to a stirred solution of 25 (91 mg, 347 µmmol) in dry DCE (1.8 mL) under an N2 atmosphere. The reaction was heated at 80°C for 24 h. After heating, the reaction mixture was quenched with saturated Na2S2O3 solution (15 mL) and extracted with CH2Cl2 (2 × 20 mL). The combined organic extracts were washed with H2O (2 × 30 mL) and brine (2 × 30 mL), dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude product (41 mg) was purified by flash column chromatography (19 : 1 hexanes/EtOAc, Rf 0.45) to yield the title compound (25 mg, 19 %) as an off-white solid; mp 171–173°C. δH (400 MHz, CDCl3) 9.01 (dd, J 8.0, 1.5, 1H), 7.82 (dd, J 7.8, 1.4, 1H), 7.51 (ddd, J 8.0, 7.2, 1.4, 1H), 7.46 (ddd, J 7.8, 7.2, 1.4, 1H), 3.95 (s, 3H). δC (101 MHz, CDCl3) 161.6 (C), 145.1 (C), 144.1 (C), 142.2 (C), 133.1 (C), 130.5 (C), 125.9 (CH), 124.5 (CH), 123.3 (CH), 122.2 (CH), 79.9 (C), 52.5 (CH3). m/z (LCMS APCI) (%) tR 3.73 min, 396.9 (100, [M + Na]+). HPLC: tR 7.43 min, 99.5 % purity at 254 nm. m/z (HRMS ESI) 374.8999; calcd for C12H8IO2S2+ [M + H]+ 374.9005.
Methyl 2-((2-(Methylthio)benzo[b]thiophen-3-yl)ethynyl)benzoate (27)
Methyl 2-iodobenzoate (116 mg, 445 µmol) was dissolved in diisopropylamine (DIPA) (2.5 mL), followed by addition of Pd(PPh3)2Cl2 (6.2 mg, 9 µmol) and CuI (5.1 mg, 27 µmol). The mixture was degassed with N2 three times. A solution of 20 (100 mg, 489 µmol) in dry THF (2 mL) was added slowly to the mixture at 45°C. The mixture was stirred at 45°C overnight. On completion, the suspension was filtered through Celite® and washed with Et2O (25 mL). Washed with H2O (2 × 20 mL) and brine (2 × 20 mL), the combined organic extracts were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography (100 % toluene, Rf 0.45) to yield the title compound (115 mg, 77 %) as a pale yellow solid; mp 88–89°C. δH (400 MHz, CDCl3) 8.02 (td, J 7.5, 1.6, 2H), 7.76 (dd, J 8.0, 1.2, 2H), 7.73 (dt, J 8.0, 0.9, 2H), 7.53 (td, J 7.6, 1.4, 1H), 7.49–7.36 (m, 2H), 7.34 (ddd, J 8.3, 7.1, 1.3, 1H), 4.00 (s, 3H), 2.71 (s, 3H). δC (101 MHz, CDCl3) 166.8 (C), 144.9 (C), 140.4 (C), 138.3 (C), 134.5 (CH), 131.9 (CH), 131.6 (C), 130.7 (CH), 128.1 (CH), 125.3 (CH), 124.7 (CH), 123.9 (C), 122.7 (CH), 121.8 (CH), 117.6 (C), 96.0 (C), 87.3 (C), 52.5 (CH3), 19.1 (CH3). m/z (LCMS ESI) (%) tR 3.74 min, 339.0 (20, [M + H]+), 361.0 (100, [M + Na]+). HPLC: tR 7.78 min, 97.6 % purity at 254 nm. m/z (HRMS ESI) 339.0513; calcd for C19H15O2S2+ [M + H]+ 339.0508.
