

Laser-Based Formation of Copper Nanoparticles in Aqueous Solution: Optical Properties, Particle Size Distributions, and Formation Kinetics*
Ashley J. Mulder A , Rhys D. Tilbury A , Phillip J. Wright A , Thomas Becker A , Massimiliano Massi A and Mark A. Buntine A BA Department of Chemistry and Curtin Institute for Functional Molecules and Interfaces, Curtin University, GPO Box U1987, Perth, WA 6845, Australia.
B Corresponding author. Email: m.buntine@curtin.edu.au
Australian Journal of Chemistry 70(11) 1212-1218 https://doi.org/10.1071/CH17363
Submitted: 30 June 2017 Accepted: 21 August 2017 Published: 15 September 2017
Abstract
We explore the formation kinetics, optical absorption spectra, and particle size distributions of copper nanoparticles (CuNPs) formed by direct laser ablation from the bulk metal via a process we refer to as Laser Ablation Synthesis in Solution (LASiS). Comparisons are made between CuNPs formed in pure water versus those formed in the presence of 1 × 10−4 M solutions of the N-donor ligands 4,4′-bipyridine (4,4′Bipy) and 1H-5-(4-pyridyl)tetrazole (T-4Py). CuNPs formed in pure water and in the presence of 4,4′Bipy display similar UV-visible absorption spectra and very similar particle size distributions. In comparison, CuNPs formed in the presence of T-4Py display significantly different absorption properties, with the surface plasmon resonance transition blue-shifted by ∼55 nm, and a much smaller and narrower particle size distribution compared with the former samples. Based on previous literature reports, it is possible to ascribe these differences to differences in the CuNP surface oxidation states for samples prepared in the presence of T-4Py. However, an analysis of the formation kinetics of all three samples indicates near-identical behaviour.
Introduction
Exploration of the chemical properties of metal nanoparticles continues to attract considerable attention due, in part, to the growing number of applications in which these materials are being deployed.[1–4] Copper-based nanoparticles, in particular, have been shown to offer new catalytic reaction pathways,[5,6] and have the potential to increase the efficiency of water-based cooling fluids in heat exchange[7,8] and reduce the coefficient of friction in lubricating oils.[9–15] These applications have commonly exploited the addition of CuO nanoparticles to achieve the desired outcomes. The thermal and chemical properties of Cu nanoparticles have been shown to be dependent on the surface oxidation states.[16–18]
In recent years, the use of laser-based methods to produce a variety of metal nanoparticles has become increasingly common.[19–24] The laser-based preparation of copper nanoparticles (CuNPs) has received attention,[25–27] in part owing to the history of copper as a coinage metal. However, unlike, for example, the laser-based formation of gold[23] and platinum[24] nanoparticles, the susceptibility of CuNPs to surface oxidation both during and following their formation has made studies of CuNP chemistry more challenging.[28–30]
Muniz-Miranda et al.[31] have demonstrated that CuNPs prepared via laser ablation in acetone do not form a surface oxide layer and are quite stable in solution, whereas those produced in pure water do have a surface oxide layer but are relatively short-lived. These authors attribute the lack of stability of these particles to ‘rapid aging through oxidation’ and fast coagulation. The authors go on to recommend, ‘In general, it is advisable to perform the ablation in solutions containing proper particle stabilizers’.[31] CuNPs formed in the presence of aqueous solutions of 1,10-phenanthroline (phen) or 4,4′-bipyridine (bipy) have a surface oxide covering and are also quite stable in solution. A key probe for the presence or absence of a CuNP surface oxide layer is to monitor the surface plasmon resonance (SPR) electronic transition. Muniz-Miranda et al. demonstrate that oxide-free CuNPs result in an approximately 26-nm blue shift in the maximum absorbance wavelength (λmax) of the SPR transition, with the SPR λmax in acetone being 588 nm whereas it is 611 nm for CuNPs generated in aqueous solutions of phen and 614 nm for those generated in bipy.[31]
A critical question that remains to be addressed is to what extent does the (aqueous) environment in which CuNPs are formed by laser ablation affect the mechanism of their formation. In the present paper, we report on studies that employ monitoring the CuNP SPR transition to probe the nanoparticle formation kinetics in pure water and in the presence of two N-donor ligands, 4,4′-bipyridine (4,4′Bipy) and 1H-5-(4-pyridyl)tetrazole (T-4Py). CuNPs are produced by a technique we refer to as Laser Ablation Synthesis in Solution (LASiS).[20,21] The technique involves focussed laser irradiation of a bulk metal target in a liquid. Previous studies from our laboratory have involved the production of gold nanoparticles (AuNPs) encapsulated by anionic[19,21] and cationic[20] surfactants in aqueous solution. A motivation for the study is to explore the fundamental chemistry involved in CuNP production in the presence of N-donor ligands via the LASiS approach. In particular, we are interested in how the N-donor ligands influence the formation of CuNPs via LASiS and to compare the formation kinetics with those determined for AuNPs.
