

History and fundamentals of molecular photochromism
David Jago A , Emma E. Gaschk A and George A. Koutsantonis A *
A , Emma E. Gaschk A and George A. Koutsantonis A *
A Chemistry, School of Molecular Science, The University of Western Australia, Crawley, WA 6009, Australia.
Australian Journal of Chemistry - https://doi.org/10.1071/CH23115
Submitted: 20 June 2023 Accepted: 2 August 2023 Published online: 6 September 2023
© 2023 The Author(s) (or their employer(s)). Published by CSIRO Publishing. This is an open access article distributed under the Creative Commons Attribution 4.0 International License (CC BY)
Abstract
Photochromic molecules reversibly change their colour upon exposure to light. The increasing need for smart materials in the real world, coupled with progress in synthetic chemistry, fast spectroscopic techniques, and theoretical power in research laboratories, have seen research in organic photochromism accelerate over the past few decades. In this Primer Review, the topic of organic photochromism is introduced. The fundamental concepts and histories are given to contextualise this field. Moreover, key photochromic molecules and selected applications are showcased to provide the interested reader with an entry to this fascinating field of science and emerging technology.
Keywords: molecular devices, nanotechnology, organic, photochemistry, photochromism, photoswitch, Primer Review, smart materials, switching.
Introduction
The exponential growth in research on photochromic molecules can be attributed to the increasing demand for stimuli-responsive molecular switches in organic electronics and smart materials. The development of dynamic systems incorporating molecular photoswitches has rapidly advanced our ability to control properties and functions. Photochromic molecules have been explored for a wide range of applications, including optoelectronic switches and data storage, biomolecules and biomimicry, imaging and detection, surface functionalisation, catalysis, ion sensing, and drug delivery and photopharmacology.1 Utilising light as a stimulus offers several advantages, including precise directional and wavelength control, the ability to probe closed systems and reduced risk of chemical contamination.
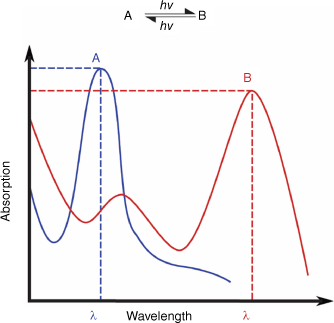
Photochromism is the reversible change of a material between two different states in response to light.2 The term ‘photochromism’ is derived from the Greek words ‘phos’ (light) and ‘chroma’ (colour). Although these molecules exhibit distinct absorption spectra (Fig. 1), the photochemical change can also lead to various other changes in chemical and electronic properties, such as emission, dipole moment, dielectric field constant, refractive index, non-linear optics, energy transfer, redox properties, conductivity, molecular structure and reactivity. It is these property changes that make photochromic materials appealing for light-responsive smart materials.
Photochromism is the reversible change of a material between two different states (A and B) in response to light, with each having its own distinct absorption spectrum.
The study of photochromism is an interdisciplinary field that encompasses concepts from organic chemistry, materials science and physics.1 Scientists from these disciplines investigate the design and synthesis of new photochromic compounds, tailor their properties, and characterise their photoresponsive behaviour using analytical techniques such as UV–Vis and time-resolved spectroscopy, X-ray crystallography, and theoretical calculations (including quantum chemistry and molecular dynamics simulations).
This Primer Review aims to provide an overview of the fundamental principles of photochromism in organic molecules, discuss key performance factors to consider when selecting photochromes for smart materials, describe the main classes of photochromes and showcase selected applications that highlight the range of tasks accessible. This Primer Review will focus on areas of academic interest in the context of organic photochromic molecules. It is salient to note that photochromic molecules are used in phototransition lenses, which are a lucrative commercial product, with sales in billions of dollars per year globally. These commercial products rely on an inorganic photochromic system consisting of silver halide crystals in a vitreous environment where light causes the electrons to move from the valence band to the conduction band.2 The academic pursuit of new systems is important to extend and to improve on the applications available to photochromic molecules.
History of organic photochromism
This historical account will cover principal organic compounds used to investigate photochromism and main mechanistic insights until the discovery of the main photochromic families used in the literature. Fig. 2 illustrates the timeline of photochromism up until the early 21st century.
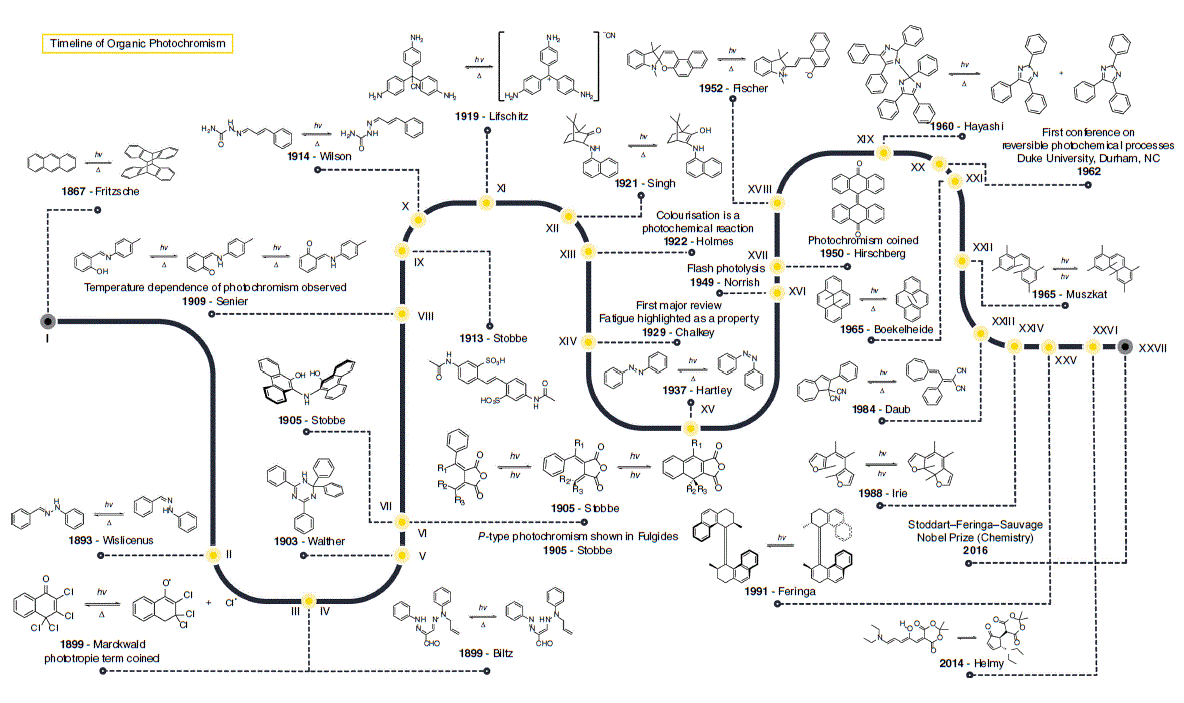
In 1867, the formation of microscopic colourless crystals were reported when a solution of anthracene was exposed to sunlight and it noted that anthracene could be regenerated by heating the melt of the molecule (I).3 The essential role that light had in turning benzaldehydephenylhydrazone (II) red and its gradual reversal in the dark was noted in the late 19th century.4 In 1899, the reversible colour change of 2,3,4,4-tetrachloro-4H-naphthalen-1-one (III) and the anhydrous hydrochloride of benzo-1,8-naphthyridine was also demonstrated.5 This physical phenomenon was termed phototropy5; however, this term is now used to describe the growth or alignment of organisms towards light. In the same year, the photochromism of osazones was also reported (IV).6
After these early descriptions of photochromic phenomena, further studies in the early 20th century were associated with the synthesis of new molecules and exploring their interaction with radiation. The first review of this area described over 200 photochromic molecules.7 However, there was very little commentary about the physical mechanism operating in these examples of photochromism.
The discovery of other photochromes continued to be reported. Thus, it was noted that 2,2,4,6-tetraphenyl-1,2-dihydro-1,3,5-triazine (V) in the solid state becomes rose coloured when exposed to UV light and back to white in the dark8; however, the mechanism of the photochromism is still unknown.9 The synthesis and photochromism of fulgides was first described in a publication in 1905,10 and the ability to use a different wavelength of light to reverse the initial photochromic response was reported a couple of years later (VI).11 A solution of 10,10′-dihydroxy-diphenylanthryl-9,9′-amine decolourised to pale yellow under exposure to sunlight, and returned to brown–red in the dark, in contrast to other molecules discovered around this time that colourised upon exposure to light (VII).12 The first in the family of photochromic salicylamides (VIII) was reported in 1909.13 The idea of thermochromism and the dependence of photochromism on temperature was also introduced in 1909 (VIII).14 In 1913, the photochromism of the stilbene derivative diacetyl-4,4′-diaminostilbene-2,2′-disulfonic acid (IX) was reported.15 In this case, the molecule switches between light yellow in sunlight and dark red in the dark, and the Group 1 and 2 salts are also photochromic. Around the same time, the unusual photochromic behaviour of cinnamaldehyde semicarbazone (X) was also reported.16 Here, upon exposure of the compound to light and subsequent storage in the dark the compound becomes yellow, but re-exposure to light then bleaches the compound colourless. A few years later, it was reported that the cyano salts of triphenylmethane (XI) become coloured under exposure of UV light and return to colourless in the dark.17 In 1921, the photochromism of napthylamino-camphor (XII) was observed from a colourless to a green solution upon exposure to light, which reverts back in the dark.18 Mechanistically, triphenylmethane sulfonic acids were used to conclude that colourisation was a photochemical phenomenon as the relationship between light intensity and rate of colourisation offered insight into the chemical kinetics of the phenomenon (XIII).19 A review in 1929 introduced the concept of photochemical fatigue based on their survey (XIV).7
By the mid-20th century, photochromism was generally understood as a chemical transformation to a thermodynamically metastable state. Many different mechanisms were starting to be proposed for the library of discovered photochromic molecules, but by the latter half of the century the scientific community was encouraged to build evidence correlating chemical constitution and photochromic mechanism.20 In 1935, evidence was presented for an additional irreversible hydrolysis step in the photolysis mechanism of leucocyanides of triphenylmethane dyes.21 Colorimeter measurements of solutions of the leucocyanides in ethanolic solutions evinced hydrolysis of the cyano to a carbinol in the presence of water. The cis–trans photoisomerisation of azobenzene was serendipitously discovered in 1937 when trying to determine its solubility by using photometric methods (XV).22 The majority of photochromic compounds studied now were discovered after 1950.
