

Synthesis of R- and S-MDMA via nucleophilic ring-opening of homochiral N-tosylaziridines
Katie D. Lewis A , Glenn A. Pullella A , Han Chern Loh A , Brian W. Skelton A , Gavin R. Flematti A and Matthew J. Piggott A *
A *
A Chemistry, School of Molecular Sciences, University of Western Australia, Perth, WA 6009, Australia.
Australian Journal of Chemistry 76(5) 299-310 https://doi.org/10.1071/CH23064
Submitted: 20 April 2023 Accepted: 20 June 2023 Published: 12 July 2023
© 2023 The Author(s) (or their employer(s)). Published by CSIRO Publishing. This is an open access article distributed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License (CC BY-NC-ND)
Abstract
Homochiral (R)- and (S)-3,4-methylenedioxymethamphetamine (MDMA) were prepared in six steps (each) from the chiral pool precursors d- and l-alanine, respectively. The key step, copper-catalysed regioselective ring-opening of an N-tosylaziridine with an aryl Grignard reagent, proceeded in high yield with complete regioselectivity. Elaboration was achieved with preservation of configurational integrity, affording R- and S-MDMA hydrochlorides with enantiopurities of >99.5%, as determined by enantioselective HPLC with fluorescence detection. Attempts to apply the synthetic methodology to the synthesis of the homochiral enantiomers of the α-phenyl analogue of MDMA (UWA-001) were thwarted by a switch in regioselectivity in the key step.
Keywords: aziridine, chiral pool, enantiopurity determination, HPLC with fluorescence detection, nucleophilic ring-opening, organic chemical synthesis, (R)- and (S)-3,4-methylenedioxymethamphetamine, R- and S-MDMA, X-ray crystal structure.
Introduction
3,4-Methylenedioxymethamphetamine (MDMA), better known by its street name ‘ecstasy’, is a popular recreational drug with a long and interesting history[1–5] (see also the primer in this special issue).[6] The best-known member of a sub-class of psychedelic drugs known as the entactogens, MDMA has prosocial, mood-elevating, anxiolytic and fear-extinguishing behavioural effects that make it an attractive adjunct to psychotherapy.[7–9] Indeed, MAPS, the Multidisciplinary Association for Psychedelic Studies, have completed two successful Phase III clinical trials of MDMA-assisted psychotherapy for post-traumatic stress disorder (PTSD) in which MDMA was found to be highly efficacious.[10,11] On the back of these trials, and in a world first, MDMA was rescheduled from S9 (drugs and poisons that may only be used for research) to S8 (medicines with strict legislative controls) by the Therapeutic Goods Administration of Australia specifically for use in PTSD.[12] MDMA also has potential application in the treatment of eating disorders,[13,14] alcohol[15–20] and other substance abuse,[21,22] anxiety,[23–27] chronic pain,[28] depression and other mood disorders,[23–27] and clinical trials for some of these indications are being planned.[11]
Like all amphetamines, MDMA is chiral (Fig. 1), and recreational ‘ecstasy’ is probably exclusively racemic. The enantiomers contribute synergistically to the subjective effects of racemic MDMA.[29,30] The S-(+)-enantiomer is the more potent psychoactive agent and psychostimulant, and is a more effective releaser of the monoamine neurotransmitters serotonin,[31,32] dopamine[7,32] and noradrenaline.[32] In non-human primates, R-MDMA was shown to release serotonin but not dopamine.[33] R-MDMA is also a low-efficacy partial agonist of 5-HT2A1 receptors,[34–37] canonically associated with psychedelia. In addition, the R-enantiomer causes release of prolactin,[33,38] and may have enhanced prosocial activity, and fewer adverse side effects compared with S-MDMA or the racemate.[39] Thus, it has been suggested that R-MDMA may be preferred over the racemate for psychotherapy.[22,39] Indeed, a small clinical trial (n = 24) in healthy volunteers is currently under way, examining the acute autonomic, biochemical and behavioural effects of racemic, R- and S-MDMA.[40] The chemistry and pharmacology of MDMA enantiomers are covered in more detail in recent excellent reviews.[5,22]

Racemic MDMA has been shown to alleviate the motor side-effects of long-term dopaminergic therapy for Parkinson’s disease (PD), both anecdotally in a single human with disabling levodopa-induced dyskinesia[41] and in related animal models.[42–47] There are also suggestions that MDMA could positively address cognitive impulsive/compulsive behaviours and visual hallucinations associated with extended use of dopaminergic agents in PD.[42,43] In addition, MDMA was reported to be selectively toxic to a Burkitt’s lymphoma cell line, perhaps via a serotonergic mechanism.[48–50] We required homochiral R- and S-MDMA for evaluation in both PD and Burkitt’s lymphoma models.