Experimental Methods
N-Donor Ligands
The structures and abbreviated names of the N-donor ligands used in this study are reported in Chart 1. 4,4′Bipy was obtained from Sigma–Aldrich and used as received without further purification. T-4Py was prepared according to previously published procedures.[32]

|
Copper Nanoparticle Formation
The LASiS experimental method employed in our laboratory has been previously reported.[19–21] Only salient details are reported here. CuNPs were prepared in a manner similar to that reported by Haram and Ahmad,[33] either in ultrapure water (purified with a Merck Millipore Synergy UV water purification system; Millipore S.A.S, Molsheim, France) or in the presence of either 4,4′Bipy or T-4Py at a concentration of 1 × 10−4 M. Laser ablation was performed for 60 min at 1064 nm using a Continuum Surelite II Nd:YAG laser operating at 10 Hz and 7.5 mJ pulse−1. The laser radiation was focussed onto a solid copper plate with a 250-mm focal length plano-convex lens. The copper substrate is high-purity, electronic-grade, oxygen-free copper alloy C101 (99.99 %). UV-visible absorption spectra of samples prepared by the method described above following 60 min of 1064-nm irradiation were recorded using a Cary 4000 UV-vis spectrometer.
Atomic Force Microscopy
All samples synthesized following 60 min of 1064-nm irradiation were prepared for atomic force microscopic (AFM) analysis. The nanoparticles were deposited via spin-coating for 4 min at 2000 rpm on a freshly cleaved mica substrate for the AFM analysis. AFM topography measurements were performed using a Dimension FastScan AFM system (Bruker, Santa Barbara, CA, USA) in tapping mode operation. AFM probes of type TESPA (Bruker) with a resonant frequency of 320 kHz, a spring constant of 42 N m−1 and a tip radius of 8 nm were used for the acquisition of all AFM data. The data were processed and analysed with the Gwyddion AFM analysis software program.[34] Processing steps included data levelling by mean plane subtraction, horizontal scar correction, and a plane level. If needed, a second-degree polynomial background subtraction was executed. The size distribution data of the imaged nanoparticles were extracted from the height distribution, i.e. the diameter, of the nanoparticles, the data then being processed in Sigmaplot™ to generate the size distributions. A representative AFM image of CuNPs formed in 4,4′Bipy is presented in Fig. 1.

|
Kinetic Studies of Copper Nanoparticle Formation
During LASiS-based formation of CuNPs, at regular intervals up to 100 min of irradiation time, a small portion of the sample being irradiated was placed in a UV-vis spectrometer (PG Instruments T90+) where its visible absorption spectrum was recorded. Errors in absorbance measurements between experiments have a maximum uncertainty of ±0.01 absorption units; the uncertainty in absorbance within a given experiment (investigating the same sample) is several orders of magnitude smaller. These time-dependent absorption measurements were used to explore the CuNP formation kinetics.