Critical in the elucidation of photochemical reactions, including photochromism, was the pioneeering work in flash photolysis that resulted in the development of time-resolved ultrafast spectroscopy (XVI).23,24 In conjunction with the reported photochromism of bianthrones in 1950 (XVII),25 the term ‘photochromism’ was also coined (XVII). In 1952, the photochromism of 1,3,3-trimethylindolinonapthospiropyran was published (XVIII).26 In 1960, it was reported that the compound hexaarylbiimidazole undergoes a rare homolytic cleavage of a C–N bond to give two triphenylimidazolyl radicals (XIX).27 The prominence of photochromism in the literature was cemented with the first conference on reversible photochemical processes at Duke University in Durham (XX). The described photochromism of dihydropyrenes was published in the mid-1960s (XXI).28 The cis-hexamethylstilbene system (XXII) was shown to unambiguously photocyclise to hexamethyldihydrophenanthrene in the mid-1960s.29 This study was pivotal towards the development of the diarylethene photochromic family. The dihydroazulene class of photochromic molecules was introduced in 1984 (XXIII).30 In 1988, it was demonstrated that replacement of the phenyl rings of a stilbene with heterocyclopentene derivatives, to give diarylethenes (XXIV), gave a molecule that switched between an open and closed form only by irradiation with an appropriate wavelength of light.31 The 2016 Nobel Prize32 in Chemistry (XXVII) recognised the importance of sterically overcrowded olefins discovered some 20 years prior (XXV),33 as chiroptical molecular switches.
Advances in spectroscopic and theoretical techniques in the late 20th and early 21st century has given the scientific community tools for the cogent understanding of photochromism. This is best illustrated by the new class of photochromic molecules, donor–acceptor Stenhouse adducts (XXVI), introduced in 2014.34 Stenhouse adducts are a testament to what has been learned about photochromic behaviour and the advances in scientific techniques. In a few short years, fundamental insights into their photochromic behaviour and potential applications have been demonstrated.35
Principle of organic photochromism
Photochromism is, in the majority of cases, a unimolecular photochemical reaction. Therefore, some basic photochemistry is required to study photochromic molecules. We will introduce the topic below in this section and describe how it relates to photochromism. For a rigorous overview of molecular photochemistry, ‘Modern Molecular Photochemistry of Organic Molecules’ is a highly recommended text.36 The general paradigm for a photochemical process is shown in Scheme 1.

The transformation from A to B requires a discrete amount of energy, E = hv, to overcome the potential energy barrier separating them. Here, molecule A absorbs at a particular wavelength and is promoted to an excited state, A*. This excited state then has the potential to transform into B. The process by which A* transforms into molecule B (box in Scheme 1) forms the basis of photophysical and photochemical processes. Some understanding of basic photophysical processes is required before we explore the photochemical processes further.
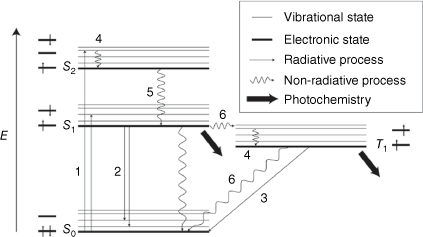
Photochemical or photochromic processes compete with the photophysical processes for excited state deactivation. These photophysical processes are shown in the state diagram, Fig. 3. An electron can be promoted to an excited state from the ground state by absorbing light. This vertical transition is governed by the Franck–Condon (FC) principle, where the nuclear positions do not change during the excitation. A consequence of this structural distortion or non-equilibrium in nuclear positions is the molecule will adapt to the new electronic configuration by molecular motion. This excited electron is metastable and can relax by several methods. These methods are vibrational relaxation, internal conversion (IC), intersystem crossing (ISC), fluorescence and phosphorescence. IC is a non-radiative transition between energy levels of the same multiplicity, and ISC is a non-radiative transition between energy states of different multiplicity. Both IC and ISC involve an energy redistribution and are irreversible. Fluorescence and phosphorescence involve the emission of a photon as an electron transition to a lower energy state. The excited state level and lifetime are factors that determine photochemical transformation. The lowest energy states are associated with photochemical processes as well as the relaxation processes.
State diagram depicting photophysical and photochemical processes. Thick lines are electronic states and are arranged vertically by relative energy (i.e. S0 is singlet ground state) and horizontally by multiplicity (i.e. T1 is triplet first excited state). Vibrational states are shown by thin lines. Radiative processes are depicted by the numbers: (1) Absorption, (2) Fluorescence, and (3) Phosphorescence. Non-radiative processes are depicted by the numbers: (4) Vibrational relaxation, (5) Internal conversion, and (6) Intersystem crossing. The thick arrows depict the states at which photochemical reactions, i.e. photochromism, take place.
Knowledge of photoreaction pathways is crucial to understanding the bond making and breaking of photoexcited molecules. Potential energy surfaces (PESs) are employed to describe the process from reactant to photoproducts through relevant excited states. A simplified PES is illustrated in Fig. 4. Ultrafast spectroscopy techniques and theoretical calculations are used to characterise excited states and intermediates, shedding light on photoreaction pathways and the shapes of PESs. When compound A absorbs a photon to give the excited state A* at the FC region, the new electron density gained by the molecule after the photoexcitation coincides with a change in nuclear positions to minimise the energy. The inertia in the ground state is conserved in the excited state, thereby opening new regions along the PES of the excited state.37
Simplified potential energy (E) surface diagram of a photochemical process (Q). The absorption of light (1) excites the molecule (A) from the ground state to an electronic excited state (A*) in the Franck–Condon region (2). Vibrational relaxation (3) and light emission (4) can reduce the energy level of the excited state. The nuclear wave packet can continue along the excited state PES (5). Along this trajectory, the wave packet can undergo internal conversion to the ground state by encountered funnels such as an avoided crossing (6) or conical intersection (7) to give the photoproduct (B or B′).
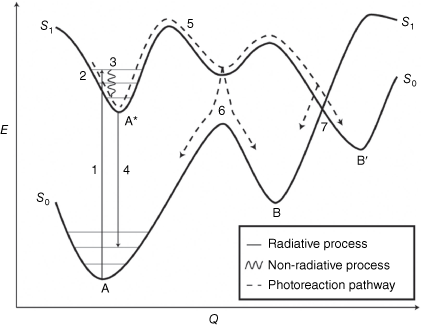
The excited state molecule can have competing pathways, as described previously, and will be guided along the PES on a trajectory known as the minimum energy path (MEP). A central mechanism for photoreactions involves funnels such as an avoided crossing or a conical intersection (CoIn), which facilitate efficient non-radiative decay of the excited state species to a new electronic state, in this case, the ground electronic state.38,39 These funnels occur when two PESs approach similar energies, causing the breakdown of the Born–Oppenheimer adiabatic approximation and leading to a mixture of electronic and nuclear degrees of freedom. These funnels act as bottlenecks not only for the radiationless deactivation but also for chemical transformation.38 From these funnels, the molecule can relax back to either the new photoproduct B (reactive pathway), or the starting compound A (non-reactive pathway).
Performance factors
Quantum yield
The quantum yield (Φ) represents the probability that a molecule undergoes a certain photophysical and photochemical process after the absorption of a photon. It measures the ratio between the number of reacted molecules (nx) and the number of absorbed photons (np). This can be represented by Eqn 1:
Quantum yields are measured using an actinometer and in general lie between 0 and 1. A quantum yield can be manipulated by synthetic modifications. Moreover, its value can also be affected by temperature, solvent and other environmental factors. The process by which the quantum yield is defined needs to be explicit. For example, a photochemical reaction can be defined either by the consumption of the reactant or by the formation of the product. However, the quantum yield for these processes will not be the same in many cases due to competing side reactions.
Photostationary state (PSS)
In a reversible photochemical reaction between two states, A and B, the PSS describes the ratio of B to A at a given wavelength. Competitive absorptions between the species A and B, and when the rates of formation and disappearance between each state become equal, exhaust the conversion at the given wavelength of light. The PSS is represented by Eqn 2:
where nx are the mol amounts of each state, Φx→y are the quantum yields of photoisomerisation, and εx are the molar absorption coefficient of the states at the wavelength.
Half-life
The half-life (t1/2) for a photochromic compound is the time needed for thermal bleaching to half the absorbance of the coloured form at a specific wavelength during a cycle. This kinetic parameter can use pulsed or continuous irradiation methods. The measurement of the kinetic rate constant for photochromic materials also quantifies the effects that temperature and solvent can have on this property.
Fatigue
The limit to the number of photochromic cycles is termed fatigue. Although the photochromic process is non-destructive, during the photochromic reaction, undesirable side reactions and oxidation can occur. Even extremely low-yielding side reactions lead to significant loss of the photochromic species as the number of colouration–decolouration cycles increases.
Main classes of photoswitches
There are two general types of photochromes: T-type and P-type. T-type photochromes have a low potential energy barrier between B and A, Scheme 1, resulting in the metastable state B spontaneously reverting to state A. In contrast, P-type photochromes have a high potential energy barrier in both reaction directions, and the conversion between the two stable states can only be initiated with light. These two types can be further subdivided into positive or negative photochromes. In positive photochromic systems, molecule A is colourless and converts into the coloured state B. Conversely, negative photochromes exist as a coloured state A that converts into the colourless state B.
The literature describes numerous families of photochromes, each with distinct characteristics that influence their suitability for various smart materials. The following descriptions will focus on selected photochrome families and briefly discuss their photoswitching mechanism.
Spiropyrans
Spiropyrans (SPs) are a class of T-type photochromes that undergo a colour change from a colourless SP form to a metastable merocyanine (MC) form (Fig. 5a). This transformation occurs when the central Cspiro–O bond is cleaved, a process that is triggered by UV light. The colour exhibited by the MC form is a result of the conjugation pathway extending throughout the entire molecule. In contrast, the conjugation in the SP form is interrupted by the central tetrahedral Cspiro atom. In the SP form, the indoline and chromene ring moieties are connected by the Cspiro carbon in an orthogonal manner.40,41 A related class of SPs are spirooxazines (SPOs, Fig. 5a), which introduces a nitrogen atom in the ethylene bridge. SPOs show improved fatigue over SP and were also used in early generation photochromic lenses.41
(a) Photoswitching of the spiropyran (SP) to merocyanine (MC). Indoline moiety highlighted in pink, chromene in black. PES diagrams for the switching of SP to MC on the (b) singlet and (c) triplet manifold. Reproduced from Kortekaas and Browne (2019)42 with permission from the Royal Society of Chemistry.
