

Approach to achieve controlled particle size synthesis of non-polar functionalised siloxane particles using a one-pot synthesis
Rowan McDonough A , Daniel Mangos A , Chris Hassam A , Jonathan Campbell A and David A. Lewis A *
A , Daniel Mangos A , Chris Hassam A , Jonathan Campbell A and David A. Lewis A *
A
Australian Journal of Chemistry 76(10) 719-729 https://doi.org/10.1071/CH23128
Submitted: 30 June 2023 Accepted: 26 September 2023 Published: 20 October 2023
Abstract
The homogeneous addition of functional silanes dissolved in a co-solvent such as ethanol has been shown to change particle growth mechanisms as well as control over particle size and dispersity, especially for non-polar silanes. Highly monodisperse size-controlled silica particles from 50 to over 1000 nm bearing thiol, vinyl, phenyl, propyl, cyanopropyl and chloropropyl R group functionality were synthesised directly from the corresponding single organosilane source. Observing particle growth over time revealed that it proceeded by a conventional Stöber process, where silane monomers and oligomers add to nucleation sites, resulting in particle growth. Scale-up of the homogeneous mixing approach produced gram quantities of particles without affecting size, size dispersion or functionality under good mixing. The presence and functionality of the surface R groups were confirmed by a simple second addition of reactive molecules to the particle synthesis liquor, resulting in theoretical maximum attachment densities.
Keywords: click, functionalisation, homogeneous, interfacial, monodisperse, nanoparticle, non-polar siloxane, one-pot, organosilane, size control, Stöber process.
Introduction
Silica nanoparticles prepared from tetraethyl orthosilicate (TEOS) are advantageous owing to their biological inertness and straightforward synthesis, providing consistent and controllable size and size distribution.1–5 However, the range of possible applications with this platform is constrained by the challenges in tailoring the surface chemistry to suit specific applications, owing to the limited reactivity of the hydroxysilanes at the surface.5 To overcome this challenge and introduce functionality, a core–shell particle can be obtained through the reaction of a functionalised siloxane, such as mercaptopropyltrimethoxysilane (MPTMS); however, this requires additional steps and can cause loss of control of the particle size and uniformity.5–8 The direct formation of particles with reactive functionalities can be achieved using trifunctional siloxane precursors such as MPTMS; however, solubility is a major challenge.5,9–11
The Stöber process has better size control and can achieve smaller particle sizes than the alternative stabilised emulsion approach. It also has the advantage that it avoids the use of surfactants that are difficult to remove after synthesis and can interfere with subsequent surface modification.
Both the Stöber and stabilised emulsion approaches rely on the siloxane monomer undergoing hydrolysis and condensation reactions, as shown in Scheme 1, with a strong dependence on pH of the relative rate of the reactions and the structures that are formed.12

Although conceptually simple and quite straightforward for TEOS, the Stöber process is based on a nucleation and growth mechanism in which the reactants remain soluble through the initial hydrolysis and condensation until, effectively, the number of nucleating species becomes constant and any further condensation is onto the particles, which ideally remain dispersed until the reaction is complete.13 By having a relatively fast nucleation process compared with particle growth, monodisperse particles are grown, the particle size increases uniformly with conversion and final particle size can be controlled by both conversion and initial reaction concentration.5,10 The limited solubility of the monomer in the aqueous alcohol solvent system necessitates the reaction solutions being fairly dilute, leading to large amounts of solvent, and the reaction can be very sensitive to a wide range of parameters, resulting in variable sizes and distributions.
The ability to apply this method to the simple preparation of a vast range of particles with different chemical surface functionalities is highly desirable because the external surface chemistry of the particle controls the end-use functional properties such as dispersion and interactions with surroundings.10,11,14 MPTMS nanoparticles have been shown to be quite straightforward to synthesise through a modified Stöber process9 as well as a stabilised emulsion approach,11 and these are suitable for subsequent modification.10 However, for less-soluble siloxane monomers such as those containing vinyl or cyano groups, direct synthesis of functional nanoparticles poses a significant challenge owing to the poor solubility of the silane in the aqueous reaction media, relying on the hydrolysis reaction of the siloxane to improve solubility, which can result in uncontrolled particle growth. Scale-up of the synthesis of these particles is also a significant challenge owing to the solubility limitations.
Conventional approaches to the Stöber method of adding the siloxane to the aqueous mixture result in a two-phase structure, which is often difficult to observe, especially with small reaction volumes. Interfacial hydrolysis increases the solubility of the silane in the water–ethanol–NH3 reaction mixture. For less polar silanes, this can result in either an emulsion forming under shaking, which does not allow particle size control or in limited conversion owing to insolubility slowing the reaction rate, which also limits particle size. This is a major impediment to the scale-up of the synthesis of particles from less polar silanes.