Results and Discussion
In a manner consistent with previous reports from our laboratory on the formation and stability of AuNPs,[19–21] CuNPs are directly prepared in aqueous solution by laser ablation.[35,36] Although the SPR peak position of all metal nanoparticles (MNPs) is affected by particle size and shape, the SPR peak position and intensity in CuNPs are reported to be particularly susceptible to oxidation at the NP surface, with the formation of Cu2O and CuO.[28–30]
The UV-vis absorption spectrum of CuNPs in pure water, produced after 60 min of 1064-nm laser irradiation (at 10 Hz, see Experimental Methods), is shown as the solid grey trace in Fig. 2. The spectrum has been normalized for the maximum intensity of the SPR transition. The SPR transition has an absorption maximum at ∼650 nm, reportedly indicative of oxidation of the nanoparticle surface.[30] Long and coworkers have demonstrated that the peak of the SPR transition shifts to ∼580 nm on removal of the CuNP oxide layer by treatment with glacial ethanoic acid.[30] All experiments reported herein were undertaken under identical conditions, with the only variable being the nature of the N-donor ligand present in solution during the laser-based production of CuNPs. We thus assume that SPR transitions for CuNPs formed in the presence of these ligands are a measure of the presence or absence of CuNP oxidation, and that CuNPs formed in pure water are oxidized.

|
Also presented in Fig. 2 are the normalized UV-vis spectra of CuNP samples prepared in 1 × 10−4 M aqueous solutions of 4,4′Bipy (long-dashed line) and T-4Py (short-dashed line). Inspection of Fig. 2 indicates that CuNPs prepared in the presence of 4,4′Bipy display an SPR peak at wavelengths of ∼630 nm, only slightly blue-shifted from that of CuNPs prepared in pure water. This suggests that the surface chemical composition and electron density are very similar for CuNPs formed in pure water or the presence of 4,4′Bipy. It is also clear that the T-4Py ligand induces a significant blue shift of the CuNP SPR peak of ∼55 nm (to ∼595 nm).
Based on the report of Long and coworkers,[30] we attribute the T-4Py spectral shift to oxidation state changes of the CuNP surface atoms. That is, the CuNP SPR spectral blue shift when the nanoparticles are formed in the presence of T-4Py is attributed to them being oxide free. This is a unique demonstration of the potentially profound influence of the ablation ligand solution on the oxide-mediated plasmonic behaviour of the laser-formed metal nanoparticle.
It has been well established that for larger MNPs (tens of nanometre diameter), the peak position of the SPR transition is influenced by nanoparticle size.[37] However, El-Sayed and coworkers have reported consistently that for AuNPs of diameters less than ∼20 nm, this effect is not expected to be significant.[38–40] Rice et al. discuss how copper and gold can be expected to behave in a similar manner owing to both metals having similar band structures and optical properties.[41] Moreover, these workers demonstrate that changes in CuNP optical properties can be primarily attributed to changes in surface oxidation rather than NP size. Sun et al. demonstrate that CuNPs synthesized with diameters of 7.5 and 12.2 nm display very similar absorption spectra.[42] Finally, El-Sayed and coworkers show that CuNPs with diameters of 12 and 30 nm display nearly identical absorption spectra.[43] Therefore, although we do not expect the T-4Py spectral blue shift evident in Fig. 2 to be attributable to significantly smaller particle size distributions for CuNPs produced via LASiS in the presence of this tetrazole-based ligand, a determination of the CuNP particle size distributions is required as confirmation.
Particle size distributions were measured via AFM. Representative particle size distributions and associated cumulative distribution functions from AFM measurements for bare CuNPs (those formed in pure water) and for those formed in the presence of both 4,4′Bipy and T-4Py are presented in Fig. 3. To improve the statistics of the particle-size distribution determinations, each histogram was generated by analysing multiple images (see Experimental Method for details).

|
The particle size distributions for bare CuNPs and those formed in the presence of 4,4′Bipy in aqueous solution are very similar, with the former having a maximum in the size distribution of 8 nm. The latter exhibit a size distribution maximum of 6 nm that is a little narrower, as indicated by the sharper cumulative distribution function. Full particle size distribution function statistics are presented in Table 1. Here, the uncertainty in the measurements represent one standard deviation from the mean. The distribution of CuNPs formed in the presence of T-4Py is somewhat smaller and narrower (see Fig. 3), with a maximum in the size distribution of 3 nm. As such, there may indeed be a correlation between the observed SPR spectral blue shift and the smaller particle size distributions of CuNPs formed in the presence of T-4Py.