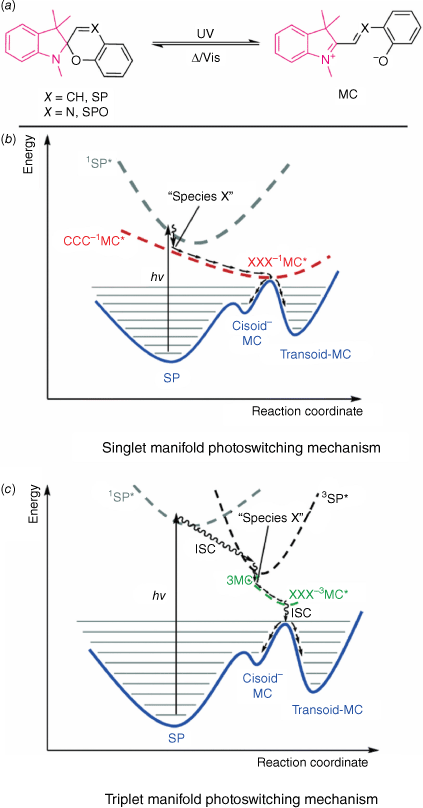
The photoisomerisation of SP to MC can be thought of as a two-step process. The first step involves the cleavage of the Cspiro–O bond to form the cis-MC, with the second step being either rotation to form the trans-MC or ring-closing to reform the SP.41 The MC can exist in either the quinoidal or zwitterionic state, depending on substitution and solvent conditions. Substitution of the chromene ring with an electron-withdrawing group, such as nitro, elongates the Cspiro–O bond, reducing the barrier to ring-opening and enabling access to the triplet excited state by n–π* transitions, facilitating formation of the MC.40,42 Such a substitution also enhances the ring-opening quantum yield, and stabilises the open MC state through resonance contributions. Switching can occur either by a singlet (Fig. 5b) or triplet manifold (Fig. 5c), with the triplet being the more efficient. In both cases, excitation to ‘Species X’ can result either in relaxation by pericyclic rearrangement to reform the SP or following of the reaction coordinate to reach MC isomers. From the cis-MC, rotation around the π–π* surface to form the more thermally stable trans-MC by non-radiative relaxation can occur. The triplet manifold involves ISC between the singlet SP excited state to the triplet SP excited state, with subsequent non-radiative relaxation through the triplet states to MC.40,42
Dihydropyrenes
Dihydropyrenes (DHPs) are a class of negative T-type photochromes, with the green coloured form being thermally stable. They switch between the more conjugated and coloured DHP and colourless cyclophanediene (CPD) forms by cleavage of the central transannular bond (Fig. 6a).
(a) Switching action of DHPs. PES diagram of DHP switching with the three-electron–three-centre bond funnel (b) or zwitterion funnel (c). Figure reproduced from Boggio-Pasqua et al. (2007)43 and Lognon et al. (2023)44 with permission. Copyright 2007 (b) and 2023 (c) American Chemical Society.
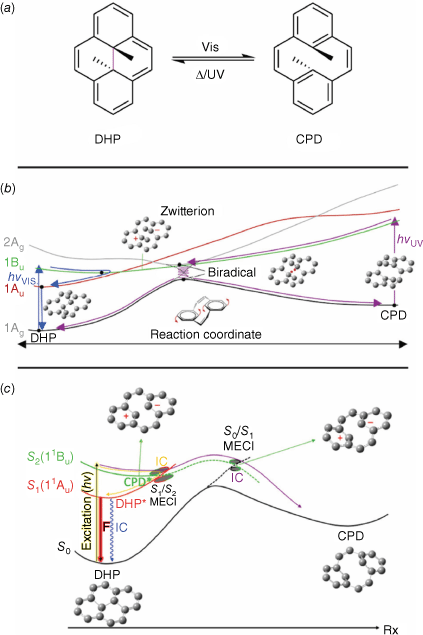
DHPs typically have poor quantum yields, owing to inefficient cleavage of the transannular bond. However, the quantum yield can be improved by synthetic modifications.45 The mechanism of switching for DHPs is complex, with many excited states (e.g. 2Ag, 1Bu and 1Au) available to be populated upon irradiation (Fig. 6b). This, in turn, is partially responsible for the inefficient quantum yields often seen with DHPs. Three singlet states are accessible to the excited DHP, the locally excited (LE, 1Au), zwitterionic (Z, 1Bu) and biradical (B, 2Ag) states. Each state has a unique reaction and decay pathway, with the LE and Z states able to decay back to the ground-state DHP. Excitation to the Z state and partial relaxation crosses with the LE state. From here, the excited DHP can undergo radiative decay to the CPD state or non-radiative decay to the DHP. Excitation to the B state facilitates a crossover to the CPD relaxation pathway by a conical intersection, with efficient IC. The B excited state is higher in energy compared to the LE and Z excited states, contributing to the inefficient overall conversion from DHP into CPD. A single decay pathway via the biradical intermediate exists for reversion from the CPD to DHP form.43 This potential energy profile has recently been revised using spin-flip time-dependent density functional theory (SF-TD-DFT) compared to the complete active space self-consistent field (CASSCF) treatment (Fig. 6c). Although the overall mechanism is similar, the main photochemical funnel responsible for the DHP to CPD isomerisation involves the zwitterion state (S2(11Bu)) rather than the energically less accessible three-electron–three-centre bond funnel.44
Diarylethenes
Diarylethenes (DAEs) are modified stilbene derivatives that switch between open and closed isomers. The most common modifications include the introduction of thiophenes in place for the aryl rings and perfluorocyclopentene moieties on the bridging ethene.46 In the coloured, closed form, the conjugation extends over the entire molecule, with conjugation localised over each aryl moiety in the open form (Fig. 7a). They are a class of P-type photochromes, and hence have a high thermal stability and fatigue resistance.46 The parent DAE can adopt either a parallel or anti-parallel conformation (Fig. 7b), with the anti-parallel conformer being able to switch to the closed cyclohexadiene (CHD) form. Due to the thermal stability of the open and closed isomers of DAEs, they are prime candidates in optical data storage.46
(a) Switching action of diarylethenes (DAEs). (b) Anti-parallel and parallel conformers of DAE. (c) Potential energy diagram of the switching of DAE. CI3 indicates the productive conical intersection between the excited S1 and ground S0 singlet states. Figure reproduced from Boggio-Pasqua et al. (2003)47 with permission. Copyright 2003 American Chemical Society.
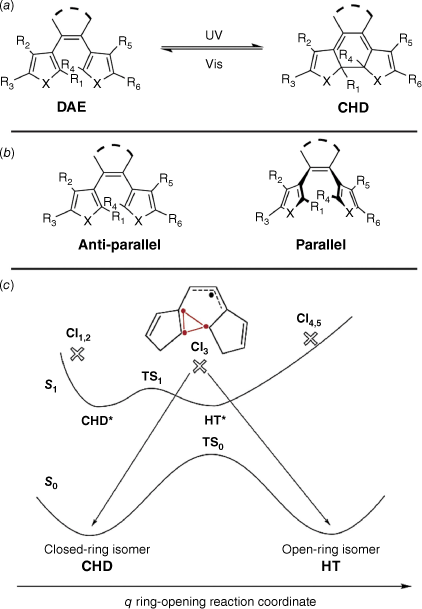
This switching action occurs by a 6π electrocyclic rearrangement. The anti-parallel conformation allows the photochemically promoted conrotatory rotation, according to Woodward–Hoffman rules, inhibited in the parallel conformer.48
Irradiation results in the negotiation of singlet energy states before relaxing to the CHD form (Fig. 7c). The energy of the DAE and CHD forms are similar, giving the CHD its characteristic thermal stability. Numerous conical intersections between the ground and excited states exist, with CI3 providing the pathway between the ring-opened and -closed minima on the ground state curve, characterised by three weakly coupled electrons and an allyl-type electron.47
Donor–acceptor Stenhouse adducts
Donor–acceptor Stenhouse adducts (DASAs), similar to DHPs, are negative T-type photochromes. Several iterations of DASAs have been synthesised with the 1st generation49 featuring a zwitterionic decolourised state and the 2nd generation having a neutral decolourised state (Fig. 8a).50 The pKa of the N substituent is crucial in determining whether the closed DASA adopts a neutral or zwitterionic decolourised isomer. In 2nd generation DASAs, the R groups of the N substituent are cyclised, lowering the pKa and favouring the neutral closed form. The photochromic response of the DASAs is dependent on the nature of the donor and acceptor groups as well as the polarity of the medium.35
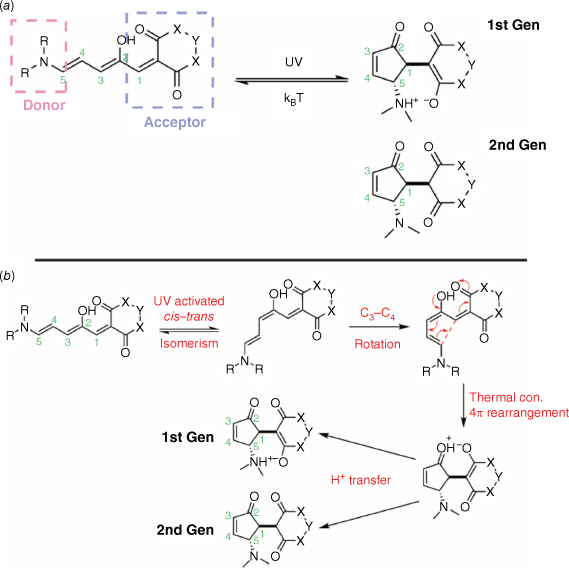
Irradiation of the open DASA promotes photoisomerisation of the C2–C3 double bond from the Z geometry to the E, directed by the H-bonding interactions of the adjacent hydroxy group with the ketone of the bis-ester ring. Subsequent isomerisation, followed by a thermal conrotatory 4π rearrangement and proton transfer, furnishes the closed DASA, with the rearrangement being the key ring-closing step (Fig. 8b). Photoswitching from the conjugated, open DASA to the closed cyclopentenone breaks the conjugation of the push–pull structure. This results in the loss of the red-shifted π–π* absorption band, and as such bi-directional photoswitching has proved problematic. Reversion to the open DASA would require a cyclopentenone with LUMO density over the C1–C5 bond, a feature not found in these DASAs.35
Fulgides
Fulgides are a class of P-type photochromes, and are derivatives of 1,3-butadiene-2,3-dicarboxylic acid. This class of compounds switches between a 1,3,5-hexatriene and cyclohexadiene structure (Fig. 9a). The exo-methylene carbon must have at least one aromatic substituent to form the hexatriene and facilitate the 6π electrocyclic rearrangement to form the cyclohexadiene.