4-Iodo-3-(2-(methylthio)benzo[b]thiophen-3-yl)-1H-isochromen-1-one (28)
I2 (128 mg, 505 µmol) was added to a stirred solution of 27 (57 mg, 168 µmol) in dry DCE (1.6 mL) under an N2 atmosphere. The reaction was heated at 50°C for 22 h. On completion, the mixture was quenched with saturated Na2S2O3 solution (15 mL) and extracted with CH2Cl2 (2 × 20 mL). The combined organic extracts were then washed with H2O (2 × 30 mL) and brine (2 × 20 mL), dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure to yield the title compound (80 mg, 100 %) as an off-white solid without further purification. δH (400 MHz, CDCl3) 8.35 (d, J 8.1, 1H), 7.90–7.83 (m, 2H), 7.83–7.75 (m, 1H), 7.64 (ddd, J 8.0, 5.5, 3.0, 1H), 7.54–7.46 (m, 1H), 7.40–7.32 (m, 2H), 2.63 (s, 3H). δC (101 MHz, CDCl3) 161.9 (C), 150.1 (C), 142.8 (C), 139.5 (C), 137.9 (C), 137.8 (C), 135.9 (CH), 131.42 (C), 131.37 (CH), 130.1 (CH), 129.9 (CH), 125.3 (CH), 124.9 (CH), 122.4 (CH), 122.1 (CH), 120.9 (C), 81.9 (C), 20.1 (CH3). One quaternary carbon is missing at 131.42 ppm in DEPTQ 13C spectrum, found in normal 13C spectrum. m/z (LCMS APCI) (%) tR 4.29 min, 450.9 (100, [M + H]+). HPLC: tR 7.35 min, 98.8 % purity at 254 nm. m/z (HRMS ESI) 450.9313; calcd for C18H12IO2S2+ [M + H]+ 450.9318.
2-(Methylthio)-3-((2-(methylthio)phenyl)ethynyl)benzo[b]thiophene (29)
2-Iodothioanisole (152 mg, 608 µmol) was dissolved in DIPA (4.0 mL), followed by addition of Pd(PPh3)2Cl2 (8.1 mg, 12 µmol) and CuI (6.6 mg, 35 µmol). The mixture was degassed with N2 three times. A solution of 20 (130 mg, 636 µmol) in dry THF (2 mL) was added to the mixture at 45°C over a period of 4 h. The mixture was stirred at rt overnight. On completion, the suspension was filtered through Celite® and washed with Et2O (25 mL). Washed with H2O (2 × 20 mL) and brine (2 × 20 mL), the combined organic extracts were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography (3 : 1 hexanes/toluene, Rf 0.35) to yield the title compound (108 mg, 55 %) as a clear yellow oil. δH (400 MHz, CDCl3) 8.02 (dd, J 8.0, 1.3, 1H), 7.72 (dt, J 8.0, 1.0, 1H), 7.59 (dd, J 7.6, 1.5, 1H), 7.43 (ddd, J 8.1, 7.2, 1.1, 1H), 7.34 (dd, J 8.3, 1.0, 1H), 7.32 (dd, J 8.4, 1.1, 1H), 7.22 (dd, J 8.0, 1.1, 1H), 7.15 (td, J 7.5, 1.2, 1H), 2.71 (s, 3H), 2.56 (s, 3H). δC (101 MHz, CDCl3) 144.3 (C), 141.5 (C), 140.2 (C), 138.4 (C), 132.7 (CH), 129.0 (CH), 125.3 (CH), 124.7 (CH), 124.5 (CH), 124.4 (CH), 122.8 (CH), 121.8 (CH), 121.6 (C), 117.8 (C), 94.4 (C), 88.7 (C), 19.2 (CH3), 15.4 (CH3). m/z (LCMS APCI) (%) tR 4.74 min, 327.0 (60, [M + H]+), 341.0 (100, [M + Na]+). HPLC: tR 8.14 min, 96.6 % purity at 254 nm. m/z (HRMS ESI) 327.0319; calcd for C18H15S3+ [M + H]+ 327.0330.