In this paper, we demonstrate that through careful control of the one-pot synthetic procedure, narrow-dispersity controlled growth particles from non-polar silanes can be achieved. We subsequently demonstrate a more robust modified addition procedure that avoids the limitations of interface formation and provides a pathway to scaling up the synthesis.
Both of these high-yielding particle synthesis methods can provide a broad range of consistent and predictable particle sizes, typically ranging from 100 nm to 1 µm, which can be controlled simply by alteration of the concentrations of reactants. This was demonstrated by producing thiol, vinyl, phenyl, propyl, cyanopropyl and chloropropyl R group-functionalised particles solely from the corresponding organosilane. The more consistent and controllable silane mixing in the homogeneous approach facilitated the reaction size being increased to litre scales, producing gram quantity of particles without loss of particle quality. 29Si–1H cross-polarisation NMR showed a high degree of condensation of the particles, demonstrating structural rigidity and suitability for use in further applications. The retention of surface functionality of the scaled-up particles was confirmed by attaching and quantifying attachment of fatty acids with thiol (11-mercaptoundecanoid acid, 11-MUA) and vinyl (undecylenic acid) to the surface of vinyl and thiol-terminated particles respectively by thiol-ene ‘click’ chemistry.
Results and discussion
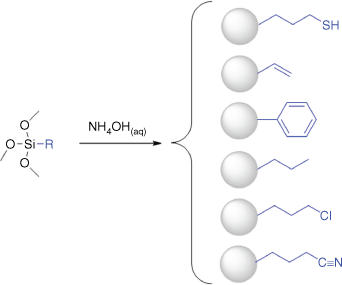
Hydrophobic R group-terminated particles were produced directly from the corresponding organosilane, using a similar approach to that for the synthesis of thiol-terminated particles reported previously.5,9–11 The general approach is shown in Scheme 2 with the different hydrophobic organosilanes and accessible surface functionalities. This was achieved using two different approaches for mixing the hydrophobic silane with the aqueous phase, as shown in Scheme 3.
Schematic of the universal one-pot method for the synthesis of hydrophobic functional particles directly from a single silane where R groups are represented in blue on right‐hand side. Adapted from D. Mangos’ PhD thesis. 15
Reaction schematic of the two one-pot single silane synthesis methods for hydrophobic particles using vinyltrimethoxysilane (VTES) as an example.
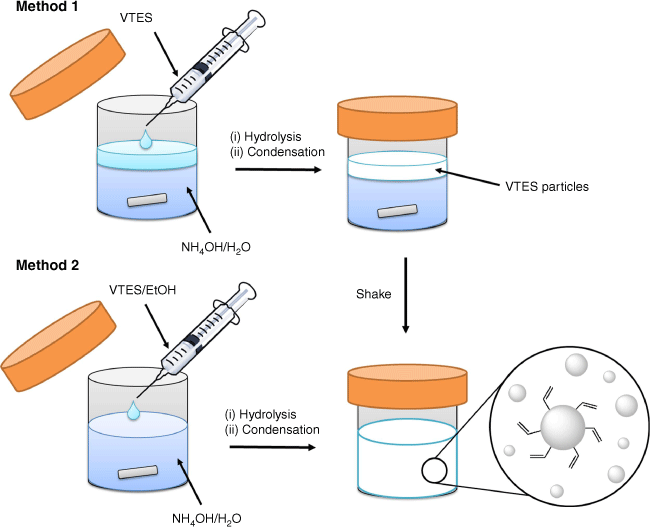
Method 1: conventional addition approach
In the conventional method, the organosilane is added directly to the aqueous ammonium hydroxide solution (Scheme 3, Method 1). Adding the silane carefully by syringe as a discreet immiscible layer either above or below the aqueous solution (depending on silane density) was found to give more consistent particle formation with less large agglomerates compared with a less careful approach. Particles form within the silane layer over a matter of minutes as evidenced by the immiscible silane layer turning milky opaque white. The solution was then vigorously shaken to disperse the particles within the aqueous phase and stirred for up to a further 3 days.
Particle growth over time was monitored by dynamic light scattering (DLS) and scanning electron microscopy (SEM) from each functional silane and is shown in Fig. 1.
Growth of particles from functional monomers (PTMS, VTES and CNpropTES) and respective sizes and maturity times, as measured by DLS and SEM.