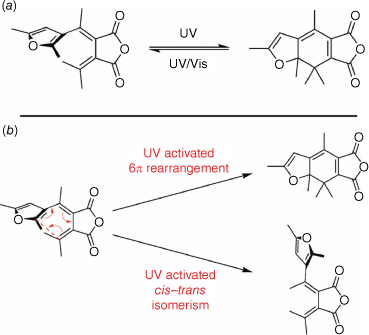
The inclusion of a heterocyclic aromatic substituent, typically furan, thermally stabilises the closed cyclohexadiene structure. The open isomer can exist in either the thermally stable E form or the Z form. Irradiation with UV light can promote either isomerisation of the E form to the Z form or the electrocyclic ring-closing, which is geometrically allowed only from the E form (Fig. 9b). UV irradiation also promotes the ring-opening of the cyclohexadiene to the hexatriene. Hence, the photostationary state comprised all three forms in various ratios, depending on the solvent and structure of the fulgide. Irradiation of the closed form with visible light exclusively promotes the ring-opening to the hexatriene open structure.51 Recent theoretical and spectroscopic investigation of the photochromic mechanism of the E form fulgide show competing CI that enable IC to the ground state that can induce either ring-closure or the E–Z isomerisation process.52,53
Azobenzenes
Azobenzenes are a class of T-type photochrome composed of two phenyl rings linked by an azo bond. They undergo cis–trans isomerism upon irradiation with UV light, with the trans isomer being the thermally stable isomer (Fig. 10a).54
(a) Cis–trans switching action of azobenzene. (b) PES for azobenzene photoswitching. Figure reproduced from Jerca et al. (2022)55 with permission. Copyright 2022, Springer Nature Limited.
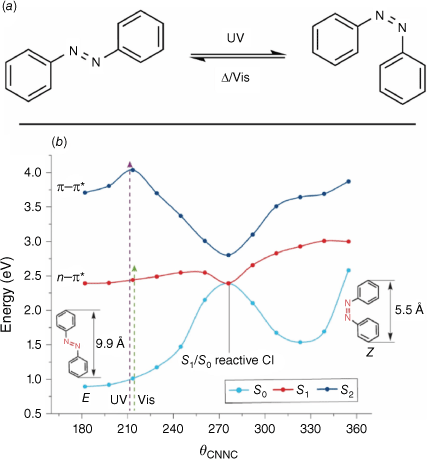
The half-life of the cis isomer is dependent on the substitution pattern of the aromatic rings. Steric modification of the azobenzene framework can increase the half-life of the cis form, a result of the increased energetic barrier to isomerisation to the trans form. Azobenzenes display a high fatigue resistance and high quantum yield, owing to the clean and efficient switching process of the isomerisation. Additionally, there is a pronounced conformational change upon switching, which can provide an additional output mechanism that may be relevant to applications.
The parent azobenzene has two absorption bands in the UV-vis region, the first being an n–π* symmetry-forbidden transition in the visible region and the second a more intense π–π* transition in the UV region. Based on these transitions, azobenzenes can be classified into three classes. The azobenzene-type class shows two absorption bands, similar to the parent. Distortion of the planarity of the azobenzene elongates the n–π* transition wavelength. The aminoazobenzenes feature substitution at the 4 position with strongly electron-donating groups. This results in a bathochromic shift of the absorption maximum of the π–π* band into the blue region of the spectrum, preserving the original n–π* transition. The pseudo-stilbene type azobenzenes have an additional substitution at the 4′ position with strongly electron-withdrawing substituents. This results in the reversal of the order of the absorption bands, and both occur in the visible region. Additionally, due to the asymmetric electron distribution, pseudo-stilbenes can display non-linear optical properties.
The isomerisation pathway for the azobenzene has been described to take place by either a torsion around the central double bond, inversion of the phenyl rings or a hula-twist.56 The isomerisation of azobenzenes takes place in the order of picoseconds to femtoseconds. The E isomer resides in a singlet S0 ground state, with the isomerisation process occurring by either an S0 → S1 or S0 → S2 transition, exhibiting anti-Kasha wavelength-dependent photochemistry (Fig. 10b).55 If photochemical excitation through the n–π* transition to the S1 state occurs, the excited species decays back to the S0 state via either a reactive or non-reactive conical intersection. Relaxation via the reactive conical intersection gives rise to the isomerisation product, whereas relaxation via the non-reactive conical intersection results in the retention of the original geometry. Photochemical excitation by the π–π* transition to the S2 state results in a myriad of pathways being available for relaxation. The excited molecule traverses through conical intersections between the S2 and S1 states and the subsequent S1–S0 conical intersections, before arriving at either the photoisomerisation product or the original state. The n–π* excitation pathway has a greater proportion of productive conical intersections, and so quantum yields via this route are greater.55,57
Dihydroazulenes
Dihydroazulenes (DHAs) are T-type photochromes that switch between the closed, colourless DHA form and the open, coloured vinylheptafulvene (VHF) upon irradiation with UV light (Fig. 11a). The VHF adopts a cis conformation upon initial opening, isomerising to the more energetically favourable trans form.58
(a) Photochromic switching action of DHAs. (b) Simulated PESs for DHA photoswitching. Modelled on five-membered ring systems for computational accessability. Figure reproduced from Boggio-Pasqua et al. (2002)59 with permission. Copyright 2002, American Chemical Society.
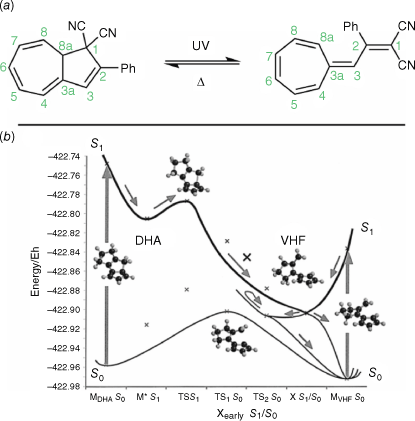
The absorption maxima and thermochromic properties can be tuned, with significant electronic differences between isomers. Switching of the DHA proceeds by a 10π electro-retrocyclisation, followed by cis–trans isomerism to the VHF. The reaction proceeds with a high quantum yield, with the back reaction being slow showing a strong correlation with solvent. Electron-withdrawing substituents in positions 1 or 3 are required for photochromism, with the absorption red-shifted with these groups in these positions. Substitution of the five-membered ring with electron-donating substituents results in a blue-shift of absorption features. Conversely, substitution of the seven-membered ring with the same substituents has an opposite effect on the absorption spectrum. The thermal ring-closing of the VHF can lead to two forms of the DHA, depending on the conformation of the transition state.
The 10π retrocyclisation is promoted by the excitation of the DHA from the S0 ground state to the S1 excited state. Relaxation through this reaction coordinate coincides with a conical intersection, leading to the VHF ground state. Furthermore, this conical intersection has a VHF-like geometry, accounting for the efficient quantum yield of the photochemical reaction (Fig. 11b). All excited states lead to the ground state with VHF geometry, further amplifying the quantum yield of ring-opening. A much steeper photochemical barrier from VHF to DHA exists, hence the photochemical reaction is essentially ‘one-way’ from DHA to VHF.59,60
Overcrowded alkenes
Overcrowded alkenes are P-type photochromes, with the switching originating from torsion around a central stilbene-like alkene bond.61 An intrinsic inversion in helicity follows cis–trans isomerism of this central stilbene moiety and gives the overcrowded alkenes their photochromic switching action (Fig. 12a). The inherent axial chirality of the overcrowded alkenes prevents inversion of chirality to the original state. Overcrowded alkenes comprised a symmetrical bottom stator and asymmetric rotor. The steric crowding around the alkene not only gives these compounds their name but prevents rotation around the central alkene linkage and imparts the chirality and helicity to the molecule.
(a) Photochromic switching of overcrowded alkenes with the rotor and stator moieties highlighted. (b) Illustration of conformations during switching. (c) PES sketch of overcrowded alkene switching. Figure reproduced from Feng and Gilson (2021)63 with permission from the Royal Society of Chemistry.
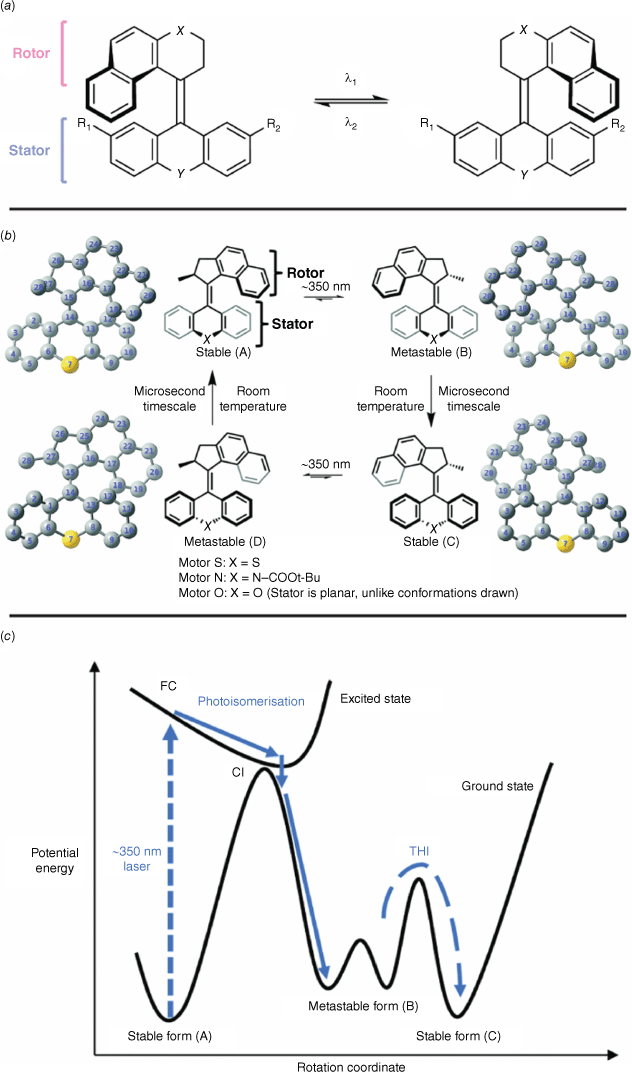
Careful substitution of the stator with electron-donating and withdrawing substituents, as well as changing the identity of X and Y along the central axis, can tune the barrier to isomerisation and hence the effective wavelength.62 The rotor rotates around the stator in a unidirectional, clockwise fashion. Switching takes place on microsecond timescales, owing to the complex negotiation of states. Initial excitation with UV light promotes the alkene A (Fig. 12b) from the ground state to the excited state, overcoming the significant energy barrier to thermal cis–trans isomerism on account of the steric crowding (Fig. 12c). This initial isomerisation step gives rise to a metastable form B (Fig. 12b) via a conical intersection of the excited and ground states (Fig. 12c). The species B then undergoes a spontaneous thermal helix inversion to form the more stable form C (Fig. 12b, c). In this step, a ‘puckering’ of the stator prompts the adoption of a more sterically favoured conformation, akin to the flapping of butterfly wings. Additional irradiation with UV light prompts another cis–trans isomerism event, with the formation of the corresponding metastable state D (Fig. 12b), before another ‘flap’ converts this state into the original conformer A. A and C, and B and D have identical energies, a consequence of the symmetrical stator.63
Selected applications of photoswitches
The applications, and the development, of new smart materials with photochromic molecules is vast. Incorporating all, or even the majority, of these into a single Primer Review is not justifiable. However, a few selected applications will be discussed below to allow the reader to gain an understanding of the diversity of photochromic molecules. A common theme emerging from applications of photochromic molecules is their interaction with secondary systems to perform a task to achieve an outcome. Complex multi-molecule systems will continue to inspire future research with these illuminating molecules.