3-Iodo-2′-(methylthio)-2,3′-bibenzo[b]thiophene (30)
I2 (124 mg, 487 µmol) was added to a stirred solution of 29 (53 mg, 162 µmol) in dry DCE (1 mL) under an N2 atmosphere. The reaction was heated at 45°C for 18 h. On completion, the reaction mixture was quenched with saturated Na2S2O3 solution (15 mL) and extracted with CH2Cl2 (2 × 20 mL). The combined organic extracts were then washed with H2O (2 × 30 mL) and brine (2 × 20 mL), dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by a silica plug (100 % toluene, Rf 0.8) to yield the title compound (60 mg, 84 %) as a brown oil. δH (400 MHz, CDCl3) 7.90–7.82 (m, 2H), 7.85–7.78 (m, 2H), 7.54–7.49 (m, 1H), 7.47–7.41 (m, 2H), 7.45–7.37 (m, 1H), 7.39–7.28 (m, 2H), 2.57 (s, 3H). δC (101 MHz, CDCl3) 141.3 (C), 141.2 (C), 140.2 (C), 139.44 (C), 139.38 (C), 135.8 (C), 129.6 (C), 126.2 (CH), 125.9 (CH), 125.5 (CH), 125.0 (CH), 124.7 (CH), 123.0 (CH), 122.5 (CH), 122.0 (CH), 84.8 (C), 20.0 (CH3). m/z (LCMS APCI) (%) tR 4.27 min, 438.9 (20, [M + H]+), 312.0 (100, [M + H – I]+). HPLC: tR 8.47 min, 97.1 % purity at 254 nm.
3-((2-Bromophenyl)ethynyl)benzo[b]thiophene-2-carbaldehyde (33)
9aa (535 mg, 1.86 mmol) was dissolved in Et3N (10 mL), followed by addition of CuI (21 mg, 111 µmol) and Pd(PPh3)2Cl2 (26 mg, 37 µmol). The RBF was then degassed and backfilled with N2 three times. Finally, 39 (360 mg, 2.0 mmol) was added dropwise under an N2 atmosphere. The reaction was stirred at rt overnight. On completion, the suspension was filtered through Celite® and washed with Et2O (75 mL). Washed with H2O (2 × 50 mL) and brine (2 × 50 mL), the organic extract was dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography (2 : 1 hexanes/CH2Cl2, Rf 0.5) to yield the title compound (487 mg, 77 %) as a pale yellow solid; mp 132–134°C. δH (400 MHz, CDCl3) 10.56 (s, 1H), 8.26 (ddd, J 7.2, 1.8, 1.0, 1H), 7.90 (ddd, J 7.2, 1.6, 1.0, 1H), 7.69 (dd, J 8.0, 1.2, 1H), 7.68 (dd, J 7.8, 1.7, 1H), 7.62–7.50 (m, 2H), 7.38 (td, J 7.6, 1.2, 1H), 7.29 (ddd, J 8.0, 7.5, 1.7, 1H). δC (101 MHz, CDCl3) 184.9 (CH), 144.3 (C), 141.2 (C), 139.5 (C), 133.9 (CH), 132.9 (CH), 130.7 (CH), 129.1 (CH), 127.5 (CH), 127.4 (C), 126.0 (C), 125.9 (CH), 125.4 (CH), 124.4 (C), 123.5 (CH), 97.3 (C), 85.1 (C). One quaternary carbon is missing at 144.3 ppm in the DEPTQ 13C spectrum, found in normal 13C spectrum. m/z (LCMS ESI) (%) tR 3.97 min, 341.0 (60, [M + H]+). HPLC: tR 8.03 min, 95.8 % purity at 254 nm. m/z (HRMS ESI) 340.9625; calcd for C17H10BrOS+ [M + H]+ 340.9630.
