

Element 70 – Ytterbium
Thomas Behrsing A , Glen B. Deacon A C and Peter C. Junk BA School of Chemistry, Monash University, Melbourne, Vic. 3800, Australia.
B College of Science and Engineering, James Cook University, Townsville, Qld 4811, Australia.
C Corresponding author. Email: glen.deacon@monash.edu
Australian Journal of Chemistry 72(12) 927-930 https://doi.org/10.1071/CH19527
Submitted: 17 October 2019 Accepted: 18 October 2019 Published: 13 November 2019
Introduction
Ytterbium is the penultimate element of the lanthanoid series. It is thus one of the heavy lanthanoid elements and lies between thulium and lutetium. Its discovery is attributed to the Swiss chemist Jean Charles Galissard de Marignac in 1878. It was the fourth element (the others are yttrium, erbium, and terbium) to be isolated from the mineral ‘ytterbite’, later named gadolinite (Fig. 1), which was obtained from the feldspar mine in the village of Ytterby on Resarö Island in the vicinity of Vaxholm in Sweden. These days, it is usually sourced from monazite though it is present in higher amounts in the less common mineral xenotime, which is a heavy rare earth mineral, substantially YPO4. The name ytterbium is the ultimate tribute to Ytterby.

|
Initially, ytterbium salts were separated from other lanthanoids by fractional crystallisation. Next, ion exchange was used, but currently counter-current solvent extraction is the method of choice.[1–6a]
Following back-extraction into aqueous solution, Yb3+ can be precipitated as the hydroxide or the oxalate to provide Yb2O3, a major commercial product, by thermolysis, or can be converted into the hydrated chloride (Scheme 1).

|
To obtain the free metal requires a powerful reductant as Yb metal is highly electropositive. The oxide can be reduced to Yb metal by metallic La, which is more electropositive than Yb or the oxide can be converted into the trifluoride with HF at 700°C under Ar, and the fluoride can be reduced by Ca.[6a] The metal has a metallic sheen (Fig. 2) but tarnishes rapidly in the air owing to oxidation.

|
Ytterbium has a limited number of uses. It can be used to improve the mechanical properties of stainless steel, and has applications in lasers, fibre optic cables, and portable X-ray devices.[1] The salts, particularly the triflate, have uses in organic synthesis, mainly as powerful Lewis acids.[2] An exciting potential use is development of photosensitized NIR emission in biological analysis and sensing.[6b]
Electronic Configuration and Oxidation States
The electronic configuration of the element is [Xe]4f146s2, and the two main oxidation states are III (4f13) and II (4f14). Thus, it is one of the three elements (with samarium and europium) long known to have an accessible +II state, though now this state has been achieved for all rare earth elements except Pm.[7]
At this stage, Yb zero-valent compounds have not been achieved, though they are known for most lanthanoids.[8] Formation of zero-valent compounds requires either a 4fn−15d16s2 configuration for the element or easy access to it, a situation not obtained for Eu or Yb, which have the most stable divalent states.
The 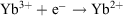 redox potential is −1.05 V (cf. Eu3+ −0.54 V, Sm3+ −1.55V).[6a] Thus, Yb2+ reduces water,
redox potential is −1.05 V (cf. Eu3+ −0.54 V, Sm3+ −1.55V).[6a] Thus, Yb2+ reduces water, 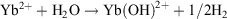 , and YbII compounds must be prepared and handled in an inert atmosphere. The redox potential is affected by the ligands attached. Thus
, and YbII compounds must be prepared and handled in an inert atmosphere. The redox potential is affected by the ligands attached. Thus 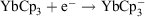 E0 −1.5 V (versus Fc+/Fc) indicates YbCp3 is somewhat harder to reduce than Yb3+ ions.[9] YbII is obviously favoured by a closed-shell configuration. It has been proposed that the redox transmetallation between Hg(C6F5)2 and Yb metal,
E0 −1.5 V (versus Fc+/Fc) indicates YbCp3 is somewhat harder to reduce than Yb3+ ions.[9] YbII is obviously favoured by a closed-shell configuration. It has been proposed that the redox transmetallation between Hg(C6F5)2 and Yb metal,
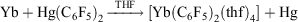
proceeds through a univalent intermediate (C6F5)Yb–Hg(C6F5)[10] and a recent theoretical study[11] provides support for this hypothesis by showing that it is energetically favoured, but no YbI compounds have been isolated.