Data storage
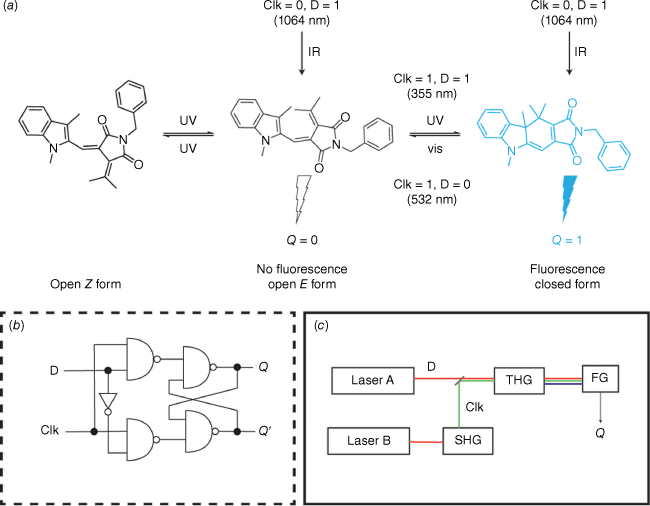
Photochromic compounds have garnered interest in logic or information processing applications.64 In these applications, the molecule’s function relies on an optically detectable output, such as absorption or emission, triggered by an external light stimulus. The integration of a fulgimide into an all-photonic molecule-based D flip-flop demonstrated that complex logic behaviour could be achieved by utilising the fluorescence of the closed form as the output state of the device (Fig. 13a).65 A D flip-flop is a data storage element commonly used in silicon circuitry to store and synchronise data (Fig. 13b). In this system, the data input (D) was provided by 1064 nm IR light from a Nd:YAG laser, whereas the clock input (Clk) was 532 nm light generated by a second-harmonic-generating (SHG) crystal. A third-harmonic-generating (THG) crystal produced 355 nm light. The output of the device (Q) was determined by the fluorescence emitted by the closed form of the fulgimide (Fig. 13c). Since the 1064 nm input does not trigger a photochromic response, Qn = Qn+1. Irradiation at 532 nm results in the open isomer and Qn+1 is set to 0 (Qn+1 = 0). By using both the 1064 nm (D) and 532 nm (Clk) light through the THG crystal, the device receives 355 nm UV light, which induces the closed isomer and fluorescence output, setting Qn+1 to 1 (Qn+1 = 1).
The photoswitch fulgimide was employed as a molecule-based D flip-flop for complex logic operations. (a) Schematic of fulgimide photoresponse and clock (Clk) and input (D) stimuli. (b) Working principle of a D flip-flop. (c) Schematic of the SHG and THG crystals to generate the required wavelengths for input, clock, stimuli and output criteria.
Phytopharmacology
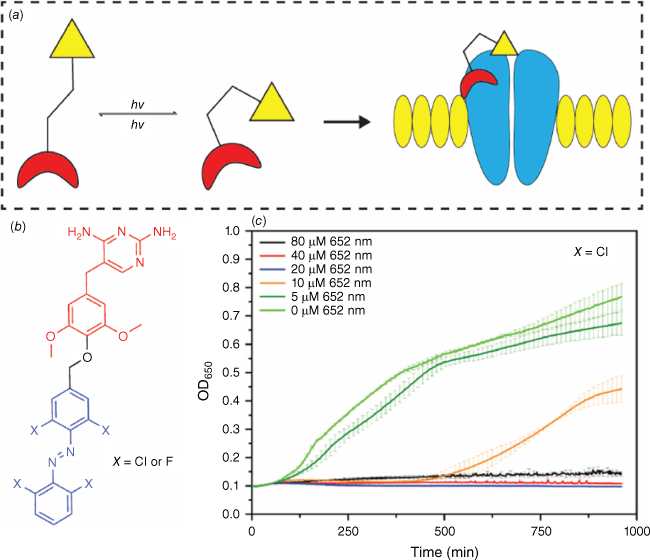
Photochromic compounds have found applications as light-activated antimicrobial agents. In this approach, a photochromic molecule is coupled with a bioactive molecule.66 Upon irradiation, the new molecule switches to its more active form, enabling the therapeutic effect (Fig. 14a). This spatial and temporal control using light allows for precise treatment. The proof of concept for a photoswitchable antibiotic was reported in 2013,67 and since then, the field has seen immense growth. Photoswitchable azobenzenes, coupled with a bactericide diaminopyrimidine, have demonstrated control over bacterial activity using green and violet light to activate and deactivate the antibacterial agent (Fig. 14b). Notably, irradiation of the compound (XCl) with red light resulted in an eight-fold difference in bacterial activity, with low-energy wavelengths being crucial for real-world therapy because of higher tissue penetration.68 Upon light irradiation, the trans-to-cis isomerisation of the azobenzene dissolves the inactive aggregates formed in the solution, leading to an increase in the molecule’s potency against bacteria (Fig. 14c).
General mechanism for a photopharmocological drug with a photoswitch (a). Photoswitchable azobenzenes coupled to diaminopyrimidine (b) showed antibacterial activity against E. coli CS1562 depicted in the bacterial growth curves (c).68
Molecular machines
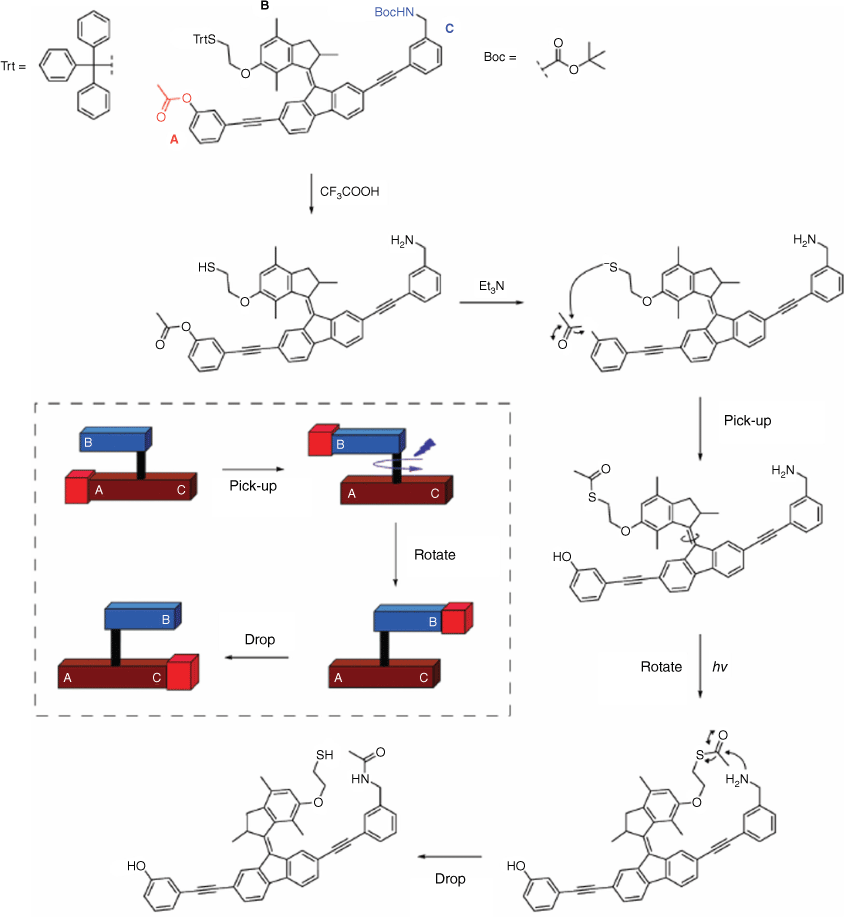
The field of research focusing on machines and devices at the molecular scale driven by light to power their functions is of significant interest.69,70 Molecular machine-based devices are designed with specific tasks in mind, such as actuation and molecular cargo transport. A recent study reported a light-driven acetyl transporter utilising a modified overcrowded alkene (Fig. 15).71 In this system, the upper part of the molecule acts as an arm or crane, capable of picking up an acetyl group from the lower part by the thiol reacting with the acetyl group at the A site of the molecule. Light is then employed to trigger the rotation of the molecule. Subsequently, the acetyl group is either released or reacted with the opposite amine side of the lower part of the molecule. The intramolecular reaction was monitored using 1H NMR and UV–Vis spectroscopy.
Structure of the acetyl transport molecular device. An intramolecular reaction and photoisomerisation leads to the pick-up and drop of an acetyl group from one site (A) of the molecule using a rotor (B) to another site (C). Insert shows an illustrative representation of the molecular device. Figure reproduced from Chen et al. (2016)71 with permission from the Royal Society of Chemistry.