Methyl 3-((2-Bromophenyl)ethynyl)benzo[b]thiophene-2-carboxylate (34)
9ac (559 mg, 1.76 mmol) was dissolved in Et3N (4.0 mL) and THF (3.0 mL), followed by addition of Pd(PPh3)4 (60.9 mg, 53 µmol) and CuI (20.1 mg, 110 µmol). The RBF was then degassed and backfilled with N2 three times. Finally, a solution of 39 (0.23 mL, 1.85 mmol) in Et3N (1.9 mL) and THF (2.9 mL) was added dropwise under an N2 atmosphere. The reaction was stirred at rt overnight. On completion, the suspension was filtered through Celite® and washed with Et2O (40 mL). Washed with H2O (2 × 40 mL) and brine (2 × 30 mL), the organic extract was dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography (1 : 1 hexanes/CH2Cl2, Rf 0.55) to yield the title compound (506 mg, 78 %) as an orange oil. δH (400 MHz, CDCl3) 8.32–8.27 (m, 1H), 7.88–7.83 (m, 1H), 7.74 (dd, J 7.8, 1.6, 1H), 7.67 (dd, J 8.1, 0.9, 1H), 7.56–7.50 (m, 2H), 7.36 (td, J 7.6, 1.2, 1H), 7.25 (ddd, J 8.0, 7.5, 1.7, 1H), 4.01 (s, 3H). δC (101 MHz, CDCl3) 162.5 (C), 140.0 (C), 139.9 (C), 134.3 (CH), 134.3 (C), 132.6 (CH), 130.2 (CH), 127.9 (CH), 127.3 (CH), 125.6 (CH), 125.5 (C), 125.3 (CH), 123.2 (C), 122.7 (CH), 96.7 (C), 87.2 (C), 52.7 (CH3). One quaternary carbon is overlapping at 125.5 ppm. m/z (LCMS ESI) (%) tR 3.94 min, 370.7 (100, [M + H]+). HPLC: tR 8.50 min, 91.1 % purity at 254 nm. m/z (HRMS ESI) 370.9737; calcd for C18H12BrO2S+ [M + H]+ 370.9736.
3-(2-Bromophenyl)-4-iodo-1H-benzo[4,5]thieno[2,3-c]pyran-1-one (35)
I2 (375 mg, 1.47 mmol) was added to a stirred solution of 34 (219 mg, 590 µmol) in dry DCE (3.0 mL) under an N2 atmosphere. The reaction mixture was heated at 65°C for 17 h. On completion, the reaction mixture was cooled to rt, quenched with saturated Na2S2O3 solution (30 mL), and extracted with CH2Cl2 (3 × 15 mL). The combined organic extracts were washed with H2O (2 × 30 mL) and brine (2 × 30 mL), dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography (3 : 2 CH2Cl2/hexanes, Rf 0.33) to yield the title compound (258 mg, 91 %). mp 218–220°C. δH (400 MHz, CDCl3) 9.34 (ddd, J 8.4, 1.3, 0.7, 1H), 8.01 (ddd, J 8.1, 1.3, 0.7, 1H), 7.73 (ddd, J 8.1, 1.1, 0.4, 1H), 7.67 (ddd, J 8.1, 7.0, 1.2, 1H), 7.61 (ddd, J 8.4, 7.1, 1.3, 1H), 7.48–7.44 (m, 2H), 7.38 (ddd, J 8.1, 7.1, 1.1, 1H). δC (101 MHz, CDCl3) 158.2 (C), 156.6 (C), 144.1 (C), 140.6 (C), 136.9 (C), 135.6 (C), 133.2 (CH), 131.8 (CH), 131.7 (CH), 129.2 (CH), 127.7 (CH), 126.2 (CH), 125.7 (CH), 124.7 (C), 123.7 (C), 123.7 (CH), 68.9 (C). HPLC: tR 7.87 min, 88.6 % purity at 254 nm. m/z (HRMS ESI) 482.8547; calcd for C17H9BrIO2S+ [M + H]+ 482.8546.