The reductive ability of YbII can be advantageous where the less reducing EuII is ineffective and the more powerful SmII leads to over-reduction. Thus [YbII(p-HC6F4N(CH2)2NR2)2(thf)2] (R = Me, Et; thf = tetrahydrofuran), formed by metathesis from YbI2, by cleavage of [Yb{N(SiMe3)2}2] with p-HC6F4NH(CH2)2NR2 and by redox transmetallation between Yb, Hg(C6F5)2, and p-HC6F4NH(CH2)2NR2 (Scheme 2) has an o-F positioned for defluorination, and underwent C–F activation on heating in toluene to form the YbIII cages, [Yb4(p-HC6F4N(CH2)2NR2)6F6] with seven- and eight-coordinate Yb atoms (Scheme 2).[12]

|
However, the EuII analogue [Eu(p-HC6F4N(CH2)2NEt2)2(thf)2] only underwent C–F activation on irradiation with light, giving [Eu4(p-HC6F4N(CH2)2NEt2)6O2F2],[13] whereas Sm metal with Hg(C6F5)2 and (p-HC6F4NH(CH2)2NMe2) gave a complex mixture probably containing much SmF3 and from which a few crystals of [Sm(p-HC6F4N(CH2)2NMe2)2F]3 were isolated with a hexanuclear (SmF)3 core and all ligands showing N,N′,F tridentate coordination.[13] Thus the redox-induced C–F activation is straightforward only for ytterbium.
Ionic Radii and Size Effects
As a result of the lanthanoid contraction, Yb2+ and Yb3+ are the second smallest in the lanthanoid series. The eight-coordinate ionic radii are 1.14 and 0.985 Å respectively with Yb3+ smaller than Y3+ (1.02 Å) of the previous period.[6a] Yb3+ is a good proxy for the more expensive Lu3+ provided redox does not intervene. In the Ln(O3SCF3)3 catalytic acylation of arenes by (MeCO)2O in nitromethane, Yb(O3SCF3)3 was the standout performer[14] (see, however, later use of Sc(O3SCF3)3[15] and Yb(O3SCF3)3/LiClO4 < Sc(O3SCF3)3/LiClO4 in reactivity[16]). More recently, a remarkable C–F activation reaction was achieved by YbI3 converting unactivated alkyl fluorides into the corresponding iodides:[17]
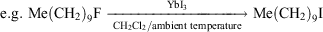
YbI3 was the most effective reagent of all LnI3 compounds examined (LnI3: Ln = La, Sm, Dy, Yb) and in competitive reactions with the corresponding chlorides and bromides, there was 100 % conversion for the fluoride and virtually no reaction of the other halides.[17]
Size is also relevant to coordination chemistry with the lanthanoid contraction affecting the behaviour of Yb3+ relative to other lanthanoids (see below). Both Yb2+ and Yb3+ are strong Lewis acids and hard acids; hence, they prefer first-row donors, notably oxy-donors. High coordination numbers of seven to ten are particularly preferred, with eight-coordinate [Yb(H2O)8]3+ the dominant species in aqueous solution, and [Yb(H2O)9]3+ is present in the hydrated triflate, bromate, and ethylsulfate.[18] High coordination numbers of eight to ten are particularly favoured with small organohydroborate and tetrahydroborate ligands, which can act as tridentate donors.[19] Low coordination numbers can be achieved with bulky donors.[20] Thus for YbII, three-coordination is observed in [Yb(OAr)(μ-OAr)]2 (OAr = OC6H2tBu2-2,6-Me-4)[21] and in monomeric [Yb{N(Dip)(Mes)}2(thf)] (Dip = C6H3iPr2-2,6; Mes = C6H2Me3-2,4,6),[22] whereas two-coordination is observed in [Yb{C(SiMe3)3}2],[23,24] but there are additional C–Yb interactions and C–Yb–C is 137.0(4)°.[23] More recently, [Yb{N(SiiPr3)2}2] was prepared and has a near-linear N–Yb–N bond (166.01(14)°) but there are six additional agostic C–Yb interactions.[7h] In the trivalent state, [Yb{N(SiMe3)2}3][25] and [Yb(OAr)3][26] provide examples of three-coordination. As to the effect of the lanthanoid contraction (0.18 Å from La3+ to Yb3+) on coordination number, decreases are well known, e.g. from [LaCp3] (coordination number (CN) = 11) to YbCp3 (CN = 9)[27] or [LaCl3(thf)2] (eight-coordinate polymer) to [YbCl3(thf)2]2 (six-coordinate dimer).[28] There are cases where no change occurs across the series particularly with use of suitable macrocyclic ligands to override the Ln contraction.[29]
