

The quantification of radical concentration in organic radical polymers: techniques and challenges
Theo A. Ellingsen A * , Stuart C. Thickett A and Rebecca O. Fuller A *
A * , Stuart C. Thickett A and Rebecca O. Fuller A *
A
Handling Editor: Curt Wentrup
Abstract
The development of new high-tech applications based on organic radical polymers has driven significant and renewed focus on these open shell macromolecules. The versatility in synthetic methods makes them highly accessible materials for a variety of researchers from different backgrounds. Although numerous overviews of the synthesis, structure and properties are available, the determination of radical concentration has been largely overlooked. This primer outlines the methods available and the non-trivial nature of the characterisation process. Although quantitative electron paramagnetic resonance and magnetometry are the gold standard for direct measurement of paramagnetism, there also exists a wide range of highly accessible complimentary methods for indirect measure such as ultraviolet–visible spectroscopy, elemental analysis and infrared spectroscopy.
Keywords: characterisation, energy storage, organic radical polymers, quantification, stable organic radicals.
Introduction
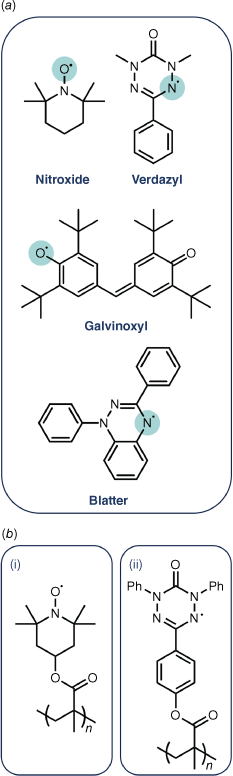
Organic radical polymers (ORPs) were first developed as a fundamental curiosity in the late 1960s.1 New, previously unconsidered technologies have driven the more recent expansion and evolution of the field into a range of disciplines. Combining the thermal and mechanical properties of a polymeric substrate with the unique nature of stable organic radical pendant groups (e.g. Fig. 1a) makes ORPs well suited for applications based in energy storage, drug delivery, catalysis and spintronics.2–7 Although the first system, poly(TEMPO methacrylate) (PTMA) (TEMPO, (2,2,6,6-tetramethylpiperidin-1-yl)oxyl) and other nitroxide-based ORPs are the most extensively studied,1,2,8–10 a plethora of new examples based on different radical pendant groups, including verdazyl,11–13 galvinoxyl3,14,15 and Blatter,16,17 are now found (e.g. Fig. 1b). A diversity of polymer backbones are used for various applications, including π-conjugated backbones which provide enhanced conductivity.18 Advances in synthetic methods underpin much of the expansion of the field into new areas. Although much has been done to review properties, synthesis and structures of ORPs, an overlooked part of this is the characterisation of radical addition to the polymer backbone. Understanding how to effectively characterise radical concentration in new systems made by these synthetically accessible routes is essential.
(a) Common examples of stable organic radicals include: nitroxide, verdazyl, Blatter and galvinoxyl. (b) Examples of previously synthesised ORPs poly(2,2,6,6-tetramethyl-piperidenyloxyl-4-yl methacrylate) (PTMA) (i) and poly(1,3,5-triphenyl-6-oxoverdazyl methacrylate) (ii).
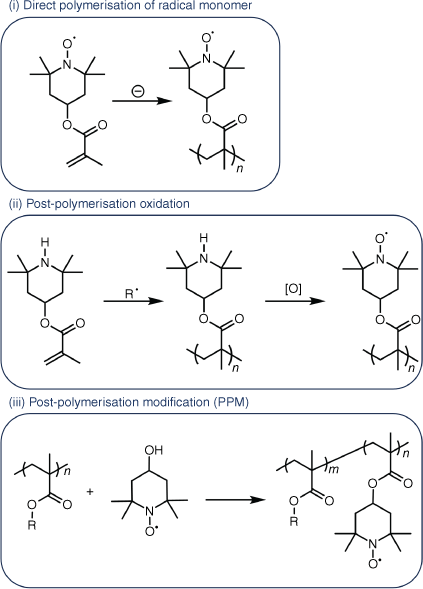
ORP radical content is related to mode of synthesis. With many reviews available, only the pertinent points to characterisation are considered.6,7,19–21 Synthesis of ORPs is broadly grouped into three categories: (i) direct polymerisation of a radical-containing monomer,1,8,12 (ii) post-polymerisation oxidation of a radical precursor-containing polymer,22,23 and (iii) post-polymerisation modification of a polymer backbone to attach a stable organic radical (Fig. 2).10,13 Polymerisation of a radical-containing monomer (Fig. 2(i)) is the most straightforward route to generate an ORP. For ORPs synthesised using this method, quantification of the radical concentration is directly related to the monomer conversion during polymerisation. The simplicity of this system is countered by the reduced scope, given that these polymers are synthesised by methods other than free-radical polymerisation, such as ionic polymerisation,1 ring-opening metathesis polymerisation,12 and olefin metathesis polymerisation.24 Approaches (ii) and (iii) offer improved control of the polymer backbone through use of a non-radical based monomer, which is compatible with free-radical polymerisation techniques including reversible addition–fragmentation chain transfer (RAFT) polymerisation. In (ii), the monomer contains a radical precursor group (Fig. 2(ii)). Following polymerisation, a post-polymerisation oxidation is used to generate the radical pendant groups. The efficiency of this step is highly variable, requiring careful quantification. Low radical concentrations are common and typically limit the usefulness of the ORP generated.10,12 Post-polymerisation modification (PPM), involves the attachment of a radical molecule to a pre-synthesised polymer by suitable functional groups (Fig. 2(iii)). This method allows fine control over both polymer and redox-active group and offers a path to independent control of both. Like method (ii) the radical concentration can be highly varied and dependent on the polymer and radical pairing.10 Accurate characterisation of radical content is essential and can be achieved using a number of methods. Given the difficulty of obtaining accurate measurements using some methods, and the variance observed between techniques, researchers need to have a clear understanding of the approach they adopt.10 This primer has been developed as a guide to the methods commonly used to determine ORP radical concentration. We focus on the accessibility of each technique, potential sources of error and the precautions needed for accurate assignment of data. We also provide direction for methods that tend to be overlooked for characterisation of ORPs, although they are often used in non-radical containing polymers and are routinely available in university chemistry departments.