3-(2-Bromophenyl)-4-iodobenzo[4,5]thieno[2,3-c]pyridine (37)
In a dry RBF, 33 (190 mg, 557 µmol) was dissolved in dry DCE (4 mL), followed by addition of anhydrous MgSO4 (201 mg, 1.67 mmol) and tert-butylamine (0.59 mL, 5.57 mmol,). The reaction mixture was stirred at 50°C for 3 h, followed by addition of tert-butylamine (0.29 mL, 2.78 mmol). The reaction mixture heated at 50°C for another 19 h. After heating, the reaction mixture was cooled to rt, diluted with CH2Cl2 (20 mL), dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure to yield 36 (223 mg) as an orange oil without purification. 36 was unstable and used in situ in the next step. Rapid analysis of 36: δH (400 MHz, CDCl3) 8.94 (s, 1H), 8.12–8.06 (m, 1H), 7.84–7.79 (m, 1H), 7.66 (td, J 7.9, 1.3, 2H), 7.49–7.40 (m, 2H), 7.36 (td, J 7.6, 1.2, 1H), 7.26–7.21 (m, 1H), 1.36 (s, 9H). δC (101 MHz, CDCl3) 149.5 (CH), 147.7 (C), 139.9 (C), 139.1 (C), 133.5 (CH), 132.8 (CH), 129.9 (CH), 127.4 (CH), 126.7 (CH), 125.6 (C), 125.3 (C), 125.1 (CH), 123.7 (CH), 122.8 (CH), 119.8 (C), 95.2 (C), 86.6 (C), 58.5 (C), 29.9 (CH3). NIS (89 mg, 394 µmol) was added slowly to a stirred solution of 36 (52 mg, 131 µmol) in dry DCE (1.3 mL) under a an N2 atmosphere. The reaction was stirred at 60°C for 18 h. After heating, the reaction mixture was cooled to rt, diluted with saturated Na2S2O3 solution (15 mL), and extracted with CH2Cl2 (2 × 15 mL). The combined organic extracts were washed with H2O (2 × 15 mL) and brine (2 × 15 mL), then dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography (100 % CH2Cl2, Rf 0.4) to yield the title compound (328 mg, 80 %) as an off-white solid; mp 192–195°C. δH (400 MHz, CDCl3) 9.59 (dd, J 8.3, 1.3, 1H), 9.14 (s, 1H), 7.98 (dd, J 8.0, 1.3, 1H), 7.75 – 7.67 (m, 2H), 7.62 (ddd, J 8.4, 7.2, 1.3, 1H), 7.47 (td, J 7.5, 1.2, 1H), 7.38 (dd, J 7.6, 1.8, 1H), 7.34 (ddd, J 8.0, 7.3, 1.8, 1H). δC (101 MHz, CDCl3) 158.4 (C), 144.6 (C), 143.7 (CH), 142.34 (C), 142.31 (C), 136.3 (C), 134.9 (C), 132.8 (CH), 131.0 (CH), 130.1 (CH), 129.6 (CH), 127.6 (CH), 126.0 (CH), 123.9 (CH), 123.41 (CH), 123.39 (C), 90.7 (C). m/z (LCMS ESI) (%) tR 4.22 min, 465.9 (100, [M + H]+). HPLC: tR 7.48 min, 99.9 % purity at 254 nm. m/z (HRMS ESI) 465.8770; calcd for C17H10BrINS+ [M + H]+ 465.8757.
1-Bromo-2-ethynylbenzene (39)
Compound 39 was synthesised according to General Procedure 1. A yellow oil (4.10 g, 85 % from 2-bromoiodobenzene) was obtained. δH (400 MHz, CDCl3) 7.60 (dd, J 8.0, 1.3, 1H), 7.53 (dd, J 7.6, 1.8, 1H), 7.28 (td, J 7.6, 1.3, 1H), 7.21 (td, J 7.7, 1.8, 1H), 3.38 (s, 1H). The spectroscopic data are consistent with those previously reported for this compound in the literature.[18]
Supplementary Material
1H and 13C NMR spectra of all newly synthesised compounds are available on the Journal’s website.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
The authors thank Monash University for stipend support for SC.
References
[1] (a) For reviews, see: B. Godoi, R. F. Schumacher, G. Zeni, Chem. Rev. 2011, 111, 2937.| Crossref | GoogleScholarGoogle Scholar | 21425870PubMed |
(b) A. K. Banerjee, M. S. Laya, E. V. Carbrera, Curr. Org. Chem. 2011, 15, 1058.
| Crossref | GoogleScholarGoogle Scholar |
(c) A. V. Dubrovskiy, A. Nataliya, N. A. Markina, R. C. Larock, Comb. Chem. High Throughput Screen. 2012, 15, 451.
| Crossref | GoogleScholarGoogle Scholar |
(d) P. T. Parvatkar, P. S. Parameswaran, G. Santosh, S. G. Tilve, Chem. – Eur. J. 2012, 18, 5460.
| Crossref | GoogleScholarGoogle Scholar |
(e) A. Palisse, S. F. Kirsh, Org. Biomol. Chem. 2012, 10, 8041.
