

Chromolactol, an Oxygenated Diterpene from the Indo-Pacific Nudibranch Goniobranchus coi: Spectroscopic and Computational Studies*
Ariyanti S. Dewi A B , Gregory K. Pierens C , Karen L. Cheney D , Joanne T. Blanchfield A and Mary J. Garson A EA School of Chemistry and Molecular Biosciences, The University of Queensland, St Lucia, Qld 4072, Australia.
B Research Center for Marine and Fisheries Product Processing and Biotechnology, Ministry of Marine Affairs and Fisheries, Jakarta 10260, Indonesia.
C Centre for Advanced Imaging, The University of Queensland, Brisbane, Qld 4072, Australia.
D School of Biological Sciences, The University of Queensland, Brisbane, Qld 4072, Australia.
E Corresponding author. Email: m.garson@uq.edu.au
Australian Journal of Chemistry 71(10) 798-803 https://doi.org/10.1071/CH18243
Submitted: 23 May 2018 Accepted: 1 July 2018 Published: 1 August 2018
Abstract
A rearranged spongian diterpene chromolactol was obtained from the mantle extract of the Indo-Pacific nudibranch Goniobranchus coi. The structure of chromolactol, either 1a or 1b, which was investigated by extensive NMR experiments and by data comparison as well as by molecular modelling studies and density functional calculations, has a different relative configuration of the 2,8-dioxabicyclo-[3.3.0]-octane ring compared with the co-metabolite norrisolide (2). A biosynthetic pathway leading to the preferred diastereomer of chromolactol (1a) is presented.
Introduction
In many nudibranchs, the loss of a protective shell is compensated by the acquisition of chemical defences from food. Nudibranchs may elaborate the chemicals, thus making them more effective as toxins or as deterrents towards predators. Alternatively, nudibranchs may biosynthesise secondary metabolites (de novo), thereby providing a defence that is independent of diet.[1,2] The distastefulness and toxicity of nudibranchs are thought to be related to colour patterns used as warning signals (aposematism).[3–5]
In terms of chemistry, among the most intensively studied nudibranchs are those of the two closely related genera Chromodoris and Goniobranchus, which have been reported to contain furanosesquiterpenes,[6,7] norditerpenes,[8–11] diterpenes,[9,11–27] sesterterpenes,[28,29] and occasionally macrolides.[30] Our group has investigated aplyroseol-type compounds from Chromodoris sp. and G. reticulata collected near Mooloolaba, south-east Queensland.[24,26] Further investigations on G. reticulata yielded chromoculatimines A and B, and a spongian-16-one derivative.[26] Other spongian diterpenes have been obtained from the extracts of G. albopunctata and Doriprismatica atromarginata.[11,23,27] Finally, chemical extracts from G. verrieri and G. splendidus yielded a diverse array of norditerpenes and diterpenes bearing gracilane skeletons.[9,10]
In the present paper, we report the chemical analysis of an extract from G. coi from Mackay, Queensland, and describe the structure elucidation of a new oxygenated diterpene chromolactol, which contains two distinct chiral domains. Our spectroscopic and computational investigations unambiguously determined the relative configuration within each chiral domain, but did not conclusively distinguish between candidate diastereomers 1a or 1b. However, biosynthetic expectations were in accordance with diastereomer 1a as the preferred structure.
Results and Discussion
The new diterpene chromolactol (Fig. 1) was isolated from the Et2O extract of a single specimen of Goniobranchus coi (sample code #1095) collected near Mackay, together with norrisolide (2),[12] cheloviolene C (3),[31] macfarlandin C (4),[32] and dendrillolide A (5).[33] The isolated metabolites were purified by normal-phase (silica) flash chromatography followed by normal-phase high-performance liquid chromatography (HPLC) separation. The structures of chromolactol and of the known metabolites were characterized by means of 2D NMR spectroscopic and mass spectrometric analysis and by comparison with literature data.