Gas adsorbent
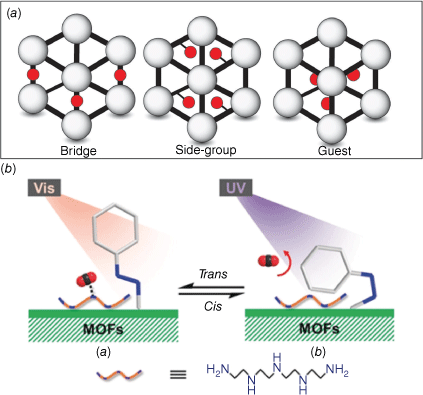
Photochromic molecules have been incorporated into metal–organic frameworks (MOFs) to enhance the functionality of these nanoporous materials.72 The photoswitch can be integrated into the framework, as a bridge, side group or as a guest (Fig. 16a). The concept of incorporating a photoswitch into a MOF was first explored in 2011 with the synthesis of a photoresponsive [Zn2(NDC)2] (CAU-5), which featured azobenzene protruding into the pores.73 A more recent example reported a photoresponsive azobenzene-modified UiO-type MOF. These MOFs were further modified with tetraethylenepentaamine with amine active sites suitable for CO2 absorption.74 The cis–trans photoisomerisation of the azobenzene moiety was able to modify the amine active sites responsible for the CO2 absorption capacity (Fig. 16b). Irradiation of the MOF with visible light triggers the trans–cis isomerisation, leading to the release of up to 45.6% of the bound CO2 from tetraethylenepentaamine. The photoresponsive MOFs also exhibited selectivity for CO2 over CH4 and N2. Irradiation with UV light triggered the cis–trans isomerisation and consequent uptake of gas in the structure.
(a) Representation of the different modes to incorporate photoswitches into MOFs (left to right), in the framework, as a side group or as a guest. (b) A photoresponsive azobenzene-modified UiO-type MOF for CO2 absorption. Figure reproduced from Jiang et al. (2019)74 with permission from the Royal Society of Chemistry.
Catalysis
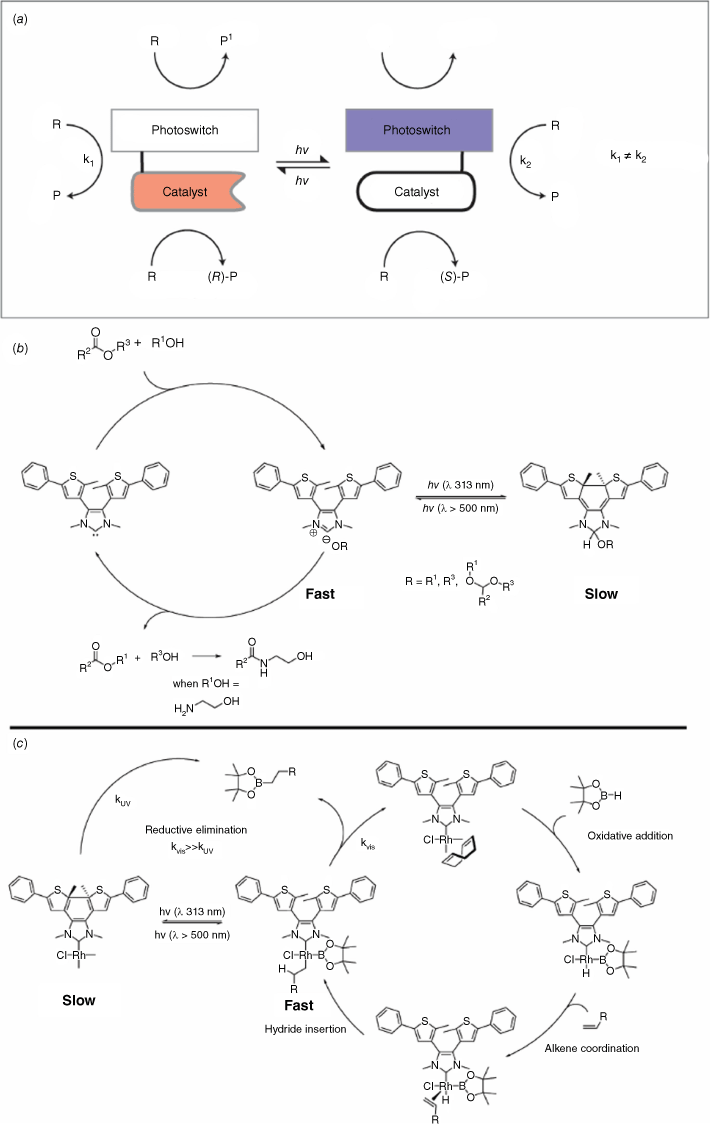
Catalysis is employed to efficiently increase the rate of chemical reactions and create functional molecules. Photoswitches are used to enhance, and in some cases control, the selectivity of chemical reactions (Fig. 17a).75,76 It is important to note that this area is conceptually different from photocatalysis, which is also light mediated.77 One of the first examples of using photoswitchable catalysis described a five-fold increase in the rate of hydrolysis of p-nitrophenyl acetate when an azobenzene-capped β-cyclodextrin was photoisomerised from the trans to the cis form using UV light.78 In this case, the trans–cis isomerisation resulted in a change in the cavity space with enhanced binding and a more favourable geometry for the hydrolysis reaction (Fig. 17a). A functional photoswitchable catalyst based on the DTE photochrome has been shown to be an ideal scaffold for activating and deactivating catalytic behaviour due to its photochemical electrocyclisation. An N-heterocyclic carbene (NHC) modified with a DTE core was reported to promote the transesterification of allyl alcohol and vinyl acetate, as well as the amidation of ethyl acetate, in its open form (Fig. 17b).79 Photoconversion of the catalyst to its closed form using UV irradiation exhibited a ten-fold decrease in catalytic activity. The changes in catalytic activity were ascribed to a decrease in the electron-donating ability of the NHC when the DTE is in its closed form. Interconversion between the open and closed isomers using visible and UV light was able to attenuate the conversion of single reactions. NMR spectroscopic 13C labelling studies showed that upon UV irradiation and photocyclisation, the closed NHC–DTE catalyst formed a less-active alcohol adduct. Building on from this study, a photochromic DAE-annulated NHC–RhI complex was shown to act as a light-tunable catalyst for alkene and alkyne hydroborations (Fig. 17c).80 Exposure of the catalyst to UV light closes the DTE moiety, leading to a decrease in the electron-donating ability of the NHC. The subsequent decrease in electron density of the Rh catalyst centre results in attenuated catalytic activity of up to an order of magnitude. The inhibition of the rate-determining reductive elimination step was postulated as the reason for the observed reduced activity of the catalyst.
(a) Representation of a photoswitchable catalyst where the catalytic activity or outcome can be tuned with light. The catalyst can be attenuated by the integrated photoswitch to change its catalytic properties. (b) An NHC–DTE catalyst for the transesterification and amidation. (c) An organometallic NHC–DTE complex catalyst for hydroboration of alkenes.
Molecular electronics
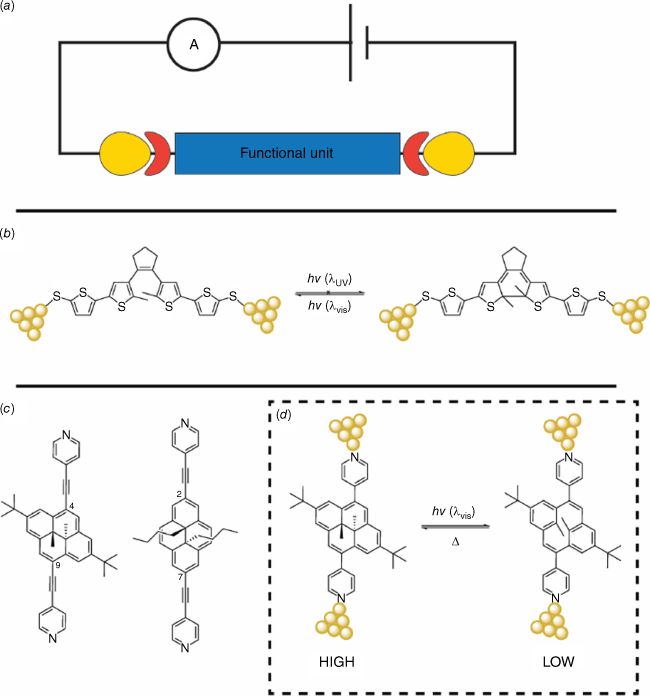
Photoswitching and studying the changes in junction transport and phenomena when a photochromic molecule is sandwiched between two electrodes are pivotal for the development of molecular optoelectronic devices (Fig. 18a).81 Molecular photoswitches have been extensively studied at the single-molecule level and using self-assembled monolayers. One of the first reported experiments involved the charge transport at the single-molecule level of a DTE molecule with modified sulfanythienyl contact groups, using the mechanically controllable break-junction (MCBJ) technique (Fig. 18b).82 The results showed an increase in resistance when the single-molecule junctions of the closed form of the DTE were irradiated with visible light, which was correlated with the opening of the molecule. However, attempts to switch the molecule back to the closed form using UV light were not successful, which was attributed to the quenching of the excited state by the gold electrodes. More recently, charge transport experiments using scanning tunneling microscope–break-junctions (STM-BJs) were conducted on several DHPs with alkynylpyridyl substituents. The results demonstrated that the single-molecule conductance depended on the substitution pattern on the DHP (either at the 2,7 or 4,9 positions), as well as the applied bias (Fig. 18c).83 Although the alkynyl-modified DHPs did not exhibit photochromic properties, the use of visible light to switch a DHP with direct pyridyl anchor groups to the CPD isomer resulted in a significant decrease in conductance, consistent with studies conducted using mechanically controlled break junctions (Fig. 18d).84
(a) Schematic of a single-molecule junction with a functional unit (i.e. photoswitch) sandwiched between two electrodes. (b) Single-molecule junctions of DTE showed switching to the open state but not to the closed state when the molecule was connected to the electrodes. (c) DHPs with 4,9 or 2,7-substituted alkynylpyridyl contact groups showed different conductances but were unable to be photochemically switched. (d) A high conductance DHP was able to be converted into a low-conducting CPD using visible light.
Conclusion
The phenomenon of photochromism has captivated scientists for over 150 years. What started as curious observations of materials changing colour when exposed to light has evolved into a well-developed field, where a combination of chemical synthesis, time-resolved spectroscopy and theory has provided a deep understanding of photochromism and enabled the rational design of photochromes for various material applications. Although several organic photochromic molecules have been extensively studied, there is still an incredible range of applications waiting to be explored using photochrome-based materials.
The future challenges for organic photochromic molecules serve as both inspiration and motivation for researchers pursuing real-world material applications. The transition from laboratory-based research systems to commercial materials will require input from future scientists and engineers. However, there are still economical and practical challenges that need to be overcome. The function and performance of photochromic molecules must be fine-tuned to meet the requirements of commercial materials. Key performance issues that still need attention include chemical degradation affecting performance lifetime, as well as the lack of control over colouration and bleaching rates and efficiency.