| Crossref | GoogleScholarGoogle Scholar |
[2] (a) For selected applications to drug discovery, see: Y. He, D. Duckett, W. Chen, Y. Y. Ling, M. D. Cameron, L. Lin, C. H. Ruiz, P. V. LoGrasso, T. M. Kamenecka, M. Koenig, Bioorg. Med. Chem. Lett. 2014, 24, 161.
| Crossref | GoogleScholarGoogle Scholar | 24332487PubMed |
(b) A. Nakhi, M. S. Rahman, S. Archana, R. Kishore, G. P. K. Seerapu, K. L. Kumar, D. Haldar, M. Pal, Bioorg. Med. Chem. Lett. 2013, 23, 4195.
| Crossref | GoogleScholarGoogle Scholar |
(c) Y. He, S. Liu, A. Menon, S. Stanford, E. Oppong, A. M. Gunawan, L. Wu, D. J. Wu, A. M. Barrios, N. Bottini, A. C. B. Cato, Z.-Y. Zhang, J. Med. Chem. 2013, 56, 4990.
(d) L.-F. Zeng, J. Xu, Y. He, R. He, L. Wu, A. M. Gunawan, Z.-Y. Zhang, ChemMedChem 2013, 8, 904.
(e) R. Rossi, A. Carpita, F. Bellina, P. Stabile, L. Mannina, Tetrahedron 2003, 59, 2067.
(f) C. T. Bui, B. L. Flynn, J. Comb. Chem. 2006, 8, 163.
| Crossref | GoogleScholarGoogle Scholar |
(g) B. L. Flynn, G. P. Flynn, E. Hamel, M. K. Jung, Bioorg. Med. Chem. Lett. 2001, 11, 2341.
| Crossref | GoogleScholarGoogle Scholar |
(h) B. L. Flynn, P. Verdier-Pinard, E. Hamel, Org. Lett. 2001, 3, 651.
| Crossref | GoogleScholarGoogle Scholar |
(i) B. L. Flynn, D. Grobelny, G. S. Gill, J. H. Chaplin, PCT Int. Appl. WO 2006084338 2006.
(j) S. A. Scott, C. T. Spencer, M. C. O’Reilly, K. A. Brown, R. R. Lavieri, C.-H. Cho, D.-I. Jung, R. C. Larock, H. A. Brown, C. W. Lindsley, ACS Chem. Biol. 2015, 10, 421.
| Crossref | GoogleScholarGoogle Scholar |
(k) M. Sala, R. Kristen, K. R. Hollinger, A. G. Thomas, R. P. Dash, C. Tallon, V. Veeravalli, L. Lovell, M. Kogler, H. Hrebabecky, E. Prochazkova, O. Nesuta, A. Donoghue, J. Lam, R. Rais, C. Rojas, B. S. Slusher, R. Nencka, J. Med. Chem. 2020, 63, 6028.
(l) S. He, P. Jain, B. Lin, M. Ferrer, Z. Hu, N. Southall, X. Hu, W. Zheng, B. Neuenswander, C.-H. Cho, Y. Chen, S. A. Worlikar, J. Aubé, R. C. Larock, F. J. Schoenen, J. J. Marugan, T. J. Liang, K. J. Frankowski, ACS Comb. Sci. 2015, 17, 641.
| Crossref | GoogleScholarGoogle Scholar |
(m) A. Nakhi, M. Shafiqur Rahman, S. Archana, R. Kishore, G. P. K. Seerapu, K. Lalith Kumar, D. Haldar, M. Pal, Bioorg. Med. Chem. Lett. 2013, 23, 4195.
| Crossref | GoogleScholarGoogle Scholar |
[3] (a) K. O. Hessian, B. L. Flynn, Org. Lett. 2006, 8, 243.
| Crossref | GoogleScholarGoogle Scholar | 16408885PubMed |
(b) R. Halim, P. J. Scammells, B. L. Flynn, J. Org. Chem. 2013, 78, 4708.
| Crossref | GoogleScholarGoogle Scholar |
(c) Q. Huang, J. A. Hunter, R. C. Larock, J. Org. Chem. 2002, 67, 3437.
| Crossref | GoogleScholarGoogle Scholar |
(d) T. Yao, R. C. Larock, J. Org. Chem. 2003, 68, 5936.