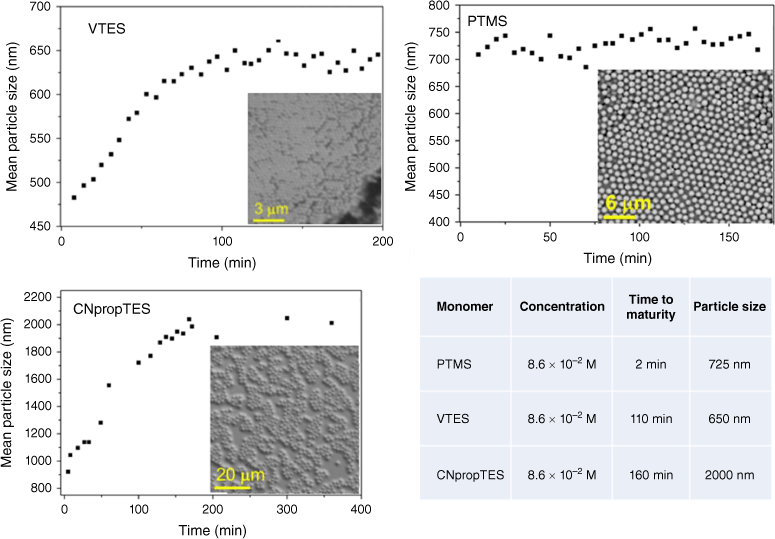
VTES and cyanopropyltriethoxysilane (CNpropTES) showed a linear increase in particle size to ~600 and 2000 nm with time over 100 and 200 s respectively, consistent with a nucleation–growth Stöber mechanism.16
The particle size with phenyltrimethoxysilane (PTMS), however, did not appear to change in size after the initial 5 min when particles appeared, indicative of an unstabilised emulsion-based process. Hydrolysis of PTMS results in an amphiphilic monomer, with self-emulsification of the hydrolysed monomer resulting in the rapid establishment of an oil-in-water emulsion in which the silane monomer can continue to hydrolyse and poly-condense to form rigid spheres.11
Particle synthesis reactions using the conventional mixing approach were performed on a 50-mL scale where the addition of the hydrophobic organosilane as a stable, immiscible layer was fairly straightforward and reproducible. However, the silane layer tended to adhere to the walls of the reaction vessel, resulting in the formation of agglomerates that had to be filtered out after particle formation, affecting the predictability of the particle size remaining as a dispersion. At larger reaction scales, addition of the silane as a discrete, stable immiscible layer proved challenging, with it often separating into droplets, resulting in poor size control and poor size uniformity.
Method 2: homogeneous mixing approach
In the homogeneous mixing approach, the organosilane is first dispersed in ethanol before being slowly added to the stirred aqueous ammonia solution (Scheme 3, Method 2). A milky solution (opaque white) was observed within minutes (>80 mM of VTES) to 7 days (<40 mM of VTES) of mixing, as solid particles formed. The dispersed silane–ethanol mixture remained homogeneous in the aqueous alcohol solvent until evidence of particle formation. This method was also applied to phenyltriethoxysilane (PTES), propyltrimethoxysilane (PropTMS) and chloropropyltriethoxysilane (ClpropTES).
Like the conventional method, the homogeneous mixing method provided silane concentration-dependent size control. Fig. 2 shows the increase in the size of the particles obtained with increasing concentration of the different hydrophobic organosilane precursors, with example SEM images of particles produced at silane concentrations of 20 and 200 mM. At silane concentrations below 20 mM, no particle formation was detected with VTES and CNpropTES.
Example SEM images of showing increase in size of particles as silane concentration is increased. Error bars are standard deviation of mean particle size.
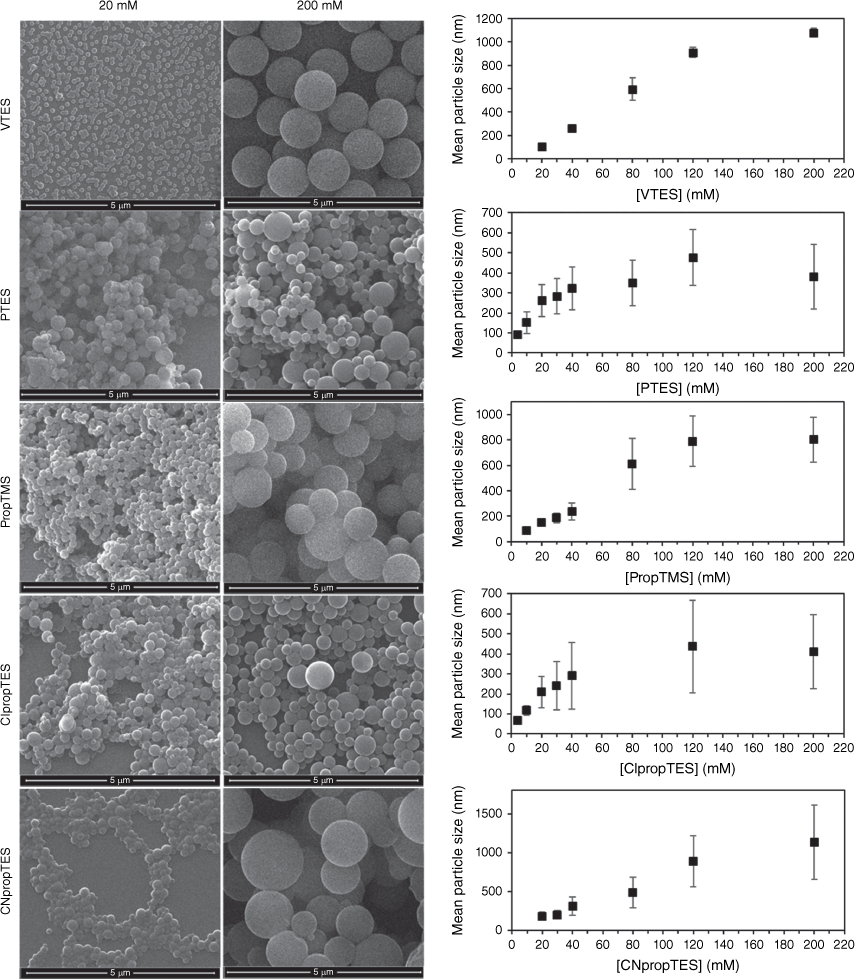
In the conventional addition method, particle sizes only as small as ~200 nm could be achieved at full conversion, whereas the homogeneous mixing method allowed <100-nm nanoparticles to be accessed for all particle types except the cyanopropyl particles, as shown in Fig. 2.