The three primary methods of ORP synthesis, using PTMA as an example. Method (i) utilises alternative techniques to free-radical initiated polymerisation such as anionic polymerisation, whereas (ii) can utilise free-radical polymerisation, and (iii) can be prepared using any polymerisation technique.
Methods for the quantification of radical concentration
Electron paramagnetic resonance
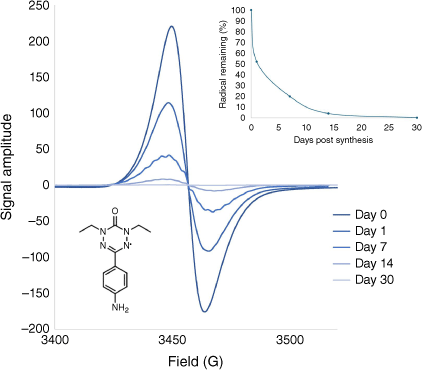
Electron paramagnetic resonance (EPR) spectroscopy, ubiquitous in the investigation of paramagnetic materials, is used to detect unpaired electrons in a variety of materials including ORPs.12,13,25–27 This tool is most often used for the qualitative measurement of spins in a solid or solution using the continuous wave (CW) mode of operation. The more powerful quantitative (q-EPR) measurements are widely adopted to the study of biological processes, as they provide a direct measure of the concentration of spins, through the comparison of the double-integral of the unknowns’ absorption, to a standard that consists of a known concentration of spins. q-EPR’s application to materials is more limited, as for many systems (e.g. coordination complexes and stable organic radicals) qualitative data provides the required information, and these measurements are simple to undertake (Fig. 3). Although the quantitative measurements offer extremely high sensitivity (1012 spins per mT of line width),28 undertaking the measurements present a limitation on accuracy and repeatability as numerous factors affect the signal amplitude, which is key to quantification.29,30 Examples of systematic errors include: cavity setup, sample size, shape and placement within the instrument, improper calibration of the instrumental variables and any alterations to these variables between analyses, such as an instrument being retuned,31 as well as the inability to use an appropriate reference sample. For ORPs the latter is significant as the standard will likely differ to the actual sample.13,25,31 Specifically, TEMPO-based reference standards are commonly used for ORP systems regardless of the composition of the polymer,12,13,25 alternatives such as metal salts have also been used.26 Differences between the dielectric constant of the reference standard and ORP are known to reduce the accuracy of the technique, unless an internal standard within the instrumental cavity is used to normalise the signal amplitude.31 As with standard qualitative EPR, the presence of dissolved oxygen in the samples can interfere with the EPR signals, adding error to q-EPR measurements.32 Furthermore, spin relaxation must be sufficient for detection; an ORP low concentration or insufficient packing density will provide a further limitation. The opposite case is also problematic for ORPs i.e. a high spin-concentration and small inter-spin distances will cause significant broadening of the EPR spectra.33 Specialised pulsed EPR or rapid scan CW-EPR are further methods that can be used28,29,31 to alleviate the artifacts.34
An example of qualitative EPR measurements being used to monitor the decomposition of a verdazyl radical (1,5-diethyl-3-(4-aminophenyl)-6-oxoverdazyl), over the course of 1 month. Measurements were performed using a MicroESR Standard benchtop (ver. 3.0, Bruker) EPR spectrometer at 298 K.
To achieve a high degree of accuracy in q-EPR, a nuanced approach is required but seldom detailed for ORPs.12,13,27 To highlight this, one groups’ efforts to minimise error associated with their q-EPR measurements involved housing their instrument in a glove box and preparing identical shortened capillary tubes so that consistent scans across different samples without detuning of the cavity was achieved.13 Furthermore, for calibration, freshly sublimed TEMPO is used as a reference standard, with the g-factor for each sample precisely measured and calibrated using a Mn2+ marker located within the instrument’s resonant cavity.12,13 The highly specialised nature of these measurements restricts their widespread adoption to ORPs.