| Crossref | GoogleScholarGoogle Scholar |
(e) C. Zhou, A. V. Dubrovsky, R. C. Larock, J. Org. Chem. 2006, 71, 1626.
| Crossref | GoogleScholarGoogle Scholar |
(f) N. Ahmed, C. Dubuc, J. Rousseau, F. Bénard, J. E. van Lier, Bioorg. Med. Chem. Lett. 2007, 17, 3212.
| Crossref | GoogleScholarGoogle Scholar |
[4] L. Aurelio, R. Volpe, R. Halim, P. J. Scammells, B. L. Flynn, Adv. Synth. Catal. 2014, 356, 1974.
| Crossref | GoogleScholarGoogle Scholar |
[5] R. Volpe, L. Aurelio, M. Gillin, E. H. Krenske, B. L. Flynn, Chem. – Eur. J. 2015, 21, 10191.
| Crossref | GoogleScholarGoogle Scholar | 26043933PubMed |
[6] Y. Yamamoto, I. D. Gridnev, N. T. Patil, T. Jin, Chem. Commun. 2009, 5075.
| Crossref | GoogleScholarGoogle Scholar |
[7] A. Gupta, B. L. Flynn, J. Org. Chem. 2016, 81, 4012.
| Crossref | GoogleScholarGoogle Scholar | 27088459PubMed |
[8] S. Mehta, J. P. Waldo, R. C. Larock, J. Org. Chem. 2009, 74, 1141.
| Crossref | GoogleScholarGoogle Scholar | 19105638PubMed |
[9] K. Gilmore, I. V. Alabugin, Chem. Rev. 2011, 111, 6513.
| Crossref | GoogleScholarGoogle Scholar | 21861478PubMed |
[10] F. Denes, C. H. Schiesser, P. Renaud, Chem. Soc. Rev. 2013, 42, 7900.
| Crossref | GoogleScholarGoogle Scholar | 23828205PubMed |
[11] (a) G. Bencivenni, T. Lanza, R. Leardini, M. Minozzi, D. Nanni, P. Spagnolo, G. Zanardi, Org. Lett. 2008, 10, 1127.
| Crossref | GoogleScholarGoogle Scholar | 18278931PubMed |
(b) S. Lu, B. Wu, S. Zhang, Y. Gong, S. Xu, RSC Adv. 2020, 10, 19083.
| Crossref | GoogleScholarGoogle Scholar |
[12] M. Hatano, K. Yamakawa, T. Kawai, T. Horibe, K. Ishihara, Angew. Chem. Int. Ed. 2016, 55, 4021.
| Crossref | GoogleScholarGoogle Scholar |
[13] K. N. Lehane, E. J. A. Moynihan, N. Brondel, S. E. Lawrence, A. R. Maguire, CrystEngComm 2007, 9, 1041.
| Crossref | GoogleScholarGoogle Scholar |
[14] M. Beshai, B. Dhudshia, R. Mills, A. N. Thadani, Tetrahedron Lett. 2008, 49, 6794.
| Crossref | GoogleScholarGoogle Scholar |
[15] Y. Chen, C. H. Cho, R. C. Larock, Org. Lett. 2009, 11, 173.
| Crossref | GoogleScholarGoogle Scholar | 19046068PubMed |
[16] L. Cuesta, I. Maluenda, T. Soler, R. Navarro, E. P. Urriolabeitia, Inorg. Chem. 2011, 50, 37.
| Crossref | GoogleScholarGoogle Scholar | 21117700PubMed |
[17] J.-Y. Chen, T.-C. Lin, S.-C. Chen, A.-J. Chen, C.-Y. Mou, F.-Y. Tsai, Tetrahedron 2009, 65, 10134.
| Crossref | GoogleScholarGoogle Scholar |
[18] M. Ghaffarzadeh, M. Bolourtchian, Z. H. Fard, M. R. Halvagar, F. Mohsenzadeh, Synth. Commun. 2006, 36, 1973.
| Crossref | GoogleScholarGoogle Scholar |
* Associate Professor Bernard Flynn is the recipient of the 2019 RACI Applied Research Award.