|
Chromolactol was isolated as a colourless oil from normal-phase HPLC. The molecular formula of C20H30O4 (m/z 333.2075 [M – H]−; calcd m/z 333.2071) was identical to those of cheloviolenes A (6) and B (7).[31] The 1H NMR of 1 revealed two acetal protons (δH 6.18 and 5.54), an exomethylene group (δH 4.92 and 4.88), three sp3 methine protons (δH 3.01, 2.76, 2.06), and three methyls at δH 0.67 (3H), 0.86 (6H). The heteronuclear multiple-bond correlation (HMBC) spectrum revealed alkene signals at δC 148.2 and 111.5, and a lactone carbonyl at δC 175.4. The exomethylene and the lactone contributed to two of the required six double-bond equivalents; therefore, the compound was tetracyclic.
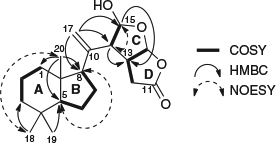
The 1H and 13C NMR data (Table 1) for the A/B ring system of chromolactol matched closely those of norrisolide (2)[12] or cheloviolene C (3);[31] thus, a perhydroindane system was inferred. This conclusion was confirmed by correlation spectroscopy (COSY) and HMBC data (Fig. 2). The relative configurations at C-5, C-8, and C-9 in chromolactol were deduced to be the same as those of 2 and 3 based on the close similarity of the NMR data. Furthermore, there were nuclear Overhauser effect spectroscopy (NOESY) correlations observed between H-5/H-8 and between Me-18/Me-20 (Fig. 2). The same NOESY correlations have been reported for 2.[12]

|
|
Rearranged spongian diterpenes frequently contain either a [3.3.0]- or a [3.2.1]-dioxabicyclooctane ring system that contains a γ-lactone (δC 168–178) or a δ-lactone (δC 165–168) motif respectively. A W-coupling between H-13 and H-15 is diagnostic of coplanar bridgehead protons within the 2,8-dioxabicyclo-[3.2.1]-octane ring system.[32] A 2,8-dioxabicyclo-[3.3.0]-octane ring system was identified in 1 by the presence of a lactone carbonyl at δC 175.4 (C-11). There were COSY correlations between H-12/H-13/H-16, and H-14/H-15, while the absence of W-coupling between H-13 and H-15 further supported the [3.3.0]-octane ring system. HMBC correlations from both H-14 and H-15 to C-13 (δC 45.2), from H-17a and H-17b to C-14 (δC 58.5) and to C-8 (δC 58.8) linked the exomethylene moiety to the perhydroindane system and to the 2,8-dioxabicyclo-[3.3.0]-octane ring (Fig. 2).
The stereochemistry of the 2,8-dioxabicyclo-[3.3.0]-octane ring system was next considered, comparing the relative configuration with those of norrisolide (2), macfarlandin C (4), and dendrillolide A (5), as well as those of cheloviolenes A (6) and B (7). The 2,8-dioxabicyclo-[3.3.0]-octane ring systems of 2, 4, and 5 have a cis configuration between H-13/H-14 and a trans configuration between H-14/H-15. Further, the absolute configuration of 3 has been determined by enantioselective synthesis.[34] Cheloviolenes A (6) and B (7), which differ from chromolactol in possessing a perhydroazulene hydrocarbon domain rather than a perhydroindane moiety, are rare examples of rearranged spongian diterpenes with a 2,8-dioxabicyclo-[3.3.0]-octane ring system that possess a trans configuration between H-13 and H-14. In the original isolation work, the relative configuration of cheloviolene A (6) was established by an X-ray study that revealed that H-14 was trans to both H-13 and H-15.[31] Confirmation of this relative configuration, together with determination of the absolute configuration of cheloviolene A, has recently been achieved by the Overman group via an 11-step enantioselective total synthesis.[35] Cheloviolene B, which was initially reported as the C-15 epimer of 6,[31] has also been the target of synthetic work by the Overman group, which led to a revision of relative configuration to that shown in 6.[31] Cheloviolenes thus have the same relative configuration in their 2,8-dioxabicyclo-[3.3.0]-octane rings, but differ only in the configuration of this bicyclic moiety relative to the perhydroazulene domain. The structure shown for 7 was first presented in the literature as that of a sponge diterpenoid chelonaplysin B by Bobzin and Faulkner;[36] however, the published 1H NMR spectra of their metabolite were shown to be identical to those of cheloviolene A.[31]