Syntheses of enantiopure or enriched MDMA have been reviewed;[5] the chiral pool precursor or key stereoselective/resolution steps are outlined in Scheme 1. Nichols, Shulgin and co-workers described the first synthesis of homochiral R- and S-MDMA using a diastereoselective reductive amination of piperonyl methyl ketone (1) with the chiral derivatisation agents R- or S-α-methylbenzylamine (2), respectively.[29,51] Escubedo and colleagues developed syntheses of both enantiomers of MDMA involving a similar diastereoselective reductive sulfonamidation of ketone 4 with the two enantiomers of Ellman’s sulfinamide, as exemplified by the conversion of R-5 to 6 with good diastereoselectivity.[52] A shorter route using the same methodology, but beginning with piperonal, was recently patented by Xin, although this claim was not supported by any experimental data.[53] The patent also proposed a prophetic diastereoselective methylation of the N-sulfinylimine derived from homopiperonal (7) and the same chiral auxiliary to give 8. Diastereoselective addition of methylmagnesium iodide to the chemoenzymatically derived O-silyl cyanohydrin 9, followed by in situ transimination with methylamine and borohydride reduction, afforded the amino alcohol 10, which was proto-dehydroxylated in two steps, providing S-MDMA.[54]

Surprisingly, a resolution of racemic MDMA via diastereomeric salts or amides has never been reported; however, Joglar and co-workers resolved racemic 3,4-di(benzyloxy)amphetamine (11) as the dibenzoyl-d-(+)-tartaric acid (12) salt, and advanced the S-enantiomer of amine 13 to S-MDMA.[55] Semi-preparative enantioselective chromatography has also been used to resolve racemic MDMA,[56,57] including a tritiated isotopologue.[57] Finally, Xin patented (and exemplified) a chiral pool synthesis of R-MDMA, beginning with levodopa methyl ester hydrochloride (14).[58]
Herein, we report our contribution to the synthesis of homochiral MDMA using amino acid-derived chirons.
Results and discussion
Nenajdenko and co-workers reported the copper-catalysed regioselective ring-opening of l-amino acid-derived N-tosylaziridines 15 by (hetero)aryl Grignard reagents to give sulfonamides 16, which were deprotected without racemisation to afford the corresponding primary amines, isolated as the hydrochlorides 17 (Scheme 2).[59]

This methodology was adapted to the synthesis of the (homochiral) enantiomers of MDMA, as depicted in Scheme 3. The required N-tosylaziridines R19 and S19 were synthesised in three steps from d- and l-alanine, respectively.[60,61] Copper-catalysed ring-opening of the aziridines by the Grignard reagent derived from 5-bromobenzodioxole (18)[62] gave the secondary sulfonamides R20 and S20 in excellent yield. The complete regioselectivity of the ring-opening step is attributed to steric hindrance at C2 of the aziridine,[63] the strongly electron-withdrawing tosyl group, which favours an SN2 mechanism,[64] and the soft nucleophilic character of the intermediate organocuprate. The reaction of activated aziridines with harder nucleophiles such as Grignard reagents (in the absence of a copper catalyst) has been shown to give a mixture of regioisomers[64,65] and lower yields.[66] Quantitative N-methylation affording R21 and S21 was followed by reductive deprotection. Unsurprisingly, given the stability of sulfonamides,[67] deprotection proved troublesome. Conditions used are summarised in Table 1.