|
An important question to ask based on the data presented thus far is ‘To what extent does the presence of the ablation ligand solutions during LASiS influence the formation kinetics of the CuNPs?’ We have previously reported the development of a kinetic model for the laser-based formation of AuNPs under our experimental conditions,[19,21] and we seek to explore the CuNP formation kinetics using this same approach.
In Fig. 4 we present the absorbance at the SPR λmax as a function of the number of laser irradiation pulses for CuNPs formed in pure water (top panel) and in the presence of 1 × 10−4 M 4,4′Bipy (middle panel) and T-4Py (bottom panel). The data points represent absorbance readings taken after a specified number of laser pulses, whereas the solid lines represent fits using our previously published kinetic model,[19,21] which is discussed below when applied to CuNP formation. A key aspect of the equivalent kinetic data previously reported for AuNP formation is that after ∼2 × 104 laser pulses, the SPR λmax absorbance increases linearly, with significant curvature in the data evident at early irradiation times.[19,21] For CuNPs, no such linearity is observed over the timeframe investigated (60000 laser pulses = 100 min), indicating (from a kinetics perspective) that a CuNP SPR λmax sink channel (or channels) is operating concurrently with a source channel (or channels). Moreover, the growth in the SPR λmax absorbance appears qualitatively similar irrespective of the nature of the LASiS ablation solution. This qualitative observation suggests that the CuNP formation kinetics are not significantly different in the presence of T-4Py compared with 4,4′Bipy or pure water.

|
Further insight into the CuNP formation chemistry is obtained by fitting the time-dependent absorbance data to an appropriate kinetic model. As discussed, we have previously reported a model that explains the production of AuNPs by our LASiS approach.[19,21] This model was used to fit the experimental data presented in Fig. 4, and the quality of the fits is evident by inspection. The basis for the model is presented in Scheme 1. In brief, the model considers (i) CuNPs of a size too small to support a SPR transition[37] (denoted CuNP(small)); (ii) ‘medium-sized’ CuNPs having a weak, but measurable SPR molar absorbance (denoted CuNP(med)); and (iii) ‘large’ CuNPs with a greater SPR molar absorbance (denoted CuNP(large)). Interconversion between each type of CuNP is allowed, in principle.

|
We have previously reported the rationale for employing a three-size kinetic model for AuNP formation.[21] Importantly, we demonstrated that a simpler two-size model invoking only AuNPs too small to support an SPR transition (AuNP(small)) and those able to support this transition (AuNP(large)) is not able to account for the experimental observations under any circumstances. Importantly, in support of this previous work, detailed analysis involving non-linear least-squares fitting of this two-size model for CuNP formation also cannot account for the experimental data presented in Fig. 4. This requires the more flexible three-size model to be employed and points to the more general applicability of the three-size kinetic model for LASiS-based MNP formation.
The kinetic model presented in Scheme 1 contains up to 12 unique CuNP transformation channels. Rate expressions, all first-order, for the formation of each type of CuNP (‘small’, ‘medium’ or ‘large’ CuNPs) immediately follow:
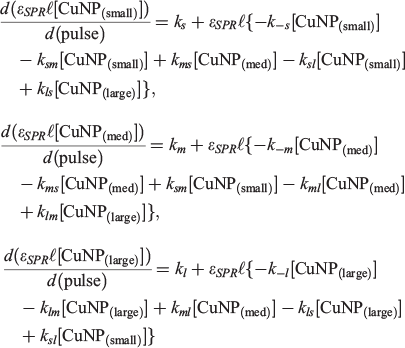
where all rate constants are defined in Scheme 1, ϵSPR refers to the absorption extinction coefficient at the wavelength of the SPR absorption maximum and l refers to an (arbitrary) absorption path length.