In chromolactol, the JH-13/H-16 of 6.2 Hz was identical to that in norrisolide (2) and cheloviolenes A (6) and B (7), and together with the NOESY correlation between the two protons indicated that H-13 and H-16 were cis-configured. The H-14 signal appeared as a broad singlet (JH-13/H-14 <1 Hz), which matched closely the equivalent signals of 6 and 7 (J values of 2.2 and 2.6 Hz respectively),[31] and thereby the relationship between H-13 and H-14 in 1 was established as trans. If H-13 and H-14 had been cis-configured, a JH-13/H-14 of 6–10 Hz would have been anticipated (cf. values of 9.4, 6.9, and 6.6 Hz for 2, 4, and 5 respectively). Considering the C-15 configuration, the JH-14/H-15 value of <1 Hz in 1 matched closely the reported values for cheloviolenes A (6) and B (7).[31] The small values of coupling constants between H-13/H-14 and H-14/H-15 likely result from the overall conformation of the 2,8-dioxabicyclo-[3.3.0]-octane ring with a hydrogen bond between the hydroxy group and the lactone. This hydrogen bonding causes each of the torsional angles of H-14–C-14–C-13–H-13 and H-14–C-14–C-15–H-15 to be close to 90°.[36]
Two candidate diastereomers of chromolactol (1a and 1b) that differ in the stereochemistry of the 2,8-dioxabicyclo-[3.3.0]-octane ring system were considered. Diastereomer 1a possesses the same configuration as cheloviolene B, whereas diastereomer 1b possesses the same configuration as cheloviolene A.
A Monte Carlo conformational search for each diastereomer 1a and 1b was undertaken with Merck Molecular Force Field (MMFF) using Macromodel[37] and selected conformers (<5 kcal mol−1 (1 cal = 4.184 J) of the global minimum: 1a, 20 conformers and 1b, 21 conformers) were optimized by density functional theory (DFT) calculations at the B3LYP/6–31+G(d,p) level with chloroform solvent (integral equation formalism-polarisable continuum model, IEF-PCM) using Gaussian software.[38] A single-point energy of the optimized conformers was calculated using M062X/6–31+G(d,p) with chloroform solvent (IEF-PCM)[39] and was used in the weighted chemical shifts. Four conformers for both 1a and 1b (M062X/6–31+G(d,p) energy <3kcal mol−1) were selected to calculate the chemical shifts using mpw1pw91/6–311+G(2d,p) with chloroform solvent (IEF-PCM). The Boltzmann-averaged 1H and 13C chemical shifts were calculated by converting the magnetic field tensors using linear scaling (1H slope: −1.0717, intercept: 31.8721 and 13C slope: −1.0417, intercept: 186.3455). Next, the mean absolute error (MAE) for both 1a and 1b was evaluated using the calculated chemical shifts, revealing that there was very little difference between the two diastereomers (1H: MAE of 0.08 ppm for both 1a and 1b isomers, and 13C: MAE of 1.8 and 1.9 ppm for 1a and 1b respectively). When the Boltzmann-averaged 1H and 13C chemical shifts were examined using DP4[40] using both 1H and 13C chemical shifts, the probability percentages were 1a 64.3 % and 1b 35.7 %; the preference for 1a was slightly improved if the 13C chemical shifts alone were analysed (68 % : 32 % for 1a : 1b).