Ytterbium Organometallics
Like organolanthanoids generally, ytterbium organometallics were late on the scene, beginning with tri(cyclopentadienyl)ytterbium,[30] a nine-coordinate monomer (see above). The divalent analogue [YbCp2] followed.[31a] The controversy as to whether the compound was red[31a–c] or green[31d] was resolved by determining the crystal structure of red [YbCp2] and green [NaYbCp3], the latter inferentially being green YbCp3.[32] Cyclopentadienyls and other π-complexes have continued to dominate organolanthanoid chemistry.[33] The divalent state has extended to the C5Me5 analogue[34] and, more recently, to the super bulky [Yb(CpBIG)2] (CpBIG = C5(C6H4Bu-4)5)[35] and [Yb(C5Ph5)2].[36] The last two have coplanar rings, slightly bent towards one another, by contrast to the open sandwich of [Yb(C5Me5)2].[37] Remarkably, the latter reacts with [Pt(Ph3P)2(C2H4)],[38a] disubstituted acetylenes,[38b] and CO.[38c]
Although ytterbium complexes with mixed cyclopentadienyl and alkyl or aryl ligands are quite common,[33] species in which alkyl or aryl groups are the sole anionic organic ligands are rarer. Ytterbium pseudo-Grignard compounds YbR(X) (e.g. R = Ph, Me; X = Br, I) were the first representatives[39] and structurally characterised examples were achieved with bulky ligands,[23,24,40] e.g. C6H3Ph2-2,6. Although Yb(Ph)I cannot be isolated, it is an excellent source of pyrazolato- and formamidinato-ytterbium complexes by reaction with 3,5-diphenylpyrazole[41a] and N,N′-diaryl-formamidines[41b] at low temperatures. Well-characterised alkyl- and aryl-ytterbium compounds include [Li(tmed3)][YbMe6] (tmed = N,N′-tetramethylethane-1,2-diamine),[42] [YbPh3(thf)3],[43] the mixed oxidation state [YbIIIPh2(thf)(π-Ph)3YbII(thf)3],[44] [Yb(C6F5)2(thf)4],[10,43b,45] and [Yb(CH2tBu)3(thf)2].[46] Synthetic routes used to prepare organoytterbium compounds include metathesis,[2,33] redox transmetallation with mercurials[10,11,43–45] and organothallium compounds,[47] protolysis for cyclopentadienyls,[33] and redox transmetallation/protolysis for cyclopentadienyls.[48]
In summary, ytterbium is one of the most versatile lanthanoid elements, with two readily accessible oxidation states, strong Lewis acidity in the trivalent state, versatile coordination chemistry in both oxidation states, and attractive organometallic chemistry. As the number of uses increases, interest in recycling will develop and it is possible that the existence of the YbII state may assist in separating it from other metals even though it has not been utilised in lanthanoid separation. Our choice of element 70 for this essay was also influenced by the recent 70th birthday of a great friend and colleague, Professor Dr Gerd Meyer, as celebrated by a special issue of Z. Anorg. Allg. Chem. 2019, 645, 870.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
The authors are grateful for support from the Australian Research Council and in particular grant DP190100798 for the present contribution and to Dr Mehdi Salehisaki for constructing Fig. 1.