As previously reported by us in relation to AuNPs,[21] at the SPR absorption maximum wavelength (λSPR) contributions from both the SPR absorption itself as well as a broader absorption (and scattering) process are active. The relative contribution of the SPR and non-SPR transitions in CuNPs can be readily determined by irradiating LASiS-prepared nanoparticle solutions at λSPR. We have previously shown that in the case of AuNPs, laser irradiation at 532 nm results in nanoparticle photofragmentation such that the SPR transition can no longer be supported.[21] Undertaking equivalent laser irradiation experiments for CuNP samples at 600 nm (5 mJ pulse−1; 10-min irradiation; 250-mm focal length plano-convex lens) completely destroys the SPR transition, leaving only the non-SPR absorption contribution. The relative contribution of the non-SPR absorbance at λmax is determined to be 073. Using the well-established notion that the SPR extinction coefficient increases with MNP size,[21,37] the CuNP absorbance at the SPR λmax can thus be expressed as:
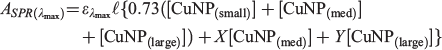
where the values of X and Y represent the extinction coefficient ratios of the SPR component of the SPR-supporting nanoparticles.
Numerical integration of these rate expressions, combined with non-linear least-squares fits to the absorbance data (including allowing the values of X and Y in the equation above to vary) yields the lines of best fit presented in Fig. 4. The most important aspect of the data fitting is that to generate the curves in Fig. 4, only four of the twelve possible transformation channels are required to fit the experimental data. However, unlike in the previous studies into AuNP formation, a single unique solution with a minimum set of parameters was not found. Rather, two indistinguishable pathways are able to account for the experimental observations. The values of the critical rate constants for each ablation solution (pure water, or N-donor ligand solutions) are reported in Table 2.

|
The data presented in Table 2 represent two four-parameter fits to the experimental results that are indistinguishable. These are identified as Scenarios 1 and 2. These two scenarios were the only identified four-parameter fits to the experimental data. No two- or three-parameter fits converged to agreement with experiment. In our previous reports on the formation kinetics of AuNPs,[19,21] we found that in all cases where surfactant (as distinct from N-donor ligands in the present study) concentrations were 1 × 10−4 M or lower, only three channels were required to fit the experimental data, viz., kl, km and k−m. Attempts to limit fits to involving only these three channels for CuNP production yielded χ2 (residual) values up to two orders of magnitude larger than those reported in Table 2. Careful analysis of the resultant fits showed poor agreement with experiment during the first 10 min of LASiS irradiation. This confirms the need to invoke a four-parameter fit to the experimental data over the entire 100-min timeframe.
In both Scenarios 1 and 2, the production of ‘large’ (kl) and ‘medium’ (km) CuNPs directly from bulk copper is operative as the source channels. There is no evidence of direct production of ‘small’ CuNPs. In Scenario 1, two sink channels are required to account for the experimental observations. These the sink channels represent the conversion of the ‘large’ (k−l) and ‘medium’ (k−m) CuNPs back to the bulk. In Scenario 2, only one sink channel involves the return of ‘medium’ CuNPs back to the bulk (k−m). However, this scenario also involves the conversion of ‘large’ CuNPs into ‘medium’ CuNPs (klm).
To within the precision reported herein, both Scenarios 1 and 2 return near-identical rate constants for all ablation solutions, with identical resultant χ2 (residual) values. The key reason we are unable to distinguish between the k−l (Scenario 1) and klm (Scenario 2) channels is that these rate constants are each almost two orders of magnitude smaller than the rate constant for the dominant sink channel, k−m (see Table 2). Irrespective of whether ‘large’ CuNPs return to the bulk (k−l) or convert into ‘medium’-sized CuNPs (klm), the rapid conversion of ‘medium’ CuNPs back to the bulk dominates. Interestingly, our previous studies into the LASiS-based formation of AuNPs demonstrated that the k−m transformation is the only operative sink channel, yet the fourth parameter is required to adequately describe CuNP formation.
An important conclusion from the kinetic analysis presented above is that, apart from demonstrating the applicability of the model to another MNP system in addition to gold, we have demonstrated that although the CuNPs formed in the presence of T-4Py have different optical (SPR transition λmax) properties and particle size distributions, the formation kinetics are essentially indistinguishable from those formed in the presence of 4,4′Bipy or in pure water. This raises interesting questions about the oxidation of copper during the NP formation process.