Although these computational data clearly indicated that chromolactol has the same partial relative configuration as the 2,8-dioxabicyclo-[3.3.0]-octane ring system of cheloviolenes A or B, it was also apparent that the NMR data alone could not distinguish between the two diastereomers 1a and 1b. The two lowest-energy conformers of 1a (together representing >98.6 % of the conformational population (ratio: 54 : 46); see Supplementary Material) differed in rotation about the C-10–C-14 bond, and hence in the orientation of the dioxabicyclooctane ring system relative to the hydrocarbon fragment. Likewise for 1b, the two most stable conformers (representing >98.1 % of the conformational population (ratio: 56 : 44)) also differed in rotation about the C-10–C-14 bond. Consequently, NOESY data could not be used to distinguish which of the two candidate diastereomers corresponded to chromolactol. This diterpene metabolite of G. coi thus represents a rare example in which computational results do not converge to a preferred stereostructure. Jiao and coworkers recently determined the partial relative configuration of two distinct chiral domains in the cyclooxygenase-2 (COX-2)-inhibitory meroterpenoid dysiarenone, but were unable to verify the overall relative configuration.[41] In our own work on the nudibranch Phyllidiella pustulosa, the antimalarial diterpene pustulosaisonitrile-1 posed significant challenges for structure elucidation owing to the C-6/C-7 stereochemical relationship and the presence of two chiral domains separated by a flexible alkane linker. The stereochemical problem was resolved by catalyst-controlled stereoselective synthesis of two key diastereomers to establish the relative configuration of the two independent chiral domains.[42]
The stereostructure initially deduced for cheloviolene A (6) has been verified by total synthesis; in the same work, the structure of cheloviolene B, which was originally assigned as the C-15 epimer of 6, was revised by the Overman group. We compared the 1H NMR data of chromolactol against those of cheloviolenes A and B. Although the chemical shift for H-15 in chromolactol (δH 5.54) matched better the equivalent signal in cheloviolene A (δH 5.52) than in cheloviolene B (δH 5.64), the chemical shift for H-13 in chromolactol (δH 3.01) matched better the equivalent signal in cheloviolene B (δH 2.98) than in cheloviolene A (δH 3.11). A comparison of the chemical shift values for (H-12)2 and H-16 was not informative.
As the Overman synthetic study provided a rigorous assignment of NMR data for the cheloviolenes, we elected to test the experimental 1H and 13C NMR data for cheloviolene A against the theoretical data for this structure using the DP4 probability approach. It was found that the calculated 1H NMR data corresponding to the stereostructure 6 matched closely with the experimental 1H NMR data of cheloviolene A (83.1 %), but in contrast, the calculated 13C NMR data corresponding to stereostructure 6 were instead in better agreement with the experimental 13C NMR data of cheloviolene B (55.7 %) rather than with the data for cheloviolene A (44.3 %). Taking both 1H and 13C NMR data into account, the probability value was 79.6 % that the