References
[1] T. Behrsing, G. B. Deacon, P. C. Junk, in Rare Earth-Based Corrosion Inhibitors (Eds M. Forsyth, B. Hinton) 2014, Ch. 1, pp. 1–37 (Woodhead Publishing: Cambridge).[2] Lanthanides: Chemistry and Use in Organic Synthesis (Ed. S. Kobayashi) Topics in Organometallic Chemistry, Vol. 2 1999 (Springer: Berlin).
[3] F. H. Spedding, A. H. Daane, The Rare Earths 1961 (Wiley: New York, NY).
[4] (a) P. L. Watson, in Rare Earth Horizons 1987 1987, pp. 5–40 (Department of Industry, Technology and Commerce: Canberra).
(b) I. R. Towner, I. R. McLeod, J. Ward, in Rare Earth Horizons 1987 1987, pp. 87–102 (Department of Industry, Technology and Commerce: Canberra).
[5] G. Meyer, Final circular, XXIX. Tage der Seltenen Erden Terrae Rarae 2019 2019 (Stockholm, Sweden). Available at: http://www.terrae-rarae.org/wp-content/uploads/2019/03/TR-2019-1st-circular-1.pdf
[6] (a) S. Cotton, Lanthanide and Actinide Chemistry 2006 (Wiley: Chichester, UK).
(b) W. D. Horrocks, J. P. Bolender, W. D. Smith, R. M. Supkowski, J. Am. Chem. Soc. 1997, 119, 5972.
[7] (a) M. N. Bochkarev, Coord. Chem. Rev. 2004, 248, 835.
| Crossref | GoogleScholarGoogle Scholar |
(b) W. J. Evans, Inorg. Chem. 2007, 46, 3435.
| Crossref | GoogleScholarGoogle Scholar |
(c) M. R. MacDonald, J. E. Bates, J. W. Ziller, F. Furche, W. J. Evans, J. Am. Chem. Soc. 2013, 135, 9857.
| Crossref | GoogleScholarGoogle Scholar |
(d) P. B. Hitchcock, M. F. Lappert, L. Maron, A. V. Protchenko, Angew. Chem. Int. Ed. 2008, 47, 1488.
| Crossref | GoogleScholarGoogle Scholar |
(e) F. Jaroschik, A. Momin, F. Nief, X. F. Le Goff, G. B. Deacon, P. C. Junk, Angew. Chem. Int. Ed. 2009, 48, 1117.
| Crossref | GoogleScholarGoogle Scholar |
(f) P. L. Arnold, F. G. N. Cloke, J. F. Nixon, Chem. Commun. 1998, 797.
| Crossref | GoogleScholarGoogle Scholar |
(g) M. E. Fieser, M. R. MacDonald, B. T. Krull, J. E. Bates, J. W. Ziller, F. Furche, W. J. Evans, J. Am. Chem. Soc. 2015, 137, 369.
| Crossref | GoogleScholarGoogle Scholar |
(h) C. A. P. Goodwin, N. F. Chilton, G. F. Vettese, E. M. Pineda, I. F. Crowe, J. W. Ziller, R. E. P. Winpenny, W. J. Evans, D. P. Mills, Inorg. Chem. 2016, 55, 10057.
| Crossref | GoogleScholarGoogle Scholar |
(i) F. Jaroschik, F. Nief, X. F. Le Goff, L. Ricard, Organometallics 2007, 26, 1123.
| Crossref | GoogleScholarGoogle Scholar |
[8] (a) F. G. N. Cloke, in Comprehensive Organometallic Chemistry II (Eds E. W. Abel, F. G. A. Stone, G. Wilkinson) 1995, Vol. 4, Ch. 1, pp. 1–9 (Pergamon: Oxford).