Magnetic susceptibility measurements
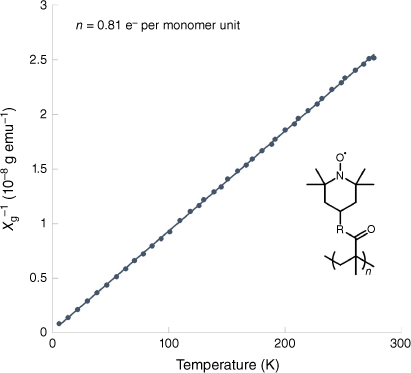
Magnetic susceptibility measurements are excellent for quantification of spin concentration in an ORP solid sample. These measurements are non-destructive and allow the effective magnetic moment to be directly measured, and degree of polymer functionalisation to be determined. Magnetometers such as superconducting quantum interference devices (SQuID),25,26,35,36 and vibrating sample magnetometers (VSMs),37 not only offer quantification at room temperature but also allow for in-depth analysis of temperature and field-dependent properties. For conducting polymers,38 magnetic exchange interactions are not often evident at room temperature or without application of a bias field, the latter is not accessible with EPR.39 The spin concentration in the ORP is often found directly from the slope of the Curie–Weiss plot (Fig. 4).25 Unlike q-EPR, which requires a specialised setup to minimise the significant errors, these instruments offer a simple setup and use standardised methodologies that greatly reduce the complexity of the measurement. Accessibility is the only major hindrance in their use for ORP characterisation. For example, in the Australian research landscape, there are only a handful of institutions maintaining magnetometers. In the absence of this instrumentation, room temperature susceptibility can still be determined with a number of modest and commonly available methods such as Gouy40 and Faraday balances,41 or even using nuclear magnetic resonance (NMR) spectroscopy through the Evans method.42 The Evans method is a technique that utilises the observed change in chemical shift of a 1H NMR peak when in the presence of a known quantity of a paramagnetic species. The change in chemical shift is proportional to the magnetic susceptibility of the compound, allowing the number of unpaired electrons per repeating unit of polymer to be calculated. Although some of these methods have decreased sensitivity and capability when compared to magnetometers, their accessibility makes them possible alternatives well suited to ORP characterisation.
A Curie–Weiss plot from a PTMA sample, figure adapted from Nishide et al.,35 with permission. Construction of a Curie–Weiss plot is achieved by plotting the inverse of the magnetic susceptibility as a function of temperature. The gradient of the slope is equal to the inverse of the Curie constant (C−1) and the number of unpaired electrons per repeating unit (n) is calculated. To calculate n, the effective magnetic moment (µeff) can be calculated (µeff = 800 µB), and the number of unpaired electrons can be determined (n = –1 + ).38 χg−1, inverse mass susceptibility; emu, electromagnetic unit.
Ultraviolet–visible absorbance spectroscopy
Ease of use, high accuracy and cost effectiveness have ensured that ultraviolet–visible absorbance spectroscopy (UV-Vis) is one of the most widely adopted methods for the determination of unknown radical concentrations in ORPs.8,11,13,23,43 Degree of polymer functionalisation can be determined by construction of a Beer–Lambert plot using an absorption that is common to the ORP and a standard, often the unbound radical (Fig. 5). An essential requirement is that the electronic transition chosen for the analysis is a characteristic absorption from the radical, which does not overlap with other absorbances associated with the polymer backbone or precursor. Nitroxide-based systems are known to be subject to significant changes in peak intensity and wavelength given the polarity of the solvent used,44 and peak intensities and wavelengths can vary following attachment to the ORP. Therefore, the commonly unbound radical reference standard is not well suited to this analysis. Any compounds with significant electronic differences (e.g. the presence of additional electron-withdrawing or electron-donating groups) to the ORP are unsuitable for use as a reference standard. The molar absorptivity must be as close as possible between the ORP and reference standard. Ideally the reference would consist of an ORP with standardised radical concentration, but the difficulties in synthesising such a system make it unviable. The limited solubility of some ORPs is a further restriction to quantification using UV-Vis.10,13
(a) Representative UV-Vis spectra of PTMA at three concentrations. The peak maxima are used to determine radical concentration using a Beer–Lambert plot (inset), constructed with a similar monomer species (tBu-TEMPO). Figure illustrative of work from Bertrand et al.23 (b) The decomposition of TEMPO upon addition of 1-hexanethiol, monitored using UV-Vis. Figure adapted from Zhang et al.45 with permission.