calculated data for stereostructure 6 matched the experimental data for cheloviolene A. In other words, these results demonstrate that the DP4 NMR chemical shift calculations do not convincingly distinguish between diastereomers containing the same relative configuration in the 2,8-dioxabicyclo-[3.3.0]-octane moiety but differing in the configuration of this domain relative to the chiral hydrocarbon fragment. The computational approach is generally considered a reliable indicator of (stereo)structure if the overall probability value is >90 %.[43]
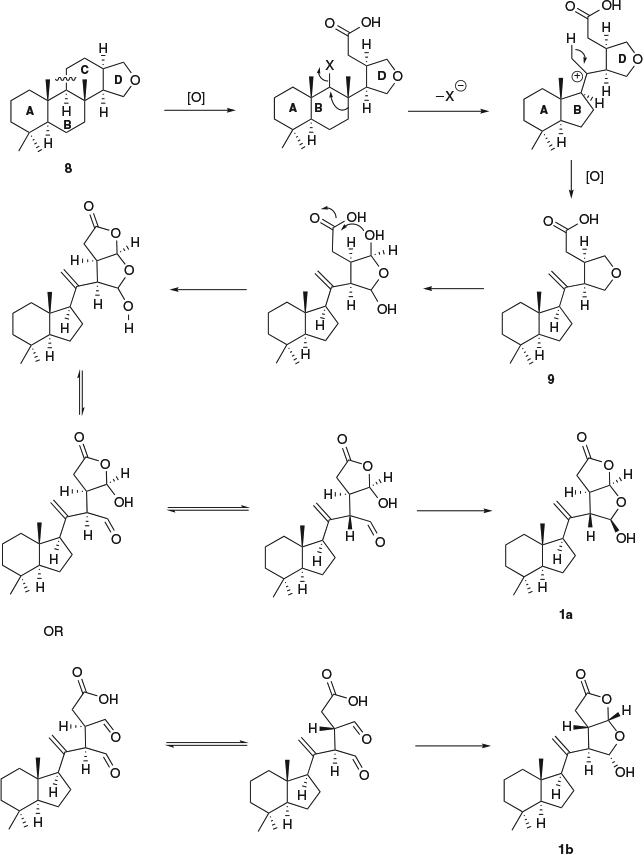
For chromolactol, diastereomer 1a may be preferred on biosynthetic grounds because it has the same configuration at C-13 and C-16 as many other oxygenated diterpenes (cf. 3). As shown in Scheme 1, the biosynthesis of 1a may proceed by opening of ring C of a spongiane precursor 8, followed by contraction of ring B to yield the norrisane skeleton 9.[12,44] Oxidation of the tetrahydrofuran ring to a lactol followed by ring opening to an aldehyde allows epimerization at C-14. In this way, the cis configuration of H-13 and H-14 that is usually associated with oxygenated diterpenes can be changed to trans. Closure of ring D produces 1a with the overall relative configuration shown. Alternatively, epimerization at C-13 followed by ring closure establishes the configuration shown in 1b.
|
Conclusions
We have reported a new rearranged spongian diterpene, chromolactol from the mantle extract of Goniobranchus coi. The relative configuration of the [3.3.0]-dioxabicyclooctane ring in chromolactol is identical to that observed in cheloviolenes A or B. Investigation of the relative configuration of chromolactol via NMR data analysis yielded two plausible diastereomers (1a, 1b). Neither the calculated MAE values nor the probability values defined by DP4 were able to differentiate conclusively between the two diastereomers. However, the overall relative configuration shown in diastereomer 1a is preferred to that of diastereomer 1b on biosynthetic grounds.
Experimental
General Experimental Procedure
Specific rotations were measured at 23°C on a Jasco P-2000 polarimeter for solutions in CHCl3 using a 1-mL cell (10-cm path length). NMR data were measured on Bruker Avance 500 and 700-MHz spectrometers (5-mm inverse probe) for solutions in CDCl3 at 298 K. Heteronuclear single quantum correlation (HSQC) and HMBC data were acquired using a 1JC–H of 145 Hz, whereas HMBC spectra were acquired using nJC–H of 8 Hz. Positive- and negative-ion electrospray mass spectra were determined using either a Bruker Esquire HCT instrument for low-resolution electrospray ionization mass spectrometry (LRESIMS) or a MicrOTOF-Q instrument for high-resolution electrospray ionization mass spectrometry (HRESIMS) with MeOH as solvent. Normal-phase HPLC (NPHPLC) was undertaken using a Waters 515 pump connected to a Gilson 132 series refractive index detector with a Waters μPorasil (10 μm, 7.8 × 300 mm) or a Phenomenex Luna (5 μm, 10 × 250 mm) column, and using isocratic elution conditions at flow rates between 1 and 2 mL min−1. Silica gel 60 G and silica TLC plates F254 were purchased from Merck. Solvents were either distilled or were of HPLC grade.