While modifying current photochromes will provide the fundamental models needed for progress in the field, the discovery of new and exciting types of photochromic molecules will expand the potential of photochromic applications. With the rapid progress in spectroscopic and theoretical techniques, as well as the versatile nature of synthetic chemistry, these obstacles can be overcome. The next generation of photochromic molecules and research is focused on investigating how these molecules interact with their surrounding environment. This is evident in the advancements in photopharmacology and molecular machines. By creating complex dynamic systems, we hope to see an influx of real-world applications that will brighten the future of photochromic materials.
Data availability
Data sharing is not applicable as no new data were generated or analysed during this study.
Conflicts of interest
G. A. Koutsantonis is a Co-Editor-in-Chief for the Australian Journal of Chemistry but did not at any stage have editor-level access to this manuscript while in peer review, as is the standard practice when handling manuscripts submitted by an editor to this journal. Australian Journal of Chemistry encourages its editors to publish in the journal and they are kept totally separate from the decision-making processes for their manuscripts. The authors have no further conflicts of interest to declare.
Acknowledgements
The authors acknowledge that this Primer Review was written on Noongar land, and that the Whadjuk Noongar people remain the spiritual and cultural custodians of their land, and continue to practice their values, languages, beliefs and knowledge. The authors pay their respects to the traditional owners of the lands on which they live and work across Western Australia and Australia. G. A. Koutsantonis is the 2022 recipient of the Royal Australian Chemical Institutes Leighton Memorial Medal.
References
3 Fritzsche M. Ueber die festen Kohlenwasserstoffe des Steinkohlentheers. [On the solid hydrocarbons of coal tar.]. J Prakt Chem 1867; 101(1): 333-343 [In German].
| Crossref | Google Scholar |
4 Wislicenus W, Reitzenstein F. Zur Kenntniss des Diketohydrindens. [About the knowledge of the diketohydrindenes.]. Justus Liebigs Ann Chem 1893; 277(3): 362-374 [In German].
| Crossref | Google Scholar |
5 Marckwald W. Ueber Phototropie. [About phototropy.]. Z Phys Chem 1899; 30U(1): 140-145 [In German].
| Crossref | Google Scholar |
6 Biltz H. Farbwechsel belichteter Substanzen. [Colour change of exposed substances.]. Z Phys Chem 1899; 30U(1): 527-528 [In German].
| Crossref | Google Scholar |
7 Chalkley L. Phototropy. Chem Rev 1929; 6(2): 217-280.
| Crossref | Google Scholar |
8 von Walther R. LVII. Zur Kenntnis der Einwirkung von Natrium auf Nitrile. [LVII. About the effect of sodium on nitriles.]. J Prakt Chem 1903; 67(1): 445-472 [In German].
| Crossref | Google Scholar |
9 Hayashi T. Photochromism of 2,2,4,6-Tetraphenyl-1,2-dihydro-1,3,5-triazine. Bull Chem Soc Jpn 1977; 50(9): 2489-2490.
| Crossref | Google Scholar |
10 Stobbe H. Die Farbe der „Fulgensäuren” und „Fulgide” (7. Abhandlung über Butadiënverbindungen). [The colour of ‘Fulgenic Acids’ and ‘Fulgides’ (7th treatise on butadiene compounds).]. Ber Dtsch Chem Ges 1905; 38(3): 3673-3682 [In German].
| Crossref | Google Scholar |
11 Stobbe H. Phototropieerscheinungen bei Fulgiden und anderen Stoffen. [Phototrophic phenomena in fulgids and other substances.]. Justus Liebigs Ann Chem 1908; 359(1–2): 1-48 [In German].
| Crossref | Google Scholar |
12 Schmidt J, Lumpp H. Gewinnung von Phenanthren-Abkömmlingen aus dem 9.9-Dichlor-10-phenanthron. [Studien in der Phenanthrenreihe. XXV. Mitteilung]. [Obtaining phenanthrene derivatives from 9.9-dichloro-10-phenanthrone. [Studies in the phenanthrene series XXV, notice].]. Ber Dtsch Chem Ges 1908; 41(3): 4215-4225.
| Crossref | Google Scholar |
13 Senier A, Shepheard FG. LIV.—Salicylidene-m-toluidine, a new phototropic compound; salicylideneamines: salicylamides. J Chem Soc Trans 1909; 95: 441-445.
| Crossref | Google Scholar |
14 Senier A, Shepheard FG. CCXIV.—Studies in phototropy and thermotropy. Part I. Arylidene- and naphthylidene-amines. J Chem Soc Trans 1909; 95: 1943-1955.
| Crossref | Google Scholar |
15 Stobbe H, Mallison H. Phototropie-Erscheinungen bei Stilben-Derivaten. [Phototropy phenomena in stilbene derivatives.]. Ber Dtsch Chem Ges 1913; 46(2): 1226-1238 [In German].
| Crossref | Google Scholar |
16 Wilson FJ, Heilbron IM, Sutherland MMJ. CCLXXI.—Contributions to our knowledge of semicarbazones. Part IV. Action of hydrogen chloride. J Chem Soc Trans 1914; 105(0): 2892-2906.
| Crossref | Google Scholar |
17 Lifschitz J. Photochemische Umlagerungen in der Triphenyl-methan-Reihe. [Photochemical rearrangements in the triphenylmethane series.]. Ber Dtsch Chem Ges 1919; 52(9): 1919-1926 [In German].
| Crossref | Google Scholar |
18 Singh BK. Studies on phototropism in solution. Part I. J Am Chem Soc 1921; 43(2): 333-334.
| Crossref | Google Scholar |
19 Holmes EO. The photochemical activity of the triphenylmethane sulfonic acids. J Am Chem Soc 1922; 44(5): 1002-1008.
| Crossref | Google Scholar |
20 Luck W, Sand H. The phenomenon of phototropy. Angew Chem Int Ed 1964; 3(8): 570-580.
| Crossref | Google Scholar |
21 Harris L, Kaminsky J, Simard RG. The absorption spectrum of malachite green leucocyanide and the mechanism of the dark reaction after photolysis. J Am Chem Soc 1935; 57(7): 1151-1154.
| Crossref | Google Scholar |
22 Hartley GS. The cis-form of azobenzene. Nature 1937; 140(3537): 281.
| Crossref | Google Scholar |
23 Norrish RGW, Porter G. Chemical reactions produced by very high light intensities. Nature 1949; 164(4172): 658.
| Crossref | Google Scholar |
24 Tamai N, Miyasaka H. Ultrafast dynamics of photochromic systems. Chem Rev 2000; 100(5): 1875-1890.
| Crossref | Google Scholar |
25 Hirshberg Y. Photochromie dans la serie de la bianthrone. [Photochromy in the bianthrone series.]. C R Acad Sci 1950; 231: 903. [In French].
| Google Scholar |
26 Fischer E, Hirshberg Y. Formation of coloured forms of spirans by low-temperature irradiation. J Chem Soc 1952; 1952: 4522-4524.
| Google Scholar |
27 Hayashi T, Maeda K. Preparation of a new phototropic substance. Bull Chem Soc Jpn 1960; 33(4): 565-566.
| Crossref | Google Scholar |
28 Blattmann HR, Meuche D, Heilbronner E, Molyneux RJ, Boekelheide V. Photoisomerization of trans-15,16-dimethyldihydropyrene. J Am Chem Soc 1965; 87(1): 130-131.
| Crossref | Google Scholar |
29 Muszkat KA, Gegiou D, Fischer E. The hexamethylstilbene–hexamethyldithydrophenanthrene interconversion, an example of a reversible photocyclization. Chem Commun 1965; 1965(19): 447-448.
| Crossref | Google Scholar |
30 Daub J, Knöchel T, Mannschreck A. Photosensitive dihydroazulenes with chromogenic properties. Angew Chem Int Ed 1984; 23(12): 960-961.
| Crossref | Google Scholar |
31 Irie M, Mohri M. Thermally irreversible photochromic systems. Reversible photocyclization of diarylethene derivatives. J Org Chem 1988; 53(4): 803-808.
| Crossref | Google Scholar |
32 Royal Swedish Academy of Sciences. The Nobel Prize in Chemistry 2016. Press release; 2016. Available at https://www.nobelprize.org/prizes/chemistry/2016/press-release/
33 Feringa BL, Jager WF, De Lange B, Meijer EW. Chiroptical molecular switch. J Am Chem Soc 1991; 113(14): 5468-5470.
| Crossref | Google Scholar |
34 Helmy S, Leibfarth FA, Oh S, Poelma JE, Hawker CJ, Read de Alaniz J. Photoswitching using visible light: a new class of organic photochromic molecules. J Am Chem Soc 2014; 136(23): 8169-8172.
| Crossref | Google Scholar |
35 Lerch MM, Szymański W, Feringa BL. The (photo)chemistry of Stenhouse photoswitches: guiding principles and system design. Chem Soc Rev 2018; 47(6): 1910-1937.
| Crossref | Google Scholar |
37 Mai S, González L. Molecular photochemistry: recent developments in theory. Angew Chem Int Ed 2020; 59(39): 16832-16846.
| Crossref | Google Scholar |
38 Boeije Y, Olivucci M. From a one-mode to a multi-mode understanding of conical intersection mediated ultrafast organic photochemical reactions. Chem Soc Rev 2023; 52(8): 2643-2687.
| Crossref | Google Scholar |
39 Domcke W, Yarkony DR. Role of conical intersections in molecular spectroscopy and photoinduced chemical dynamics. Annu Rev Phys Chem 2012; 63: 325-352.
| Crossref | Google Scholar |
40 Klajn R. Spiropyran-based dynamic materials. Chem Soc Rev 2014; 43(1): 148-184.
| Crossref | Google Scholar |
41 Minkin VI. Photo-, thermo-, solvato-, and electrochromic spiroheterocyclic compounds. Chem Rev 2004; 104(5): 2751-2776.
| Crossref | Google Scholar |
42 Kortekaas L, Browne WR. The evolution of spiropyran: fundamentals and progress of an extraordinarily versatile photochrome. Chem Soc Rev 2019; 48(12): 3406-3424.
| Crossref | Google Scholar |
43 Boggio-Pasqua M, Bearpark MJ, Robb MA. Toward a mechanistic understanding of the photochromism of dimethyldihydropyrenes. J Org Chem 2007; 72(12): 4497-4503.
| Crossref | Google Scholar |
44 Lognon E, Sarkar R, Heitz M-C, Boggio-Pasqua M. Dihydropyrene/cyclophanediene photoswitching mechanism revisited with spin-flip time-dependent density functional theory: nature of the photoisomerization funnel at stake! J Phys Chem A 2023; 127(13): 2921-2935.