Recall that CuNPs formed in the presence of an aqueous solution of T-4Py display optical absorption behaviour very similar to those reported to be formed in acetone.[27] The acetone-formed CuNPs are said to be oxide free. Thus, it is possible to conclude that those formed here in the presence of T-4Py are also oxide free. By contrast, CuNPs formed in pure water or in the presence of 4,4′Bipy display optical properties consistent with reports of oxidation having occurred.[31] The significantly smaller particle size distribution determined for CuNPs formed in the presence of T-4Py compared with those formed in pure water or the presence of 4,4′Bipy would seem consistent with different CuNP surface oxidation chemistry occurring. However, the near invariance in the formation kinetics of all CuNP samples reported herein suggests that there is no significant difference in the formation chemistry. We are undertaking further studies to unambiguously characterize the CuNP oxidation chemistry as a function of LASiS ablation solution.
Conclusions
We have explored the formation mechanism and properties of CuNPs formed by direct laser ablation from the bulk metal via a process we refer to as laser ablation synthesis in solution. Differences in the optical properties and particle size distributions of CuNP samples prepared in the presence of a 1 × 10−4 M solution of T-4Py compared with those prepared in pure water and a 1 × 10−4 M solution of 4,4′Bipy are clear. Based on literature reports, these differences may be attributable to changes in the CuNP surface oxidation chemistry under the former conditions. However, analysis of the CuNP formation kinetics under all experimental conditions shows, within experimental uncertainty, identical formation rates. Apart from demonstrating that our kinetic model previously developed to explore the formation kinetics of AuNPs is more generally applicable, this analysis suggests that different CuNP oxidation chemistry during laser-based production is not operative. Further studies are under way to better characterize this issue.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
The authors gratefully acknowledge the financial support of the Australian Research Council and Curtin University for equipment used in this study. AJM, RDT, and PJW each gratefully acknowledge scholarship support in the form of an Australian Postgraduate Award. We acknowledge helpful discussions with Dr J. R. Gascooke (Flinders University).
References
[1] B. Sebo, N. Huang, Y. M. Liu, Q. D. Tai, L. L. Liang, H. Hu, S. Xu, X.-Z. Zhao, Electrochim. Acta 2013, 112, 458.| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXhvFSku7nJ&md5=c5e540739b66ce613829691748d2fc25CAS |
[2] H. S. Noh, E. H. Cho, H. M. Kim, Y. D. Han, J. Joo, Org. Electron. 2013, 14, 278.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXptlyhsw%3D%3D&md5=cf13e78378ffd4d819e998f37aaaccf6CAS |
[3] H. Y. Chung, P. T. Leung, D. P. Tsai, Opt. Express 2013, 21, 26483.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXlsV2ntLg%3D&md5=ca69b0780c6b22fcf288da18a72cc3f7CAS |
[4] Y. Liu, B. J. J. Austen, T. Cornwell, R. D. Tilbury, M. A. Buntine, A. P. O’Mullane, D. W. M. Arrigan, Electrochem. Commun. 2017, 77, 24.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2sXjtFeju7g%3D&md5=9a0d90a6af092a976c6aa2eea8333764CAS |
[5] R. Kaur, C. Giordano, M. Gradzielski, S. K. Mehta, Chem. Asian J. 2014, 9, 189.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXhs1SrtLnE&md5=ef8ae17db75b14972bcdae50a34ee7deCAS |
[6] Z. Y. Wang, A. von dem Bussche, P. K. Kabadi, A. B. Kane, R. H. Hurt, ACS Nano 2013, 7, 8715.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXhsVCgtbjE&md5=495de9d85fc59a6d39649c969709049bCAS |
[7] S. S. Khaleduzzaman, R. Saidur, I. M. Mahbubul, T. A. Ward, M. R. Sohel, I. M. Shahrul, J. Selvaraj, M. M. Rahman, Ind. Eng. Chem. Res. 2014, 53, 10512.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXovVGmtrw%3D&md5=67e30dcf84802e719715e02d9fa041bdCAS |
[8] B. Rimbault, C. T. Nguyen, N. Galanis, Int. J. Therm. Sci. 2014, 84, 275.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXht1SlsL%2FL&md5=789aac5a62a12888fb9521aff3643bd5CAS |
[9] H. Ghaednia, R. L. Jackson, J. M. Khodadadi, J. Exp. Nanosci. 2015, 10, 1.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXnsFGkt78%3D&md5=6eac1a37615200e73e9b64baca8ec78aCAS |
[10] V. S. Jatti, T. P. Singh, J. Mech. Sci. Technol. 2015, 29, 793.