Biological Material
Goniobranchus coi (#1095) (0.68 g) was collected from the Mackay region, Queensland, in October 2014, frozen, and stored at −20°C until extraction. The specimen was dissected into mantle and gut before extraction.
Extraction and Purification
The mantle tissue of G. coi (Mackay #1095; 0.49 g) was extracted in acetone (7 × 3 mL). The extract was reduced to an aqueous suspension, extracted with Et2O (3 × 3 mL), dried over anhydrous Na2SO4, and concentrated under N2 to give crude extracts. The mantle extract (5.3 mg) was subjected to NPHPLC (20 % EtOAc/hexanes) and yielded dendrillolide A (0.6 mg), macfarlandin C (1.3 mg), norrisolide (1.3 mg), cheloviolene C (0.2 mg), and chromolactol 1a (0.2 mg). Using similar methods, the gut extract (5.2 mg) afforded norrisolide (2.0 mg), macfarlandin C (1.8 mg), cheloviolene C (1.3 mg), and dendrillolide A (0.1 mg).
Chromolactol (1a)
Colourless oil; [α]D − 3 (c 0.04 CHCl3). 1H NMR (CDCl3, 700 MHz) and 13C NMR (CDCl3, 700 MHz) data are presented in Table 1. m/z (HRESIMS) 333.2071 [M – H]−; calcd for C20H29O4: 333.2075.
Supplementary Material
An image of Goniobranchus coi, copies of 1D and 2D NMR spectra of chromolactol (1) in CDCl3, and details of computational studies are available on the Journal’s website.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
We thank the Australia Research Council and The University of Queensland (Promoting Women Fellowship to K.L.C.) for financial support, the Australian government for a Leadership Award and Allison Sudradjat Prize to A.S.D. We are grateful for copies of the 1H NMR spectra of cheloviolenes A and B provided by Professor Larry Overman (UC Irvine). The assistance of Dr T. Le and Peter Josh (MS) is acknowledged. We thank Dr A. Roberts-Thomson (Qld Sustainable Sealife Ltd) for sample collection undertaken under permits issued by the Great Barrier Reef Marine Park Authority.
References
[1] D. J. Faulkner, T. F. Molinski, R. J. Andersen, E. J. Dumdei, E. D. De Silva, Comp. Biochem. Physiol. 1990, 97C, 233.| Crossref | GoogleScholarGoogle Scholar |
[2] D. J. Faulkner, M. T. Ghiselin, Mar. Ecol. Prog. Ser. 1983, 13, 295.
| Crossref | GoogleScholarGoogle Scholar |
[3] R. Ritson-Williams, V. J. Paul, Mar. Ecol. Prog. Ser. 2007, 340, 29.
| Crossref | GoogleScholarGoogle Scholar |
[4] A. E. Winters, N. F. Green, N. G. Wilson, M. J. How, M. J. Garson, N. J. Marshall, K. L. Cheney, Proc. Roy. Soc. B. 2017, 284, 20170926.
| Crossref | GoogleScholarGoogle Scholar |
[5] F. Cortesi, K. L. Cheney, J. Evol. Biol. 2010, 23, 1509.
| Crossref | GoogleScholarGoogle Scholar |
[6] J. E. Hochlowski, D. J. Faulkner, Tetrahedron Lett. 1981, 22, 271.
| Crossref | GoogleScholarGoogle Scholar |
[7] G. R. Schulte, P. J. Scheuer, Tetrahedron 1982, 38, 1857.
| Crossref | GoogleScholarGoogle Scholar |
[8] L. C. Forster, G. K. Pierens, A. M. White, K. L. Cheney, P. Dewapriya, R. J. Capon, M. J. Garson, ACS Omega 2017, 2, 2672.