When the reaction was performed on a 1-mL scale, the particle size dispersion was low at low silane concentration and increased with increasing silane concentration. This observed poor size uniformity at high silane concentrations and small reaction scale demonstrates the importance of consistent mixing in obtaining uniform particles.
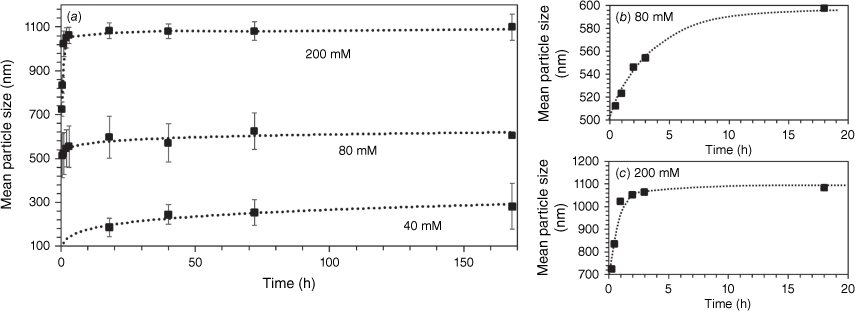
The rate of growth of VTES particles was strongly dependent on concentration, as shown in Fig. 3. At high VTES concentration (200 mM), particles are formed within minutes from hydrolysis and condensation of large nuclei, which rapidly grow by addition of further VTES monomers and oligomers to form solid spheres with a diameter of 725 nm within 15 min of reaction, as seen in Fig. 4. These particles continue to grow rapidly, resulting in a monodisperse dispersion of 1080-nm particles after just 4 h of reaction. At moderate VTES concentration (80 mM), slower particle growth is observed, with a maximum size of 600 nm reached after 18 h. At low VTES concentration (40 mM), solid particle formation (186 nm) is not observed until 18 h of reaction, with fully formed particles with a diameter of 280 nm produced after 7 days of reaction. The growth kinetics are consistent with the conventional mechanism where particle growth follows a monomer addition or aggregation growth model, resulting in the uniform growth of nuclei until all monomer in the solution is depleted.
(a) Mean particle size by SEM analysis of the growth of vinyl particles synthesised at different VTES concentrations over time magnified for the first 18 h for (b) 80 mM, and (c) 200 mM of VTES. Error bars are standard deviation of mean particle size.
SEM images of the growth of vinyl-terminated particles prepared with 200 mM of VTES over time.

It should be noted that all silanes examined, including PTES, displayed time-dependent growth, indicating a Stöber mechanism, which was contrary to the results observed from the conventional addition approach. It is hypothesised that this is due to the more homogeneous hydrolysis of the silanes resulting in greater solubility compared with interfacial processes in the conventional addition case. Similarly, the smaller dispersity and consistent size can be attributed to a fast nucleation step compared with particle growth.
To produce large amounts of nanoparticles for industrial or commercial applications, it is important for a method of nanoparticle preparation to be scalable.4 The scale of a particle synthesis reaction can significantly affect the dispersity and shape of the resultant particles and poses a significant challenge.
In the conventional mixing method, the immiscible oil phase was found to be less stable in large volumes, resulting in partitioning of the silane phase at the air surface or the walls of the reaction vessel. As a result, the particle size achieved was not predictable and less uniform owing to the effective concentrations being inconsistent between the droplets of the different phases.