Synthesis of R- and S-MDMA. ))), sonication. The wavy bonds indicate a single configuration, defined by the compound labels for each enantiomeric series.
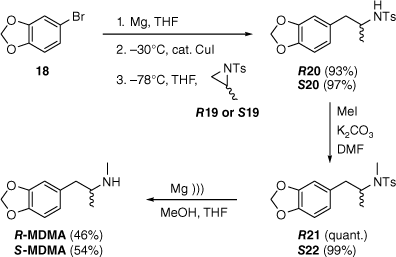
Sonication with magnesium in methanol was reported to be sufficiently gentle to avoid racemisation or ring-opening during deprotection of chiral N-tosylaziridines,[68] and this methodology was later adapted to deprotection of secondary sulfonamides related to the current work (Scheme 2).[59] When these conditions were applied to the tertiary sulfonamides S21 and R21 (Scheme 3), deprotection was clean, but stalled for no obvious reason, and neither addition of fresh magnesium nor extended sonication helped the reactions progress. As a result, yields of the free base MDMA enantiomers were moderate. However, in each instance, >40% of starting sulfonamide was recovered after chromatography. Deprotection of S21 with sodium naphthalenide radical anion[69] was also inefficient, and photolytic reductive cleavage of R21[70,71] gave an even lower yield of R-MDMA, which was surprising given that quantitative yields have been reported with similar substrates.[72] On a positive note, all of these methods retained the configurational integrity of the stereocentre (Table 2). It is likely that the efficiency of the synthesis presented here could be improved by judicious choice of a more easily removed aziridine-activating group, such as nosyl,[73] or phosphoryl.[74]
| ApproachA | Free base [α]D | Hydrochloride [α]D | Ref. | ||
|---|---|---|---|---|---|
| R-MDMA | S-MDMA | R-MDMA | S-MDMA | ||
| (a) | ND | ND | [α]D −18.2° | [α]D +17.2° | Shulgin[29,51] |
| (a) | ND | ND | [α]D −17.5° (c 1, H2O) | [α]D +17.4° (c 1, H2O) | Nichols[29,51] |
| (b) | ND | ND | [α]D −12.4° (c 0.6, H2O) | [α]D +14.2° (c 0.6, H2O) | Escubedo[52] |
| (c) | ND | ND | ND | ND | Xin[53] |
| (d) | ND | ND | ND | +17.9° (c 1.00, H2O) | Effenberger[54] |
| (e) | ND | ND | ND | +15.2° (c 0.79, H2O) | Joglar[55] |
| Enantioselective HPLC | ND | ND | ND | ND | Moreau[56] |
| Enantioselective HPLC | −6.7° (c 0.1, EtOH)B | +9.4° (c 0.1, EtOH)B | ND | ND | Hashimoto[57] |
| (e) | ND | ND | ND | ND | Xin[58] |
| Present work | −34.5° (c 1.1, CHCl3)C | +32.7° (c 1.2, CHCl3)C | −19.3° (c 1.1, H2O) | +18.3° (c 1.0, H2O) | Present work |
| −33.7° (c 1.0, CHCl3)D | +33.1° (c 1.0, CHCl3)E | ||||
| +33.0° (c 1.0, CHCl3)F | |||||
ND, no specific rotation data reported.
ARefer to Scheme 1.
BNot specified whether the specific rotations reported were obtained for MDMA free base, MDMA hydrochloride, or MDMA trifluoroacetate (the latter is included as the eluent utilised by Hashimoto et al. contained trifluoracetic acid), or indeed some other salt of MDMA.[57]
CR- and S-MDMA prepared from sulfonamides 21 using Mg in THF/MeOH with sonication.
DR-MDMA prepared from R21 using 1,4-dimethoxybenzene and NaBH4 in MeCN/EtOH under UV irradiation (λ 254 nm).
ES-MDMA prepared from S21 using Mg in MeOH with sonication.
FS-MDMA prepared from S21 using sodium naphthalenide in DME.