| Crossref | GoogleScholarGoogle Scholar |
[9] A. M. White, G. K. Pierens, L. C. Forster, A. E. Winters, K. L. Cheney, M. J. Garson, J. Nat. Prod. 2016, 79, 477.
| Crossref | GoogleScholarGoogle Scholar |
[10] Y. Hirayama, P. L. Katavic, A. M. White, G. K. Pierens, L. K. Lambert, A. E. Winters, H. Kigoshi, M. Kita, M. J. Garson, Aust. J. Chem. 2016, 69, 136.
| Crossref | GoogleScholarGoogle Scholar |
[11] K. W. L. Yong, I. W. Mudianta, K. L. Cheney, E. Mollo, J. T. Blanchfield, M. J. Garson, J. Nat. Prod. 2015, 78, 421.
| Crossref | GoogleScholarGoogle Scholar |
[12] J. E. Hochlowski, D. J. Faulkner, G. K. Matsumoto, J. Clardy, J. Org. Chem. 1983, 48, 1141.
| Crossref | GoogleScholarGoogle Scholar |
[13] M. B. Ksebati, F. J. Schmitz, J. Org. Chem. 1987, 52, 3766.
| Crossref | GoogleScholarGoogle Scholar |
[14] P. Karuso, in Bioorganic Marine Chemistry (Ed. P. J. Scheuer) 1987, pp. 31–60 (Springer-Verlag: Berlin).
[15] S. C. Bobzin, D. J. Faulkner, J. Org. Chem. 1989, 54, 3902.
| Crossref | GoogleScholarGoogle Scholar |
[16] E. J. Dumdei, E. D. De Silva, R. J. Andersen, M. I. Choudhary, J. Clardy, J. Am. Chem. Soc. 1989, 111, 2712.
| Crossref | GoogleScholarGoogle Scholar |
[17] G. Cimino, A. Crispino, M. Gavagnin, G. Sodano, J. Nat. Prod. 1990, 53, 102.
| Crossref | GoogleScholarGoogle Scholar |
[18] S. A. Morris, E. D. de Silva, R. J. Andersen, Can. J. Chem. 1991, 69, 768.
| Crossref | GoogleScholarGoogle Scholar |
[19] E. D. de Silva, S. A. Morris, S. Miao, E. J. Dumdei, R. J. Andersen, J. Nat. Prod. 1991, 54, 993.
| Crossref | GoogleScholarGoogle Scholar |
[20] M. Gavagnin, R. R. Vardaro, C. Avila, G. Cimino, J. Ortea, J. Nat. Prod. 1992, 55, 368.
| Crossref | GoogleScholarGoogle Scholar |
[21] J. Pika, D. J. Faulkner, Tetrahedron 1995, 51, 8189.
| Crossref | GoogleScholarGoogle Scholar |
[22] T. Miyamoto, K. Sakamoto, K. Arao, T. Komori, R. Higuchi, T. Sasaki, Tetrahedron 1996, 52, 8187.
| Crossref | GoogleScholarGoogle Scholar |
[23] M. J. Somerville, E. Mollo, G. Cimino, W. Rungprom, M. J. Garson, J. Nat. Prod. 2006, 69, 1086.
| Crossref | GoogleScholarGoogle Scholar |
[24] K. W. L. Yong, A. A. Salim, M. J. Garson, Tetrahedron 2008, 64, 6733.
| Crossref | GoogleScholarGoogle Scholar |
[25] M. Agena, C. Tanaka, N. Hanif, M. Yasumoto-Hirose, J. Tanaka, Tetrahedron 2009, 65, 1495.
| Crossref | GoogleScholarGoogle Scholar |
[26] Suciati, L. K. Lambert, M. J. Garson, Aust. J. Chem. 2011, 64, 757.
| Crossref | GoogleScholarGoogle Scholar |
[27] P. L. Katavic, P. Jumaryatno, J. N. A. Hooper, J. T. Blanchfield, M. J. Garson, Aust. J. Chem. 2012, 65, 531.