Pre-mixing the silane in ethanol was found to make the silane addition significantly more consistent and controllable, resulting in much more reliable and reproducible particle sizes and uniformity. The presence of 10% ethanol in the reaction solution also significantly increased the solubility of the silane and formed particles, minimising the production of aggregate structures.
To investigate the scalability of the homogeneous particle preparation method, vinyl and phenyl-terminated particles were synthesised at a silane concentration of 200 mM in reaction volumes from 1 to 100 mL. Ammonia and ethanol concentrations were held at 0.64 and 3.4 M respectively. Fig. 5 shows that vinyl and phenyl-terminated particles with very broad size distributions were synthesised at the 1-mL reaction scale; however, increasing the reaction volume to 10 mL resulted in a significant reduction in size distribution of both vinyl and phenyl-terminated particles and also increased the average size. As the scale was further increased to 100 mL, the particles became increasingly monodisperse. The large size distribution of particle sizes produced at a 1-mL reaction scale is attributed to inhomogeneous mixing within the 2-mL Eppendorf tube reaction vessel, owing to the small stirrer bar at bottom of tube not creating sufficient or consistent mixing turbulence. When the silane concentration is low, small particles condense over a period of days and inhomogeneous mixing is less detrimental to the size distribution as the components are mixed and added to the growing particle very slowly by diffusion. However, at high silane concentrations, where particles form within minutes, homogeneous mixing is critical for the consistent addition and condensation of silane monomers and oligomers to the growing particle. Inhomogeneous mixing results in a concentration gradient of the silane monomers, oligomers and growing particles, leading to different growth rates in particles and subsequently a broad range of particle sizes.
Reaction scale effect on size and size distribution of vinyl and phenyl-terminated particles. Error bars are standard deviation of mean particle size.

The scalability of the synthesis of thiol-terminated particles was also investigated to produce large quantities of particles (Table 1). The samples all showed similar Z-average diameters and low polydispersity index (PDI) values up to a 3-L scale. This indicated that the particle diameters remained controlled, and that the formation of aggregates or large particles was minimised. This suggested that the homogeneity of the reaction mixture was no longer an issue even at increased scales and allowed synthesis of large batches of particles.
| Reaction volume (mL) | Z-average diameter (nm) | PDI | |
|---|---|---|---|
| 40 | 172 | 0.158 | |
| 400 | 157 | 0.03 | |
| 800 | 165 | 0.023 | |
| 1000 | 164 | 0.034 | |
| 2000 | 176 | 0.048 | |
| 3000 | 164 | 0.026 |
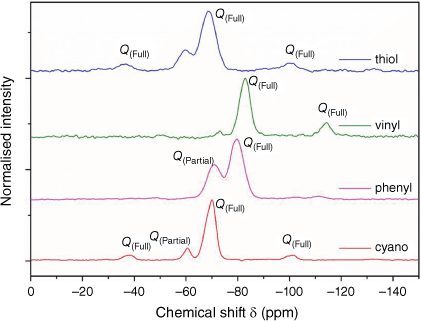
Cross-polarisation 29Si–1H solid state NMR was used to investigate the degree of condensation of the different particle systems. The silicon nuclei are either fully condensed (QFull) or partially condensed (QPartial), where one silanol arm on the monomer has not completely condensed, only forming two Si–O–Si bonds, and leaving an unreacted Si–OH bond.
Fig. 6 shows the cross-polarisation NMR of several species of silica particles where the chemical shift (resonant frequency) and therefore, relaxation time for silicon nuclei in Si–O–Si and Si–OH states varies quite significantly, depending on the nature of the ‘organo’ functional group. For the propyl-functionalised silanes such as the cyano and thiol, there are similar shifts for the silicon nuclei, where the equidistant satellites that are symmetrical around the Q(Full) peak are a product of the cross-polarisation process. Quantitative information regarding the condensation states for these silanes is shown below in Table 2. In order to quantitatively integrate the condensation states shown by NMR, a contact time of approximately three times the relaxation time of the least sensitive nucleus was required.
29Si cross polarisation NMR of thiol, vinyl, phenyl and cyano functionalised particles; Q(Full) refers to a completely condensed silicon atom, and Q(Partial) refers to the silicon atom bonded to one OH group (Si–OH).
| Monomer | Chemical environment | Chemical shift (ppm) | Integration | Degree of condensation (%) | |
|---|---|---|---|---|---|
| MPTMS | Si–O–Si | 68.6 | 11.4 | 85 | |
| Si–OH | 59.7 | 2.7 | |||
| VTES | Si–O–Si | 83.0 | 1.0 | 100 | |
| Si–OH | ND | – | |||
| PTMS | Si–O–Si | 79.7 | 36.0 | 91 | |
| Si–OH | 80.0 | 13.0 | |||
| CNpropTES | Si–O–Si | 70.1 | 18.0 | 90 | |
| Si–OH | 60.6 | 2.0 |
ND, not detected.