(b) F. G. N. Cloke, Chem. Soc. Rev. 1993, 22, 17.
| Crossref | GoogleScholarGoogle Scholar |
[9] A. M. Bond, G. B. Deacon, R. H. Newnham, Organometallics 1986, 5, 2312.
| Crossref | GoogleScholarGoogle Scholar |
[10] G. B. Deacon, W. D. Raverty, D. G. Vince, J. Organomet. Chem. 1977, 135, 103.
| Crossref | GoogleScholarGoogle Scholar |
[11] J. Lefèvre, G. B. Deacon, P. C. Junk, L. Maron, Chem. Commun. 2015, 15173.
| Crossref | GoogleScholarGoogle Scholar |
[12] G. B. Deacon, C. M. Forsyth, P. C. Junk, J. Wang, Chem. – Eur. J. 2009, 15, 2082.
[13] G. B. Deacon, P. C. Junk, R. P. Kelly, J. Wang, Dalton Trans. 2016, 1422.
| Crossref | GoogleScholarGoogle Scholar | 26673146PubMed |
[14] A. Kawada, S. Mitamura, S. Kobayashi, J. Chem. Soc. Chem. Commun. 1993, 1157.
| Crossref | GoogleScholarGoogle Scholar |
[15] A. Kawada, S. Mitamura, S. Kobayashi, Synlett 1994, 545.
| Crossref | GoogleScholarGoogle Scholar |
[16] A. Kawada, S. Mitamura, S. Kobayashi, Chem. Commun. 1996, 183.
| Crossref | GoogleScholarGoogle Scholar |
[17] M. Träff, M. Janjetovic, L. Ta, G. Hilmersson, Angew. Chem. Int. Ed. 2013, 52, 12073.
| Crossref | GoogleScholarGoogle Scholar |
[18] S. Cotton, in Comprehensive Coordination Chemistry II (Eds J. M. McCleverty, T. J. Meyer) 2004, Vol. 3, Ch. 3.2, pp. 93–188 (Pergamon: Oxford).
[19] (a) X. Chen, S. Lim, C. E. Plečnik, S. Liu, B. Du, E. A. Meyers, S. G. Shore, Inorg. Chem. 2005, 44, 6052.
| Crossref | GoogleScholarGoogle Scholar | 16097825PubMed |
(b) F. Yuan, Y. Zhu, L. Xiong, J. Organomet. Chem. 2006, 691, 3377.
| Crossref | GoogleScholarGoogle Scholar |
[20] F. Ortu, D. P. Mills, in Handbook on the Physics and Chemistry of Rare Earths (Eds J.-C. G. Bünzli, V. K. Pecharsky) 2019, Vol. 55, Ch. 306, pp. 1–87 (North Holland: Amsterdam).
[21] (a) J. R. van den Hende, P. B. Hitchcock, S. A. Holmes, M. F. Lappert, J. Chem. Soc. Chem. Commun. 1994, 1413.
| Crossref | GoogleScholarGoogle Scholar |
(b) J. R. van den Hende, P. B. Hitchcock, S. A. Holmes, M. F. Lappert, J. Chem. Soc., Dalton Trans. 1995, 1435.
| Crossref | GoogleScholarGoogle Scholar |
[22] C. N. de Bruin-Dickason, A. J. Boutland, D. Dange, G. B. Deacon, C. Jones, Dalton Trans. 2018, 9512.