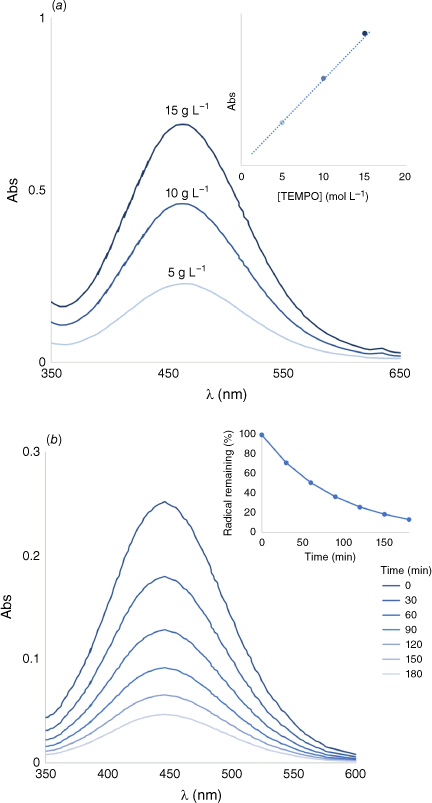
For PTMA, high accuracy has been reported when determining the radical concentration using UV-Vis.8 The absorbance of PTMA and 4-acetoxy-2,2,6,6-tetramethylpiperidine-N-oxyl at ~460 nm was measured at concentrations of 5 and 10 g L−1 in dichloromethane for both samples. The calculated molar extinction coefficient for PTMA and the monomer radical samples, when compared, gave a reported value of 100.26% functionalisation of the PTMA. Given that this ORP was synthesised using a direct polymerisation method (Fig. 2(i)), the monomer sample used as the standard is structurally similar to the ORP being analysed. Therefore, the monomer is an adequate reference for a system where known concentrations cannot be used. The good solubility exhibited by the PTMA sample also ensured suitability to UV-Vis analysis. The availability of UV-Vis makes it an attractive method for the characterisation of ORP radical concentration; however, the correct precautions must be taken to ensure validity of the obtained data. Given this technique does not directly measure the spin concentration, ideally it should be reported validated alongside other techniques for radical concentration quantification.
Alternative quantification techniques
There are a wide range of alternative methods that can be used to determine the radical concentration in an ORP. A variety of analytical techniques including iodometric titration,46 Kjeldahl nitrogen analysis22 and elemental analysis (EA) have been used.22,26,47,48 The two former methods require significant sample preparation and handling, and have only been applied to nitroxide-based ORPs. Kjeldahl nitrogen analysis has not been widely used, and may not be applied to pendant groups with significant nitrogen content, given radical concentration must be inferred from the nitrogen content found in the ORP. Using EA, the functionalisation can be calculated by comparing the observed composition with the theoretical composition with 100% functionalisation. This technique was used to confirm the quantitative functionalisation of a nitroxide-based ORP.48 However, using EA for ORP characterisation involves non-standard application of the technique with the potential for result anomalies. Usually, the acceptable difference between the calculated and experimental values in EA is ±0.4%, but in ORP analysis the difference between the calculated and experimental values is the metric for determining polymer radical content. For this reason, it is assumed that differences between the calculated and experimentally determined elemental values are due to the functionalisation of the polymer, but impurities can unknowingly alter the results. Impurities such as residual solvent may be indistinguishable from radical molecules in the experimental sample, and as such the composition of elements may be misleading if purity has not been confirmed with an alternative method such as 1H NMR analysis.
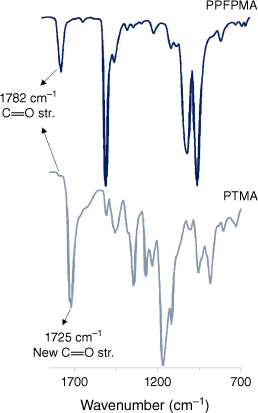
Fourier Transform–infrared (FT-IR) spectroscopy has also been used to quantify the concentration of radicals in ORPs. For PTMA, the relative intensity of the carbonyl stretches is examined and used to determine the degree of functionalisation within the polymer (Fig. 6).10 In polymer samples, broadening of FT-IR signals is often observed, meaning peak area is often more suitable than peak intensity for this calculation. As both solid and solution analysis is available, this method is also applicable for insoluble ORPs. For this to be useful, vibrations of interest in the base polymer cannot overlap with those from the newly formed bonds from radical attachment. This limits this semi-quantitative method to certain polymer types, such as acrylate-based ORPs, and only to PPM (Fig. 2(iii)) prepared ORPs. FTI-R quantification will not be useful to ORP systems with transient radical pendant groups. Like UV-Vis spectroscopy, FT-IR instruments are widely available and offer an alternative to more specialised techniques, but ideally results obtained would be supported through a direct measurement of unpaired spin concentration using q-EPR or magnetometry. FT-IR spectroscopy also has the potential to be developed for the in situ monitoring of PPM reactions used to prepare ORPs.