We previously investigated analogues of MDMA with selective toxicity toward Burkitt’s lymphoma cell lines.[48,49] These studies revealed an order of magnitude increase in the cytotoxic potency of the α-phenyl analogue of MDMA (UWA-001; the R-enantiomer is shown in Scheme 4) relative to MDMA. Hence, we were interested in the homochiral enantiomers of UWA-001, and the ring-opening methodology was applied to the N-tosylaziridines (R22 and S22) derived in two steps from phenylglycine.[75–77] However, surprisingly, the copper-catalysed reaction of the Grignard reagent derived from 18 with S22 did not give the expected α-phenylsulfonamide R23; instead, the regioisomer 24, arising from nucleophilic attack at the more sterically hindered tertiary carbon of the aziridine S22, was the only isolated product that could be identified.
Attempted synthesis of R-UWA-001. The wavy bond represents a single but undefined configuration.
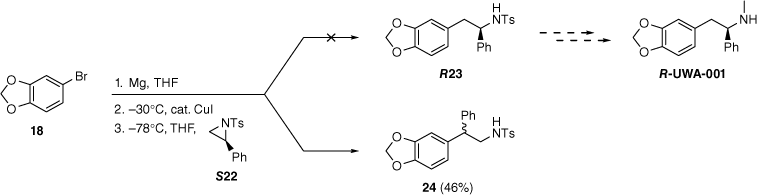
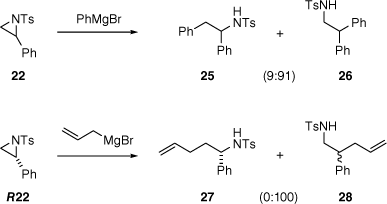
The absolute configuration of 24 was not determined, but it was optically active and no indication of diastereomeric signals was detected in a 1H NMR spectrum in the presence of the chiral shift reagent europium tris[3-(heptafluoropropylhydroxymethylene)]-(+)-camphorate. Thus, to the limit of 1H NMR sensitivity, 24 appears to be a single enantiomer. Toshimitsu and co-workers previously observed that ring-opening of racemic 22 with four equivalents of phenylmagnesium bromide, and no copper catalyst, favoured nucleophilic attack on the benzylic carbon, giving 26 in preference to 25 (Scheme 5).[78] When enantioenriched R22 was treated with allylmagnesium bromide, the regioselectivity was complete and the enantiopurity of the aziridine (94% e.e.) was preserved in the product 28. However, the configuration of 28 was not determined, so there is still more to learn about the stereochemistry and mechanism of the ring-opening of 2-arylaziridines by Grignard reagents.
Regioselectivity in nucleophilic ring-opening of N-tosylaziridine 22.[78] The wavy bond represents a single but undefined configuration.
Structural characterisation of MDMA hydrochloride and evidence for enantiopurity
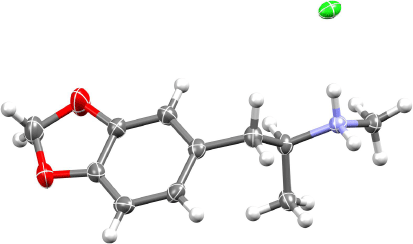
Table 2 summarises specific rotation data for (R)- and (S)-MDMA prepared by various methods. Reductive aminations, such as used in Nichols and co-workers’ first synthesis of enantioenriched MDMA,[29,51] (Scheme 1a) are unlikely to be 100% diastereoselective, but they did not report diastereomeric ratios. They recrystallised the hydrochlorides of 3 (Scheme 1) and its enantiomer to enrich the major diastereomer in each case. Following cleavage of the chiral auxiliaries, the enantiomeric hydrochlorides were also recrystallised twice, which presumably further enriched the enantiopurity. The specific rotations they obtained are close to those acquired in the current work (Table 2). At the time, there were no authentic specific rotations for Nichols et al. to compare with, and thus the configurations of the isolated MDMA hydrochlorides were uncertain. Interestingly, they stated that ‘it was anticipated that these enantiomers had the R-(−) and S-(+) absolute configurations, by analogy to earlier studies.[79] Recently, single-crystal X-ray crystallographic studies have confirmed this (manuscript in preparation).’[51] Subsequent work has shown the prediction to be correct; however, it seems that the promised X-ray crystallography manuscript never manifested. The X-ray crystal structure of S-MDMA.HCl obtained in the current work is depicted in Fig. 2. As expected, aside from the configuration, the crystal structure of the enantiomer is identical (see Supplementary Fig. S1).