| Crossref | GoogleScholarGoogle Scholar | 29964281PubMed |
[23] C. Eaborn, P. B. Hitchcock, K. Izod, J. D. Smith, J. Am. Chem. Soc. 1994, 116, 12071.
| Crossref | GoogleScholarGoogle Scholar |
[24] C. Eaborn, P. B. Hitchcock, K. Izod, Z.-R. Lu, J. D. Smith, Organometallics 1996, 15, 4783.
| Crossref | GoogleScholarGoogle Scholar |
[25] D. C. Bradley, J. S. Ghotra, F. A. Hart, J. Chem. Soc., Dalton Trans. 1973, 1021.
| Crossref | GoogleScholarGoogle Scholar |
[26] M. F. Lappert, A. Singh, R. G. Smith, in Inorganic Syntheses 1990, Vol. 27, pp. 164–168 (John Wiley & Sons: New York) and references therein.
[27] G. B. Deacon, P. MacKinnon, R. S. Dickson, G. N. Pain, B. O. West, Appl. Organomet. Chem. 1990, 4, 439.
| Crossref | GoogleScholarGoogle Scholar |
[28] G. B. Deacon, T. Feng, P. C. Junk, B. W. Skelton, A. H. White, A. N. Sobolev, Aust. J. Chem. 1998, 51, 75.and references therein
| Crossref | GoogleScholarGoogle Scholar |
[29] (a) P. Bernhardt, B. M. Flanagan, M. J. Riley, Aust. J. Chem. 2000, 53, 229.
| Crossref | GoogleScholarGoogle Scholar |
(b) P. Bernhardt, B. M. Flanagan, M. J. Riley, Aust. J. Chem. 2001, 54, 229.
| Crossref | GoogleScholarGoogle Scholar |
(c) M. Seitz, A. G. Oliver, K. N. Raymond, J. Am. Chem. Soc. 2007, 129, 11153.
| Crossref | GoogleScholarGoogle Scholar |
[30] J. M. Birmingham, G. Wilkinson, J. Am. Chem. Soc. 1956, 78, 42.
| Crossref | GoogleScholarGoogle Scholar |
[31] (a) E. O. Fischer, H. Fischer, Angew. Chem. Int. Ed. Engl. 1964, 3, 132.
| Crossref | GoogleScholarGoogle Scholar |
(b) R. G. Hayes, J. L. Thomas, Inorg. Chem. 1969, 8, 2521.
| Crossref | GoogleScholarGoogle Scholar |
(c) G. B. Deacon, A. J. Koplick, T. D. Tuong, Aust. J. Chem. 1984, 37, 517.
| Crossref | GoogleScholarGoogle Scholar |
(d) F. Calderazzo, R. Pappalardo, S. Losi, J. Inorg. Nucl. Chem. 1966, 28, 987.
| Crossref | GoogleScholarGoogle Scholar |
[32] C. Apostolidis, G. B. Deacon, E. Dornberger, F. T. Edelmann, B. Kanellakopulos, P. MacKinnon, D. Stalke, Chem. Commun. 1997, 1047.
| Crossref | GoogleScholarGoogle Scholar |
[33] F. T. Edelmann, in Comprehensive Organometallic Chemistry III (Eds D. M. P. Mingos, R. H. Crabtree) 2007, Vol. 4, Ch. 4.01, pp. 1–190 (Elsevier: Amsterdam) (and earlier editions I and II).