Future directions
There exists a variety of potentially useful techniques for ORP characterisation that are yet to be fully realised. Cyclic voltammetry and NMR spectroscopy are two unexpected examples, as they are routinely associated with other parts of ORP characterisation. Although cyclic voltammetry is not commonly used for quantification, the peak–current response is directly proportional to the concentration of the redox-active species, as explained by the Randles–Ševčík equation.49 By determining the concentration of radical using the peak–current response in a voltammogram of an ORP sample, the degree of functionalisation of an ORP could feasibly be calculated as part of the standard electron transfer investigations undertaken for ORPs. Surprisingly, although NMR spectroscopy is routinely used to characterise polymers and monitor PPM reactions to determine the degree of functionalisation in non-radical systems it has not been used to quantify the functionalisation of an ORP.50 Examples of the use of NMR spectroscopy on stable organic radicals can be found. These include monitoring of reactions,51 determining paramagnetic susceptibility using the Evans method52 and decomposition studies53; however, no examples are found of the application of NMR spectroscopy to elucidate radical concentration in ORPs. This may be a result of the expectation that the radicals’ unpaired spins produce paramagnetic line-broadening. As the unpaired electron only resides in specific molecular orbitals, it may still be possible to monitor reactions for specific atoms not involved in this molecular orbital. For example, 19F NMR spectroscopy could be used to determine the degree of functionalisation in a poly(pentafluorophenyl methacrylate) polymer used for PPM with the TEMPO radical.10
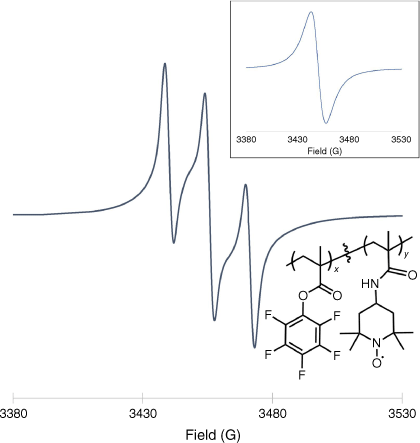
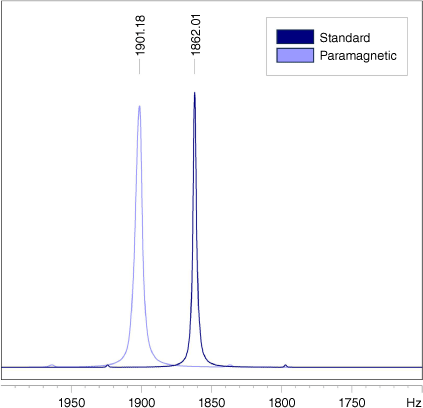
To demonstrate the usefulness of the less conventional methods for ORP characterisation, we synthesised a common nitroxide-based ORP, poly(TEMPO methacrylamide) (PTMAM), using PPM (see the ‘Experimental details’ section in the Supplementary material). The EPR spectrum of the product is shown below (Fig. 7). We have employed FT-IR spectroscopy (Supplementary Fig. S1), the Evans method (Fig. 8) and quantitative 19F NMR spectroscopy (Supplementary Fig. S2) to determine the TEMPO concentration in the ORP (Table 1). Detailed calculations are provided in the Supplementary material as a guide. Using the Evans method, the magnetic susceptibility could be calculated by measuring the change in frequency of a standard peak upon addition of a known amount of ORP (Fig. 8). Quantitative 19F NMR spectroscopy can be used to monitor the reaction and indirectly determine the functionalisation of the ORP by measuring the amount of free pentafluorophenol in solution, which is formed following radical attachment to the initial polymer backbone. Using a trifluorotoluene standard, the concentration of pentafluorophenol was determined, and assumed to be directly proportional to the concentration of TEMPO attached to the polymer.
The EPR of a dilute solution of PTMAM in degassed chloroform. Inset shows the solid-state EPR spectrum of the same sample. Measurements were recorded using a MicroESR Standard benchtop (ver. 3.0, Bruker) EPR spectrometer at 298 K.
A demonstration of the peak shift observed when using the Evans method to determine the paramagnetic susceptibility of the synthesised polymer, using acetone as a reference standard.
| Q-19F-NMR | Evans method | FT-IR | ||
|---|---|---|---|---|
| Percentage attachment (%) | 48 | 52 | 48 |
Each of the three methods are not without their limitations,54 and they must be reported together to gain confidence in the obtained values of attachment. Quantitative 19F NMR spectroscopy is a valuable tool for the in situ monitoring of the specific PPM reaction reported, however, its uses are limited for other reactions involving alternative polymers or pendant groups. This technique is further hindered by the presence of catalysts such as triethylamine, which are often used in the PPM attachment of hydroxides, as they can cause hydrolysis and detachment of pentafluorophenol. Under these conditions, the concentration of pentafluorophenol cannot be assumed to be directly proportional to the radical amount on the polymer backbone. The Evans method is effective for room-temperature and solution-state magnetic susceptibility measurements. However, it is less versatile than other magnetometers, particularly for obtaining temperature-dependent magnetic susceptibility data. This limitation arises from the need for significant corrections due to changes in solvent density at different temperatures when constructing a Curie–Weiss plot. Additionally, the Evans method is restricted to solution-state compounds and is therefore unsuitable for insoluble ORPs. As previously mentioned, the use of FT-IR spectroscopy is limited to PPM preparation of ORPs and cannot be employed in PPM reactions where the vibrations of interest overlap with new vibrations resulting from the radical attachment.