The order of most to least condensed systems based on quantitative integration was shown to be vinyl > cyano > thiol > phenyl. There are a variety of different factors that could affect the reactivity and condensation rates of the silanol groups on the functionalised silica precursors, including the impact of the functionalisation on the relative rates of the initial, secondary and tertiary condensation reactions and the relative solubility.
The presence of functional R groups in the mature particles was confirmed by ATR-FTIR (attenuated total reflectance FTIR) by identification of characteristic absorption peaks (see the ‘ATR-FTIR analysis of particles’ section in the Supplementary material) and thermogravimetric analysis (TGA) by close correlation between the experimental and the expected theoretical mass loss for each species (see the ‘TGA analysis of particles’ section in the Supplementary material).
To demonstrate the retention of functionality of the particles grown from a single silane source, as well as the utility of specifically and controllably adding new surface functionality, long-chain fatty acids were attached to the surface of thiol and vinyl-terminated particles. For the thiol-terminated particles, this was achieved using a one-pot method, where 10-undecylenic acid was directly grafted onto particles by a thiol-ene ‘click’ reaction within the particle synthesis reaction mixture, as shown schematically in Scheme 4. 11-MUA was attached to the surface of vinyl-terminated particles that were separated and washed after preparation. The attachment density of these molecular surface decorations was determined by the quantitative ATR-FTIR technique reported by Mangos et al.10 See the ‘Construction of calibration curves for surface attachment density’ section in the Supplementary material for details on calibration curve construction.

Attachment of undecylenic acid to thiol-terminated particles
Undecylenic acid was attached directly to the thiol-terminated particles within the particle synthesis reaction vessel, without prior cleaning and redispersion of the particles. Fig. 7 shows the ATR-FTIR spectrum of washed and dried undecylenic acid-functionalised particles after the thiol-ene ‘click’ reaction, with a peak at ~1700 cm−1 due to the C=O stretch of the undecylenic carbonyl group. This shows that, using this preparation method, it is possible to rapidly produce particles with high densities of a wide range of potential functionalities in little as a few days with minimal workup. Using the ATR-FTIR quantification method, the surface attachment density of undecylenic on the thiol particle was found to be 5.7 surface groups nm–2, which is at the upper limit of available surface sites.17,18
ATR-FTIR spectra of COOH groups (5.7 undecylenic acid attachments nm–2) normalised to the Si–O–Si signal.
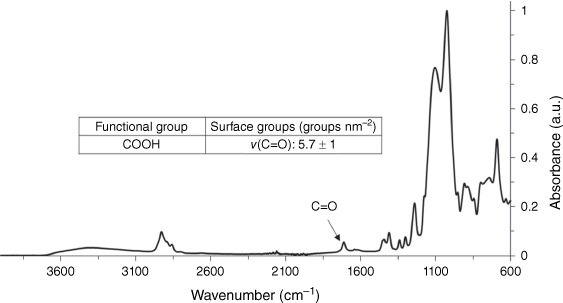
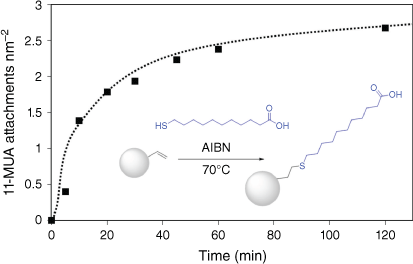
Fig. 8 shows the increase in 11-MUA attachment over 120 min of reaction time, where the reaction appears to begin to plateau. This may be due to the relatively small excess of 11-MUA compared with vinyl groups on the particle surface, equating to a 100-fold excess, assuming 5 vinyl groups nm–2 as in the case of thiol-terminated particles. An attachment density of 2.7 11-MUA is achieved after 2 h of reaction. By increasing both reaction time and 11-MUA excess, it is likely that attachment densities of ~5 11-MUA nm–2 could be achieved. However, this demonstrates that, like the thiol particles, the terminal vinyl groups can also be chemically accessed and modified with thiol groups by thiol-ene ‘click’ chemistry.
11-MUA attachment to the surface of 200-nm vinyl-terminated particles by thiol-ene ‘click’ reaction tracked by quantitative ATR-FTIR analysis; (inset) reaction scheme for attachment of 11-MUA to vinyl-terminated particles.
These results demonstrate that the methods reported in this work enable scaled-up, one-pot and size-controllable synthesis of monodisperse particles with hydrophobic R group from a single silane source that retain their functionality and can be further modified to give fine control over surface functionality.