| Crossref | Google Scholar |
45 Mitchell RH. The metacyclophanediene–dihydropyrene photochromic π switch. Eur J Org Chem 1999; 1999(11): 2695-2703.
| Crossref | Google Scholar |
46 Irie M, Fukaminato T, Matsuda K, Kobatake S. Photochromism of diarylethene molecules and crystals: memories, switches, and actuators. Chem Rev 2014; 114(24): 12174-12277.
| Crossref | Google Scholar |
47 Boggio-Pasqua M, Ravaglia M, Bearpark MJ, Garavelli M, Robb MA. Can diarylethene photochromism be explained by a reaction path alone? A CASSCF study with model MMVB dynamics. J Phys Chem A 2003; 107(50): 11139-11152.
| Crossref | Google Scholar |
48 Nakamura S, Irie M. Thermally irreversible photochromic systems. A theoretical study. J Org Chem 1988; 53(26): 6136-6138.
| Crossref | Google Scholar |
49 Helmy S, Oh S, Leibfarth FA, Hawker CJ, Read de Alaniz J. Design and synthesis of donor–acceptor stenhouse adducts: a visible light photoswitch derived from furfural. J Org Chem 2014; 79(23): 11316-11329.
| Crossref | Google Scholar |
50 Hemmer JR, Poelma SO, Treat N, Page ZA, Dolinski ND, Diaz YJ, Tomlinson W, Clark KD, Hooper JP, Hawker C, Read de Alaniz J. Tunable visible and near infrared photoswitches. J Am Chem Soc 2016; 138(42): 13960-13966.
| Crossref | Google Scholar |
51 Yokoyama Y. Fulgides for memories and switches. Chem Rev 2000; 100(5): 1717-1740.
| Crossref | Google Scholar |
52 Peng L-Y, Li Z-W, Fang Q, Xie B-B, Xia S-H, Cui G. Combined QM (MS-CASPT2)/MM studies on photocyclization and photoisomerization of a fulgide derivative in toluene solution. Phys Chem Chem Phys 2022; 24(48): 29918-29926.
| Crossref | Google Scholar |
53 Siewertsen R, Renth F, Temps F, Sönnichsen F. Parallel ultrafast E–C ring closure and E–Z isomerisation in a photochromic furylfulgide studied by femtosecond time-resolved spectroscopy. Phys Chem Chem Phys 2009; 11(28): 5952-5961.
| Crossref | Google Scholar |
54 Bandara HMD, Burdette SC. Photoisomerization in different classes of azobenzene. Chem Soc Rev 2012; 41(5): 1809-1825.
| Crossref | Google Scholar |
55 Jerca FA, Jerca VV, Hoogenboom R. Advances and opportunities in the exciting world of azobenzenes. Nat Rev Chem 2022; 6(1): 51-69.
| Crossref | Google Scholar |
56 Quick M, Dobryakov AL, Gerecke M, Richter C, Berndt F, Ioffe IN, Granovsky AA, Mahrwald R, Ernsting NP, Kovalenko SA. Photoisomerization dynamics and pathways of trans- and cis-azobenzene in solution from broadband femtosecond spectroscopies and calculations. J Phys Chem B 2014; 118(29): 8756-8771.
| Crossref | Google Scholar |
57 Nenov A, Borrego-Varillas R, Oriana A, Ganzer L, Segatta F, Conti I, Segarra-Marti J, Omachi J, Dapor M, Taioli S, Manzoni C, Mukamel S, Cerullo G, Garavelli M. UV-Light-Induced Vibrational Coherences: The Key to Understand Kasha Rule Violation in trans-Azobenzene. J Phys Chem Lett 2018; 9(7): 1534-1541.
| Crossref | Google Scholar |
58 Broman SL, Nielsen MB. Dihydroazulene: from controlling photochromism to molecular electronics devices. Phys Chem Chem Phys 2014; 16(39): 21172-21182.
| Crossref | Google Scholar |
59 Boggio-Pasqua M, Bearpark MJ, Hunt PA, Robb MA. Dihydroazulene/vinylheptafulvene photochromism: a model for one-way photochemistry via a conical intersection. J Am Chem Soc 2002; 124(7): 1456-1470.
| Crossref | Google Scholar |
60 Abedi M, Pápai M, Mikkelsen KV, Henriksen NE, Møller KB. Mechanism of photoinduced dihydroazulene ring-opening reaction. J Phys Chem Lett 2019; 10(14): 3944-3949.
| Crossref | Google Scholar |
61 Sheng J, Danowski W, Crespi S, Guinart A, Chen X, Stähler C, Feringa BL. Designing P-type bi-stable overcrowded alkene-based chiroptical photoswitches. Chem Sci 2023; 14(16): 4328-4336.
| Crossref | Google Scholar |
62 Feringa BL, van Delden RA, Koumura N, Geertsema EM. Chiroptical molecular switches. Chem Rev 2000; 100(5): 1789-1816.
| Crossref | Google Scholar |
63 Feng M, Gilson MK. Mechanistic analysis of light-driven overcrowded alkene-based molecular motors by multiscale molecular simulations. Phys Chem Chem Phys 2021; 23(14): 8525-8540.
| Crossref | Google Scholar |
64 Andréasson J, Pischel U. Molecules with a sense of logic: a progress report. Chem Soc Rev 2015; 44(5): 1053-1069.
| Crossref | Google Scholar |
65 Remón P, Bälter M, Li SM, Andréasson J, Pischel U. An all-photonic molecule-based D flip-flop. J Am Chem Soc 2011; 133(51): 20742-20745.
| Crossref | Google Scholar |
66 Hüll K, Morstein J, Trauner D. In Vivo Photopharmacology. Chem Rev 2018; 118(21): 10710-10747.
| Crossref | Google Scholar |
67 Velema WA, van der Berg JP, Hansen MJ, Szymanski W, Driessen AJM, Feringa BL. Optical control of antibacterial activity. Nat Chem 2013; 5(11): 924-928.
| Crossref | Google Scholar |
68 Wegener M, Hansen MJ, Driessen AJM, Szymanski W, Feringa BL. Photocontrol of antibacterial activity: shifting from UV to red light activation. J Am Chem Soc 2017; 139(49): 17979-17986.
| Crossref | Google Scholar |
69 Corra S, Curcio M, Baroncini M, Silvi S, Credi A. Photoactivated artificial molecular machines that can perform tasks. Adv Mater 2020; 32(20): 1906064.
| Crossref | Google Scholar |
70 Baroncini M, Silvi S, Credi A. Photo- and redox-driven artificial molecular motors. Chem Rev 2020; 120(1): 200-268.
| Crossref | Google Scholar |
71 Chen J, Wezenberg SJ, Feringa BL. Intramolecular transport of small-molecule cargo in a nanoscale device operated by light. Chem Commun 2016; 52(41): 6765-6768.
| Crossref | Google Scholar |
72 Rice AM, Martin CR, Galitskiy VA, Berseneva AA, Leith GA, Shustova NB. Photophysics modulation in photoswitchable metal–organic frameworks. Chem Rev 2020; 120(16): 8790-8813.
| Crossref | Google Scholar |
73 Modrow A, Zargarani D, Herges R, Stock N. The first porous MOF with photoswitchable linker molecules. Dalton Trans 2011; 40(16): 4217-4222.
| Crossref | Google Scholar |
74 Jiang Y, Tan P, Qi SC, Liu XQ, Yan JH, Fan F, Sun LB. Metal–organic frameworks with target-specific active sites switched by photoresponsive motifs: efficient adsorbents for tailorable CO2 capture. Angew Chem Int Ed 2019; 58(20): 6600-6604.
| Crossref | Google Scholar |
75 Kondo M, Nakamura K, Krishnan CG, Sasai H, Takizawa S. Photoswitchable chiral organocatalysts: photocontrol of enantioselective reactions. Chem Rec 2023; 23: e202300040.
| Crossref | Google Scholar |
76 Wang JB, Feringa BL. Dynamic control of chiral space in a catalytic asymmetric reaction using a molecular motor. Science 2011; 331(6023): 1429-1432.
| Crossref | Google Scholar |
77 Neilson BM, Bielawski CW. Illuminating photoswitchable catalysis. ACS Catal 2013; 3(8): 1874-1885.
| Crossref | Google Scholar |
78 Ueno A, Takahashi K, Osa T. Photocontrol of catalytic activity of capped cyclodextrin. J Chem Soc Chem Commun [3] 1981; 94-96.
| Crossref | Google Scholar |
79 Neilson BM, Bielawski CW. Photoswitchable organocatalysis: using light to modulate the catalytic activities of N-heterocyclic carbenes. J Am Chem Soc 2012; 134(30): 12693-12699.
| Crossref | Google Scholar |
80 Neilson BM, Bielawski CW. Photoswitchable metal-mediated catalysis: remotely tuned alkene and alkyne hydroborations. Organometallics 2013; 32(10): 3121-3128.
| Crossref | Google Scholar |
81 Huang XH, Li T. Recent progress in the development of molecular-scale electronics based on photoswitchable molecules. J Mater Chem C 2020; 8(3): 821-848.
| Crossref | Google Scholar |
82 Dulić D, van der Molen SJ, Kudernac T, Jonkman HT, de Jong JJD, Bowden TN, van Esch J, Feringa BL, van Wees BJ. One-way optoelectronic switching of photochromic molecules on gold. Phys Rev Lett 2003; 91(20): 207402.
| Crossref | Google Scholar |
83 Roemer M, Gillespie A, Jago D, Costa-Milan D, Alqahtani J, Hurtado-Gallego J, Sadeghi H, Lambert CJ, Spackman PR, Sobolev AN, Skelton BW, Grosjean A, Walkey M, Kampmann S, Vezzoli A, Simpson PV, Massi M, Planje I, Rubio-Bollinger G, Agraït N, Higgins SJ, Sangtarash S, Piggott MJ, Nichols RJ, Koutsantonis GA. 2,7-and 4,9-Dialkynyldihydropyrene molecular switches: syntheses, properties, and charge transport in single-molecule junctions. J Am Chem Soc 2022; 144: 12698-12714.
| Crossref | Google Scholar |
84 Roldan D, Kaliginedi V, Cobo S, Kolivoska V, Bucher C, Hong W, Royal G, Wandlowski T. Charge transport in photoswitchable dimethyldihydropyrene-type single-molecule junctions. J Am Chem Soc 2013; 135(16): 5974-5977.
| Crossref | Google Scholar |