Conclusions and outlook
Interest in the development of new high-tech materials with ORPs has gained significant momentum in recent years. The pursuit of novel systems for downstream applications highlights the need to ensure the accurate determination of the final ORP radical concentration. This primer is aimed to provide an approachable and comprehensive overview of how to characterise the degree of radical functionalisation in an ORP, outlining standard methodology and highlighting some of the experimental pitfalls. Direct measurement of paramagnetism using q-EPR or magnetometry represents the gold standard for accuracy, but can be limited by accessibility to this equipment. There are a host of more accessible alternative spectroscopies available for characterisation, including UV-Vis, FT-IR and NMR spectroscopy as well as elemental analysis. Utilisation of multiple techniques can provide further validation of results, facilitating a more accurate measurement of radical concentration in ORPs.
Data availability
All data generated have been included in the paper or Supplementary material. Analysis has been performed on published materials that can be accessed by permission or under licence.
Declaration of funding
This work was supported under the Australian Research Council’s Future Fellowship (project number FT220100096). T. A. Ellingsen was a recipient of an Australian Research Training Program (RTP) scholarship.
Acknowledgements
T. A. Ellingsen acknowledges the Australian Government for the provision of a RTP scholarship, and the Royal Australian Chemical Institute for the Masson Memorial Award. S. C. Thickett acknowledges the Australian Research Council for the provision of a Future Fellowship. Theo A. Ellingsen is the recipient of the 2023 Masson Memorial Award from the Royal Australian Chemical Institute.
References
1 Hayes. Griffith O, Keana JFW, Rottschaefer S, Warlick TA. Preparation and magnetic resonance of nitroxide polymers. J Am Chem Soc 1967; 89: 5072.
| Crossref | Google Scholar |
2 Nakahara K, Iwasa S, Satoh M, Morioka Y, Iriyama J, Suguro M, Hasegawa E. Rechargeable batteries with organic radical cathodes. Chem Phys Lett 2002; 359: 351-354.
| Crossref | Google Scholar |
3 Suga T, Ohshiro H, Sugita S, Oyaizu K, Nishide H. Emerging N-type redox-active radical polymer for a totally organic polymer-based rechargeable battery. Adv Mater 2009; 21: 1627-1630.
| Crossref | Google Scholar |
4 Suga T, Sugita S, Ohshiro H, Oyaizu K, Nishide H. p- and n-Type bipolar redox-active radical polymer: toward totally organic polymer-based rechargeable devices with variable configuration. Adv Mater 2011; 23: 751-754.
| Crossref | Google Scholar | PubMed |
5 Poizot P, Dolhem F, Gaubicher J. Progress in all-organic rechargeable batteries using cationic and anionic configurations: toward low-cost and greener storage solutions? Curr Opin Electrochem 2018; 9: 70-80.
| Crossref | Google Scholar |
6 Zhang K, Monteiro MJ, Jia Z. Stable organic radical polymers: synthesis and applications. Polym Chem 2016; 7: 5589-5614.
| Crossref | Google Scholar |
7 Tan Y, Hsu S-N, Tahir H, Dou L, Savoie BM, Boudouris BW. Electronic and spintronic open-shell macromolecules, quo vadis? J Am Chem Soc 2022; 144: 626-647.
| Crossref | Google Scholar | PubMed |
8 Bugnon L, Morton CJH, Novak P, Vetter J, Nesvadba P. Synthesis of poly(4-methacryloyloxy-TEMPO) via group-transfer polymerization and its evaluation in organic radical battery. Chem Mater 2007; 19: 2910-2914.
| Crossref | Google Scholar |
9 MacCorquodale F, Crayston JA, Walton JC, Worsfold DJ. Synthesis and electrochemical characterisation of poly(tempoacrylate). Tetrahedron Lett 1990; 31: 771-774.
| Crossref | Google Scholar |
10 Xue W, Mutlu H, Theato P. Post-polymerization modification of polymeric active esters towards TEMPO containing polymers: a systematic study. Eur Polym J 2020; 130: 109660.
| Crossref | Google Scholar |
11 Price JT, Paquette JA, Harrison CS, Bauld R, Fanchini G, Gilroy JB. 6-Oxoverdazyl radical polymers with tunable electrochemical properties. Polym, Chem 2014; 5: 5223-5226.
| Crossref | Google Scholar |
12 Paquette JA, Ezugwu S, Yadav V, Fanchini G, Gilroy JB. Synthesis, characterization, and thin-film properties of 6-oxoverdazyl polymers prepared by ring-opening metathesis polymerization. J Polym Sci – A. Polym Chem 2016; 54: 1803-1813.
| Crossref | Google Scholar |
13 Magnan F, Dhindsa JS, Anghel M, Bazylewski P, Fanchini G, Gilroy JB. A divergent strategy for the synthesis of redox-active verdazyl radical polymers. Polym Chem 2021; 12: 2786-2797.
| Crossref | Google Scholar |
14 Jähnert T, Häupler B, Janoschka T, Hager MD, Schubert US. Synthesis and charge–discharge studies of poly(ethynylphenyl)galvinoxyles and their use in organic radical batteries with aqueous electrolytes. Macromol Chem Phys 2013; 214: 2616-2623.