Conclusion
Two approaches of mixing hydrophobic silanes with the aqueous reaction mixture to form functionalised and easily modified particles using a range of non-polar silane monomers without affecting quality or functionality are explored:
conventional mixing, where addition results in an initially immiscible layer of the silane above or below the aqueous phase (depending on the density of the silane);
homogeneous mixing, where the silane is pre-mixed with a co-solvent (ethanol) to increase the solubility of the organosilane in the aqueous phase and particles form directly within the aqueous media.
Both of these high-yielding particle synthesis methods can provide a broad range of consistent and predictable particle sizes typically ranging from 100 nm to 1 µm that can be controlled simply by alteration of the concentrations of reactants.
The method of addition of functional silanes was demonstrated to have a significant effect on both the mechanism of particle growth and control over particle size and dispersity. By premixing the silane in ethanol prior to addition to the reaction mixture, the formation of a biphasic system was avoided and low-dispersity particles bearing thiol, vinyl, phenyl, propyl, cyanopropyl and chloropropyl R group functionality could be prepared directly from the corresponding organosilane using a simple one-pot method following a Stöber nucleation–growth mechanism. Particle size could be controlled by both initial silane concentration and reaction time, enabling a very wide range of particle sizes to be accessed.
Although careful addition of the pure silane to the reaction mixture can result in fairly low-dispersity particles, less-polar silanes such as PTMS formed an unstabilised emulsion, with loss of control over particle size and dispersity. The homogeneous addition overcame this issue, enabling a much wider range of particle sizes to be accessed in a more controllable way.
Consistent mixing of the silane in the homogeneous silane addition approach allowed the reaction to be scaled to produce gram-scale quantities of the particles without loss of size control, size uniformity or surface functionality.
Retention of surface functionality was demonstrated by attaching long chain fatty acids to the surface of thiol and vinyl-terminated particles. This could be achieved by simple addition of the fatty acid to the particle synthesis liquor, without clean-up steps using thiol-ene ‘click’ chemistry. Surface attachment was determined by quantitative ATR-FTIR to be 5.7 undecylenic acid groups nm–2 on the thiol-terminated particles and 2.7 11-MUA nm–2 on the vinyl terminated particles.
This demonstrates that the one-pot method can be used to simply produce large quantities of particles that can rapidly and controllably be post-functionalised to give a wide range of useful surface functional properties.
Experimental
Synthesis of hydrophobic particles
Stable colloidal suspensions of vinyl, phenyl and cyanopropyl-functionalised particles were prepared by careful dropwise addition of VTES, PTMS and CNpropTES (14–340 mM) as a discreet immiscible layer to a solution of 28% ammonia (3.60 mL) in deionised water (46.4 mL). The solution was stirred gently at 300 rpm so as to not disturb the layers for 30 min before being shaken and stirred vigorously at 800 rpm for 3 h.
For preparation of thiol, vinyl, propyl, phenyl, chloropropyl and cynaopropyl-functionalised silica nanoparticles of different particle sizes, the concentration of MPTMS, VTES, PropTMS, PTES, ClpropTES and CNpropTES was altered between 4 and 200 mM in 3.4 M of ethanol and 0.64 M of NH4OH. Mixtures were stirred for 7 days and 1-mL samples were taken at different time points. Samples were washed three times with 1 mL of ethanol followed by centrifugation and dried in a vacuum oven (Memmert VO400) (1 day, 70°C, 2000 Pa).
AIBN (0.2 mmol, 40 mg) was dissolved in ethanol (99.8%, 20 mL) and undecylenic acid (3.4 mmol, 0.7 mL) was then added to the aqueous dispersion of thiol-terminated particles (250 mL) and heated under reflux. After 2 h, the solution was returned to room temperature and sodium chloride solution (6 M, 20 mL) was added to the solutions of COOH particles to induce particle aggregation. After the particles collapsed, the sample was filtered under vacuum through a Sartorius Stedim filter (0.45 μm, PTFE).
Vinyl particles with an average diameter of 200 nm (100 mg) were mixed with 11-MUA (0.36 mmol, 80 mg) with an excess of 100 11-MUA nm–2 of vinyl particle surface with AIBN (0.05 mmol, 10 mg) in acetone and heated to 70°C. Aliquots were taken at different times over 120 min to track the reaction. The aliquots were washed thoroughly (eight times) with acetone followed by centrifugation and then dried for ATR-FTIR analysis.
Fourier-transform infrared spectroscopy
FTIR spectra of particles were measured using a Nicolet 870 spectrometer with an ATR diamond crystal from 400 to 4500 cm−1.
Dynamic light scattering
DLS measurements were performed using a Malvern Zetasizer ZS in a non-invasive backscattering (NIBS) configuration. All runs were performed in triplicate in monomodal mode, at 25°C, in 1-cm path length cuvettes. Attenuator index, path length, number of scans and mean position were determined automatically in NIBS configuration.