| Crossref | GoogleScholarGoogle Scholar |
[11] M. V. Thottackkad, P. K. Rajendrakumar, K. P. Nair, Ind. Lubr. Tribol. 2014, 66, 289.
| Crossref | GoogleScholarGoogle Scholar |
[12] J. Zhou, Z. Wu, Z. Zhang, W. Liu, Q. Xue, Tribol. Lett. 2000, 8, 213.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXitlegsA%3D%3D&md5=f53f57cc704b659c31fa8366a2024b79CAS |
[13] A. Hernandez Battez, R. Gonzalez, J. L. Viesca, J. E. Fernandez, J. M. D. Fernandez, A. Machado, R. Chou, J. Riba, Wear 2008, 265, 422.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXlvVSms7k%3D&md5=97a7bba5fce01dca545ea6bb4c754f6cCAS |
[14] A. Hernandez Battez, J. L. Viesca, R. Gonzalez, D. Blanco, E. Asedegbega, A. Osorio, Wear 2010, 268, 325.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVGmsrfF&md5=5c602d5889b3c9055f63b9daa6554c21CAS |
[15] M. Zhang, X. B. Wang, W. M. Liu, X. S. Fu, Ind. Lubr. Tribol. 2009, 61, 311.
| Crossref | GoogleScholarGoogle Scholar |
[16] D. R. Clary, G. Mills, J. Phys. Chem. C 2011, 115, 14656.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXoslWiu74%3D&md5=3f552a0221301134b4ef425ddd0da26dCAS |
[17] D. R. Clary, G. Mills, J. Phys. Chem. C 2011, 115, 1767.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmslersA%3D%3D&md5=a801157d45be08d4b2b0b594fde5369dCAS |
[18] O. Pena-Rodriguez, U. Pal, J. Opt. Soc. Am. B 2011, 28, 2735.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsVWmurnM&md5=1199bdcd892ef9e81800ebfceefcf323CAS |
[19] Y. Y. Fong, J. R. Gascooke, G. F. Metha, M. A. Buntine, Aust. J. Chem. 2012, 65, 97.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XisFaqtLY%3D&md5=a82ca7edd50d92606279a1d7f25b82feCAS |
[20] Y. Y. Fong, J. R. Gascooke, B. R. Visser, H. H. Harris, B. C. C. Cowie, L. Thomsen, G. F. Metha, M. A. Buntine, Langmuir 2013, 29, 12452.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXhsVWjs73M&md5=357a201dfc5d72fda75bbe09b6df35a1CAS |
[21] Y. Y. Fong, J. R. Gascooke, B. R. Visser, G. F. Metha, M. A. Buntine, J. Phys. Chem. C 2010, 114, 15931.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXmtVyntLs%3D&md5=3e42e8f21487dcb2e9e3a81de156cb80CAS |
[22] Y. Y. Fong, B. R. Visser, J. R. Gascooke, B. C. C. Cowie, L. Thomsen, G. F. Metha, M. A. Buntine, H. H. Harris, Langmuir 2011, 27, 8099.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXnt1Ggu7o%3D&md5=6f1c790b5401eef0936e224470c486e6CAS |
[23] F. Mafune, J. Kohno, Y. Takeda, T. Kondow, J. Phys. Chem. B 2003, 107, 12589.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXotlarsbk%3D&md5=4030b540e57289d3558dbb054922f61aCAS |
[24] F. Mafune, J. Y. Kohno, Y. Takeda, T. Kondow, J. Phys. Chem. B 2003, 107, 4218.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXivFagsbc%3D&md5=dbbd522927e80884c47764942e0b3a6cCAS |
[25] V. S. Burakov, N. V. Tarasenko, A. V. Butsen, V. A. Rozantsev, M. I. Nedel’ko, Eur. Phys. J. Appl. Phys. 2005, 30, 107.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXls1aksLk%3D&md5=5cbe6e99ced3102fed68f968e2f30462CAS |
[26] P. V. Kazakevich, A. V. Simakin, V. V. Voronov, G. A. Shafeev, Appl. Surf. Sci. 2006, 252, 4373.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XkvFWls7g%3D&md5=322a62e97480d0a1b3b47323c093d00eCAS |