| Crossref | Google Scholar |
15 Shi Z, Wang J, Teraguchi M, Aoki T, Kaneko T. Helix-sense-selective polymerization of 3,5-bis(hydroxymethyl)phenylacetylene rigidly bearing galvinoxyl residues and their chiroptical properties. Polymers 2019; 11: 1877.
| Crossref | Google Scholar | PubMed |
16 Saal A, Friebe C, Schubert US. Polymeric Blatter’s radical via CuAAC and ROMP. Macromol Chem Phys 2021; 222: 2100194.
| Crossref | Google Scholar |
17 Saal A, Friebe C, Schubert US. Blatter radical as a polymeric active material in organic batteries. J Power Sources 2022; 524: 231061.
| Crossref | Google Scholar |
18 Liang Y, Chen Z, Jing Y, Rong Y, Facchetti A, Yao Y. Heavily n-dopable π-conjugated redox polymers with ultrafast energy storage capability. J Am Chem Soc 2015; 137: 4956-4959.
| Crossref | Google Scholar | PubMed |
20 Hansen K-A, Blinco JP. Nitroxide radical polymers – a versatile material class for high-tech applications. Polym Chem 2018; 9: 1479-1516.
| Crossref | Google Scholar |
21 Ellingsen TA, Hoffmann N, Olivier WJ, Thickett SC, Silvester DS, Fuller RO. Stable organic radicals and their untapped potential in ionic liquids. Aust J Chem 2022; 75: 893-898.
| Crossref | Google Scholar |
22 Kurosaki T, Lee KW, Okawara M. Polymers having stable radicals. I. Synthesis of nitroxyl polymers from 4-methacryloyl derivatives of 2,2,6,6-tetramethylpiperidine. J Polym Sci A-1: Polym Chem 1972; 10: 3295-3310.
| Crossref | Google Scholar |
23 Bertrand O, Ernould B, Boujioui F, Vlad A, Gohy J-F. Synthesis of polymer precursors of electroactive materials by SET-LRP. Polym Chem 2015; 6: 6067-6072.
| Crossref | Google Scholar |
24 Fujii A, Ishida T, Koga N, Iwamura H. Syntheses and magnetic properties of poly(phenylacetylenes) carrying a (1-oxido-3-oxy-4,4,5,5-tetramethyl-2-imidazolin-2-yl) group at the meta or para position of the phenyl ring. Macromolecules 1991; 24: 1077-1082.
| Crossref | Google Scholar |
25 Suga T, Konishi H, Nishide H. Photocrosslinked nitroxide polymer cathode-active materials for application in an organic-based paper battery. Chem Commun 2007; 2007(17): 1730-1732.
| Crossref | Google Scholar | PubMed |
26 Janoschka T, Teichler A, Krieg A, Hager MD, Schubert US. Polymerization of free secondary amine bearing monomers by RAFT polymerization and other controlled radical techniques. J Polym Sci – A. Polym Chem 2012; 50: 1394-1407.
| Crossref | Google Scholar |
27 Daniel DT, Oevermann S, Mitra S, Rudolf K, Heuer A, Eichel R-A, Winter M, Diddens D, Brunklaus G, Granwehr J. Multimodal investigation of electronic transport in PTMA and its impact on organic radical battery performance. Sci Rep 2023; 13: 10934.
| Crossref | Google Scholar | PubMed |
28 Möser J, Lips K, Tseytlin M, Eaton GR, Eaton SS, Schnegg A. Using rapid-scan EPR to improve the detection limit of quantitative EPR by more than one order of magnitude. J Magn Reson 2017; 281: 17-25.
| Crossref | Google Scholar | PubMed |
30 Burns DT, Flockhart BD. Application of quantitative EPR. Philos Trans A Math Phys Eng Sci 1990; 333: 37-48.
| Crossref | Google Scholar |
31 Nagy V. Quantitative EPR: some of the most difficult problems. Appl Magn Reson 1994; 6: 259-285.
| Crossref | Google Scholar |
32 Ingalls RB, Pearson GA. A basis for the determination of dissolved oxygen by electron spin resonance spectroscopy. Anal Chim Acta 1961; 25: 566-569.
| Crossref | Google Scholar |
33 Daniel DT, Mitra S, Eichel R-A, Diddens D, Granwehr J. Machine learning isotropic g values of radical polymers. J Chem Theory Comput 2024; 20: 2592-2604.
| Crossref | Google Scholar | PubMed |
34 Kulikov I, Vereshchagin AA, Lukianov DA, Levin OV, Behrends J. A nitroxide-containing cathode material for organic radical batteries studied with pulsed EPR spectroscopy. J Magn Reson Open 2023; 16–17: 100134.
| Crossref | Google Scholar |
35 Nishide H, Iwasa S, Pu Y-J, Suga T, Nakahara K, Satoh M. Organic radical battery: nitroxide polymers as a cathode-active material. Electrochim Acta 2004; 50: 827-831.