Scanning electron microscopy
Silicon wafers (University Wafer, P/B, SSP, 100 orientation) were cleaned by sonication in ethanol, then rinsed with ethanol and dried under a stream of N2 gas. The particle solutions were then drop-cast onto the wafers and allowed to dry. The samples were placed in a vacuum oven and dried at 60°C for at least 1 h at 2000 Pa, to ensure removal of solvent. Before measurement, the particle samples were coated with a 2-nm platinum film using a Quorumtech K575X sputter coater. The measurements were made using an Inspect FEI F50 SEM.
Acknowledgements
D. A. Lewis is the recipient of 2022 RACI Weickhardt Medal for Distinguished Contribution to Economic Advancement.
References
1 Gao W, Rigout M, Owens H. The structural coloration of textile materials using self-assembled silica nanoparticles. J Nanopart Res 2017; 19(9): 303.
| Crossref | Google Scholar |
2 Bhakta S, Dixit CK, Bist I, et al. Sodium hydroxide catalyzed monodispersed high surface area silica nanoparticles. Mater Res Express 2016; 3(7): 075025.
| Crossref | Google Scholar | PubMed |
3 Li SB, Wang J, Zhao S, et al. Effect of surface modification and medium on the rheological properties of silica nanoparticle suspensions. Ceram Int 2016; 42(6): 7767-7773.
| Crossref | Google Scholar |
4 Hoang CV, Thoai DN, Cam N, et al. Large-scale synthesis of nanosilica from silica sand for plant stimulant applications. ACS Omega 2022; 7(45): 41687-41695.
| Crossref | Google Scholar | PubMed |
5 Nakamura M, Ishimura K. One-pot synthesis and characterization of three kinds of thiol-organosilica nanoparticles. Langmuir 2008; 24: 5099-5108.
| Crossref | Google Scholar | PubMed |
6 Takeda Y, Komori Y, Yoshitake H. Direct Stöber synthesis of monodisperse silica particles functionalized with mercapto-, vinyl- and aminopropylsilanes in alcohol–water mixed solvents. Colloids Surf A Physicochem Eng Asp 2013; 422: 68-74.
| Crossref | Google Scholar |
7 Effati E, Pourabbas B. One-pot synthesis of sub-50 nm vinyl- and acrylate-modified silica nanoparticles. Powder Technol 2012; 219: 276-283.
| Crossref | Google Scholar |
8 Qhobosheane M, Santra S, Zhang P, et al. Biochemically functionalized silica nanoparticles. Analyst 2001; 126(8): 1274-1278.
| Crossref | Google Scholar | PubMed |
9 Toster J, Lewis D. Investigation of roughness periodicity on the hydrophobic properties of surfaces. Aust J Chem 2015; 68: 1228-1232.
| Crossref | Google Scholar |
10 Mangos DN, Nakanishi T, Lewis DA. A simple method for the quantification of molecular decorations on silica particles. Sci Technol Adv Mater 2014; 15(1): 015002.
| Crossref | Google Scholar |
11 Lu Z, Sun L, Nguyen K, Gao C, Yin Y. Formation mechanism and size control in one-pot synthesis of mercapto-silica colloidal spheres. Langmuir 2011; 27: 3372-3380.
| Crossref | Google Scholar | PubMed |
12 Stöber W, Fink A, Bohn E. Controlled growth of monodisperse silica spheres in the micron size range. J Colloid Interface Sci 1968; 26: 62-69.
| Crossref | Google Scholar |
13 Han Y, Lu Z, Teng Z, et al. Unraveling the growth mechanism of silica particles in the Stöber method: in situ seeded growth model. Langmuir 2017; 33(23): 5879-5890.
| Crossref | Google Scholar | PubMed |
14 Lim J-H, Ha S-W, Lee J-K. Precise size-control of silica nanoparticles via alkoxy exchange equilibrium of tetraethyl orthosilicate (TEOS) in the mixed alcohol solution. Bull Korean Chem Soc 2012; 33(3): 1067-1070.
| Crossref | Google Scholar |
16 Belton DJ, Deschaume O, Perry CC. An overview of the fundamentals of the chemistry of silica with relevance to biosilicification and technological advances. FEBS J 2012; 279(10): 1710-1720.
| Crossref | Google Scholar | PubMed |
17 Zhuravlev LT. The surface chemistry of amorphous silica. Zhuravlev model. Colloids Surf A Physicochem Eng Asp 2000; 173: 1-38.
| Crossref | Google Scholar |
18 Davydov VY, Kiselev AV, Zhuravlev LT. Study of the surface and bulk hydroxyl groups of silica by infrared spectra and D2O-exchange. Trans Faraday Soc 1964; 60: 2254-2264.
| Google Scholar |