[27] R. M. Tilaki, A. I. Zad, S. M. Mahdavi, Appl. Phys. A: Mater. Sci. Process. 2007, 88, 415.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXmsVOjsbc%3D&md5=01c7399c278280728d1a48fc57bea6e0CAS |
[28] G. H. Chan, J. Zhao, E. M. Hicks, G. C. Schatz, R. P. Van Duyne, Nano Lett. 2007, 7, 1947.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXmtlaqsbo%3D&md5=bfdd57dfb1fc3742b44d12b2b6e8fe36CAS |
[29] F. Gonzalez-Posada, R. Sellappan, B. Vanpoucke, D. Chakarov, RSC Adv. 2014, 4, 20659.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXos1ejtbg%3D&md5=b5d6d95c9b7728eb21f557c0a03b35e3CAS |
[30] L. X. Qin, C. Jing, Y. Li, D. W. Li, Y. T. Long, Chem. Commun. 2012, 1511.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XmtFyksQ%3D%3D&md5=ba1b10aa89638573a7e0f2b4a1277833CAS |
[31] M. Muniz-Miranda, C. Gellini, E. Giorgetti, J. Phys. Chem. C 2011, 115, 5021.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXosl2jsA%3D%3D&md5=a8a71a288aeb4a9a79aa7be656fe0631CAS |
[32] P. J. Wright, S. Muzzioli, M. V. Werrett, P. Raiteri, B. W. Skelton, D. S. Silvester, S. Stagni, M. Massi, Organometallics 2012, 31, 7566.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhsFOltrnP&md5=a2d1e1b4e2d5de322c83d4715f981bf2CAS |
[33] N. S. Basheer, B. R. Kumar, A. Kurian, S. D. George, Appl. Phys. B: Lasers Opt. 2013, 113, 581.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXhvFWrtbzJ&md5=7103bdf112041dc92a400aeef204b53dCAS |
[34] D. Necas, P. Klapetek, Cent. Eur. J. Phys. 2012, 10, 181.
[35] N. Haram, N. Ahmad, Appl. Phys. A: Mater. Sci. Process. 2013, 111, 1131.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXns1GrtLY%3D&md5=21bf6f73de9d8ca659798efe7329a7c1CAS |
[36] B. Kumar, R. K. Thareja, Phys. Plasmas 2013, 20, 053503.
| Crossref | GoogleScholarGoogle Scholar |
[37] C. F. Bohren, D. R. Huffman, Absorption and Scattering of Light by Small Particles 1983 (Wiley-Interscience: New York, NY).
[38] S. Link, M. A. Ei-Sayed, Annu. Rev. Phys. Chem. 2003, 54, 331.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXntFSgsLw%3D&md5=aaf29ef37d5c6c23d3bfe4aeebe7305cCAS |
[39] S. Link, M. A. El-Sayed, J. Phys. Chem. B 1999, 103, 8410.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXlslGntrs%3D&md5=ea34953ad439dcfe5ffc704efec74c69CAS |
[40] S. Link, M. A. El-Sayed, Int. Rev. Phys. Chem. 2000, 19, 409.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXlsFOntrs%3D&md5=023c80433c8a26159800144ea8840e82CAS |
[41] K. P. Rice, E. J. Walker, M. P. Stoykovich, A. E. Saunders, J. Phys. Chem. C 2011, 115, 1793.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmslemsQ%3D%3D&md5=86ceba828da4f70cf2e9dd62ee0180c5CAS |
[42] Q. C. Sun, Y. C. Ding, S. M. Goodman, H. H. Funke, P. Nagpal, Nanoscale 2014, 6, 12450.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXhsFKrurfM&md5=f6f425c36c53054c2721971d2e293327CAS |
[43] Q. Darugar, W. Qian, M. A. El-Sayed, M. P. Pileni, J. Phys. Chem. B 2006, 110, 143.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXht12rtb7N&md5=1af4a66dcda7eb7356c75fdd49d3b19eCAS |
* Mark A. Buntine was the recipient of the 2016 RACI Leighton Memorial Medal.