| Crossref | Google Scholar |
36 Qu J, Katsumata T, Satoh M, Wada J, Masuda T. Macromolecules 2007; 40: 3136-3144.
| Crossref |
37 Jobelius H, Wagner N, Schnakenburg G, Meyer A. Verdazyls as possible building blocks for multifunctional molecular materials: a case study on 1,5-diphenyl-3-(p-iodophenyl)-verdazyl focusing on magnetism, electron transfer and the applicability of the Sonogashira–Hagihara Reaction. Molecules 2018; 23: 1758.
| Crossref | Google Scholar | PubMed |
38 Mugiraneza S, Hallas AM. Tutorial: a beginner’s guide to interpreting magnetic susceptibility data with the Curie–Weiss law. Commun Phys 2022; 5: 95.
| Crossref | Google Scholar |
39 Cai H, Tang H, Wang T, Xu C, Xie J, Fu M, Luo X, Hu Z, Zhang Y, Deng Y, Li G, Liu C, Huang F, Cao Y. An n-type open-shell conjugated polymer with high-spin ground-state and high intrinsic electrical conductivity. Angew Chem Int Ed 2024; 63: e202402375.
| Crossref | Google Scholar |
40 Saunderson A. A permanent magnet Gouy balance. Phys Educ 1968; 3: 272-273.
| Crossref | Google Scholar |
41 Morris BL, Wold A. Faraday balance for measuring magnetic susceptibility. Rev Sci Instrum 1968; 39: 1937-1941.
| Crossref | Google Scholar |
42 Evans DF. 400. The determination of the paramagnetic susceptibility of substances in solution by nuclear magnetic resonance. J Chem Soc 1959; 59: 2003-2005.
| Crossref | Google Scholar | PubMed |
43 Aqil M, Aqil A, Ouhib F, El Idrissi A, Detrembleur C, Jérôme C. RAFT polymerization of an alkoxyamine bearing acrylate, towards a well-defined redox active polyacrylate. RSC Adv 2015; 5: 85035-85038.
| Crossref | Google Scholar |
44 Falbo E, Fusè M, Lazzari F, Mancini G, Barone V. Integration of quantum chemistry, statistical mechanics, and artificial intelligence for computational spectroscopy: the UV–Vis spectrum of TEMPO radical in different solvents. J Chem Theory Comput 2022; 18: 6203-6216.
| Crossref | Google Scholar | PubMed |
45 Zhang X, Huang S, Podgórski M, Han X, Claudino M, Bowman CN. Assessment of TEMPO as a thermally activatable base generator and its use in initiation of thermally-triggered thiol-Michael addition polymerizations. Polym Chem 2018; 9: 4294-4302.
| Crossref | Google Scholar | PubMed |
46 Endo T, Takuma K, Takata T, Hirose C. Synthesis and polymerization of 4-(glycidyloxy)-2,2,6,6-tetramethylpiperidine-1-oxyl. Macromolecules 1993; 26: 3227-3229.
| Crossref | Google Scholar |
47 Zhang X, Li H, Li L, Lu G, Zhang S, Gu L, Xia Y, Huang X. Polyallene with pendant nitroxyl radicals. Polymer 2008; 49: 3393-3398.
| Crossref | Google Scholar |
48 Nguyen TP, Easley AD, Kang N, Khan S, Lim S-M, Rezenom YH, Wang S, Tran DK, Fan J, Letteri RA, He X, Su L, Yu C-H, Lutkenhaus JL, Wooley KL. Polypeptide organic radical batteries. Nature 2021; 593: 61-66.
| Crossref | Google Scholar | PubMed |
49 Rial-Rodríguez E, Williams JD, Eggenweiler H-M, Fuchss T, Sommer A, Kappe CO, Cantillo D. Development of an open-source flow-through cyclic voltammetry cell for real-time inline reaction analytics. React Chem Eng 2024; 9: 26-30.
| Crossref | Google Scholar |
50 Eberhardt M, Mruk R, Zentel R, Théato P. Synthesis of pentafluorophenyl(meth)acrylate polymers: new precursor polymers for the synthesis of multifunctional materials. Eur Polym J 2005; 41: 1569-1575.
| Crossref | Google Scholar |
51 Gitzhofer E, Vileno B, Bouquey M, Chan-Seng D. Model reactions for the evaluation of poly- and multifunctional molecules as potential interfacial agents for the compatibilization of polyethylene/poly(ethylene-co-vinyl alcohol) blends. Polym Chem 2023; 14: 934-942.
| Crossref | Google Scholar |
52 Lin C-G, Hutin M, Busche C, Bell NL, Long D-L, Cronin L. Elucidating the paramagnetic interactions of an inorganic–organic hybrid radical-functionalized Mn-Anderson cluster. Dalton Trans 2021; 50: 2350-2353.
| Crossref | Google Scholar | PubMed |
53 Moffat KA, Hamer GK, Georges MK. Stable free radical polymerization process: kinetic and mechanistic study of the thermal decomposition of MB-TMP monitored by NMR and ESR spectroscopy. Macromolecules 1999; 32: 1004-1012.
| Crossref | Google Scholar |
54 Ostfeld D, Cohen IA. A cautionary note on the use of the Evans method for magnetic moments. J Chem Educ 1972; 49: 829.
| Crossref | Google Scholar |