[34] T. D. Tilley, R. A. Andersen, B. Spencer, H. Ruben, A. Zalkin, D. H. Templeton, Inorg. Chem. 1980, 19, 2999.
| Crossref | GoogleScholarGoogle Scholar |
[35] C. Ruspic, J. R. Moss, M. Schürmann, S. Harder, Angew. Chem. Int. Ed. 2008, 47, 2121.
| Crossref | GoogleScholarGoogle Scholar |
[36] G. B. Deacon, C. M. Forsyth, F. Jaroschik, P. C. Junk, D. L. Kay, A. F. Masters, T. Maschmeyer, J. Wang, L. D. Field, Organometallics 2008, 27, 4772.
| Crossref | GoogleScholarGoogle Scholar |
[37] (a) M. Schultz, C. J. Burns, D. J. Schwartz, R. A. Andersen, Organometallics 2000, 19, 781.
| Crossref | GoogleScholarGoogle Scholar |
(b) R. A. Andersen, R. Blom, J. M. Boncella, C. J. Burns, H. V. Volden, Acta Chem. Scand. A 1987, 41a, 24.and references therein
| Crossref | GoogleScholarGoogle Scholar |
[38] (a) C. J. Burns, R. A. Andersen, J. Am. Chem. Soc. 1987, 109, 915.
| Crossref | GoogleScholarGoogle Scholar |
(b) C. J. Burns, R. A. Andersen, J. Am. Chem. Soc. 1987, 109, 941.
| Crossref | GoogleScholarGoogle Scholar |
(c) M. Schultz, C. J. Burns, D. J. Schwartz, R. A. Andersen, Organometallics 2001, 20, 5690.
| Crossref | GoogleScholarGoogle Scholar |
[39] (a) D. F. Evans, G. V. Fazakerley, R. F. Phillips, J. Chem. Soc. Chem. Commun. 1970, 244.
| Crossref | GoogleScholarGoogle Scholar |
(b) D. F. Evans, G. V. Fazakerley, R. F. Phillips, J. Chem. Soc. A 1971, 1931.
| Crossref | GoogleScholarGoogle Scholar |
[40] M. Heckmann, M. Niemeyer, J. Am. Chem. Soc. 2000, 122, 4227.
| Crossref | GoogleScholarGoogle Scholar |
[41] (a) M. Wiecko, G. B. Deacon, P. C. Junk, Chem. Commun. 2010, 5076.
| Crossref | GoogleScholarGoogle Scholar |
(b) S. H. Ali, G. B. Deacon, P. C. Junk, S. Hamidi, M. Wiecko, J. Wang, Chem. – Eur. J. 2018, 24, 230.
| Crossref | GoogleScholarGoogle Scholar |
[42] H. Schumann, J. Müller, N. Bruncks, H. Lauke, J. Pickardt, H. Schwarz, K. Eckart, Organometallics 1984, 3, 69.
| Crossref | GoogleScholarGoogle Scholar |
[43] (a) L. N. Bochkarev, T. A. Zheleznova, A. V. Safronova, M. S. Drozdov, S. F. Zhil’tsov, L. N. Zakharov, G. K. Fukin, S. Ya. Khorshev, Russ. Chem. Bull. 1998, 47, 165.
(b) G. B. Deacon, C. M. Forsyth, Organometallics 2003, 22, 1349.
| Crossref | GoogleScholarGoogle Scholar |
[44] M. N. Bochkarev, V. V. Khramenkov, Y. F. Rad’kov, L. N. Zhakharov, Y. T. Struchkov, J. Organomet. Chem. 1992, 429, 27.
| Crossref | GoogleScholarGoogle Scholar |
[45] G. B. Deacon, D. G. Vince, J. Organomet. Chem. 1976, 112, C1.
| Crossref | GoogleScholarGoogle Scholar |
[46] M. Niemeyer, Z. Anorg. Allg. Chem. 2000, 626, 1027.
| Crossref | GoogleScholarGoogle Scholar |
[47] G. B. Deacon, G. N. Pain, T. D. Tuong, in Synthetic Methods of Inorganic and Organometallic Chemistry (Eds W. A. Herrmann, F. T. Edelmann) 1997, Vol. 6, pp. 62–65, 86–87 (Thieme: Stuttgart).
[48] G. B. Deacon, C. M. Forsyth, S. Nickel, J. Organomet. Chem. 2002, 647, 50.
| Crossref | GoogleScholarGoogle Scholar |