| Crossref | GoogleScholarGoogle Scholar |
[28] J. E. Hochlowski, D. J. Faulkner, L. S. Bass, J. Clardy, J. Org. Chem. 1983, 48, 1738.
| Crossref | GoogleScholarGoogle Scholar |
[29] T. Miyamoto, K. Sakamoto, H. Amano, R. Higuchi, T. Komori, T. Sasaki, Tetrahedron Lett. 1992, 33, 5811.
| Crossref | GoogleScholarGoogle Scholar |
[30] D. G. Corley, R. Herb, R. E. Moore, P. J. Scheuer, V. J. Paul, J. Org. Chem. 1988, 53, 3644.
| Crossref | GoogleScholarGoogle Scholar |
[31] P. R. Bergquist, B. F. Bowden, R. C. Cambie, P. A. Craw, P. Karuso, A. Poiner, W. C. Taylor, Aust. J. Chem. 1993, 46, 623.
| Crossref | GoogleScholarGoogle Scholar |
[32] T. F. Molinski, D. J. Faulkner, C.-H. He, G. D. Van Duyne, J. Clardy, J. Org. Chem. 1986, 51, 4564.
| Crossref | GoogleScholarGoogle Scholar |
[33] S. C. Bobzin, D. J. Faulkner, J. Org. Chem. 1989, 54, 5727.
| Crossref | GoogleScholarGoogle Scholar |
[34] T. P. Brady, S. H. Kim, K. Wen, C. Kim, E. A. Theodorakis, Chem. – Eur. J. 2005, 11, 7175.
| Crossref | GoogleScholarGoogle Scholar |
[35] M. R. Garnsey, Y. Slutskyy, C. R. Jamison, P. Zhao, J. Lee, Y. H. Rhee, L. E. Overman, J. Org. Chem. 2018, 83, 6958.
| Crossref | GoogleScholarGoogle Scholar |
[36] S. C. Bobzin, D. J. Faulkner, J. Nat. Prod. 1991, 54, 225.
| Crossref | GoogleScholarGoogle Scholar |
[37] Schrödinger, MacroModel: Release 2018-1 2018 (Schrödinger: New York, NY).
[38] M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox, Gaussian 09, Revision B. 01 2013 (Gaussian Inc.: Wallingford, CT).
[39] J. Tomasi, B. Mennucci, R. Cammi, Chem. Rev. 2005, 105, 2999.
| Crossref | GoogleScholarGoogle Scholar |
[40] K. Ermanis, K. E. B. Parkes, T. Agback, J. M. Goodman, Org. Biomol. Chem. 2017, 15, 8998.
| Crossref | GoogleScholarGoogle Scholar |
[41] W.-H. Jiao, B.-H. Cheng, G.-D. Chen, G.-H. Shi, J. Li, T.-Y. Hu, H.-W. Lin, Org. Lett. 2018, 20, 3092.
| Crossref | GoogleScholarGoogle Scholar |
[42] A. M. White, K. Dao, D. Vrubliauskas, Z. A. Könst, G. K. Pierens, A. Mándi, K. T. Andrews, T. S. Skinner-Adams, M. E. Clarke, P. T. Narbutas, D. C.-M. Sim, K. L. Cheney, T. Kurtán, M. J. Garson, C. D. Vanderwal, J. Org. Chem. 2017, 82, 13313.
| Crossref | GoogleScholarGoogle Scholar |
[43] N. Grimblat, M. M. Zanardi, A. M. Sarotti, J. Org. Chem. 2015, 80, 12526.
| Crossref | GoogleScholarGoogle Scholar |
[44] R. A. Keyzers, P. T. Northcote, M. T. Davies-Coleman, Nat. Prod. Rep. 2006, 23, 321.
| Crossref | GoogleScholarGoogle Scholar |
* Mary J. Garson is the inaugural recipient of the Margaret Sheil Women in Chemistry Leadership award of the RACI.