Representation of the X-ray crystal structure of S-MDMA hydrochloride. Displacement ellipsoids for non-H atoms are drawn at 50% probability. Grey = C, white = H, blue = N, red = O. The crystal structure of the R-enantiomer (Supplementary Fig. S1) is identical except for absolute configuration. Crystallographic data have been deposited in the Cambridge Structural Database: CCDC 1825534 (S) and 1825511 (R). The X-ray crystal structure of racemic MDMA.HCl has been reported previously.[80]
The purity of both homochiral MDMA.HCl samples was first assessed by quantitative 1H NMR spectroscopy (Table 3) according to methodology outlined by the European Network of Forensic Science Institutes.[81] Subsequent assessment of the enantiopurity was attempted by enantioselective HPLC–UV at several wavelengths. However, for fairly concentrated (400–1000 μg/mL) samples of both R- and S-MDMA.HCl, traces of the opposite enantiomer (S and R, respectively) could not be reliably detected at all concentrations at any of the wavelengths assessed (Supplementary Fig. S2).
| Q 1H NMR purityB (%) | LOD (μg/mL) | LOQ (μg/mL) | MDMA conc. (μg/mL)C | Enantiomeric impurity (μg/mL) | Enantiopurity (%) | |
|---|---|---|---|---|---|---|
| R-MDMAA | 97.7 | 0.035 | 0.12 | 391 | 0.62 | 99.8 |
| S-MDMAA | 99.4 | 0.039 | 0.13 | 398 | 1.4 | 99.6 |
Q, quantitative; LOD, limit of detection; LOQ, limit of quantification.
ASample concentration 400 μg/mL.
B1,3,5-Trimethoxybenzene was used as the internal standard.
CConcentration of both enantiomers in the sample; calculated by factoring in wt-% purity (determined by Q 1H NMR) of each sample.
Sensitivity was enhanced by using fluorometric detection (excitation 285 nm, emission 320 nm)[82] (Supplementary Fig. S3). Quantification of the trace enantiomeric impurity by HPLC is best achieved when the minor enantiomer elutes before the major,[83] and this order was achieved for the S-MDMA sample (Supplementary Fig. S3b). Fortunately, a recent study suggests that the disadvantages of a non-ideal elution order, as for the R-MDMA sample (Supplementary Fig. S3a), do not prevent our objective of assessing enantiopurity to 0.1% accuracy.[84]
To quantify the enantiomeric impurity in each sample, measurements were carried out for (±)-MDMA hydrochloride over the concentration range 0.1–20 μg/mL (n = 5) using fluorescence detection. Calibration curves using peak areas as a function of concentration were obtained for each enantiomer from unweighted least-squares linear regression analysis of the data (Supplementary Fig. S4). Coefficients of determination (R2) were greater than 0.997 for both calibration curves. Limits of detection (LOD) and quantification (LOQ) were estimated from signal-to-noise ratios.[85] Enantiopurities of both R- and S-MDMA were thus, unequivocally, shown to be >99.5% (Table 3).
Conclusion
A novel synthesis of R and S-MDMA, taking advantage of N-tosylaziridine chirons readily available from the chiral pool, was developed. The efficiency in terms of number of steps, overall yield and enantiopurity of the target amphetamines compare favourably with previously reported methods, with further gains possible through use of more easily removed aziridine-activating groups. Indeed, during our revisions of this manuscript, Sherwood and colleagues reported similar syntheses of the homochiral enantiomers of MDMA and the corresponding primary amine MDA (3,4-methylenedioxyamphetamine) from N-Boc aziridines (Boc, tert-butoxycarbonyl).[86] The methodology reported herein should be adaptable to the synthesis of other N-alkylamphetamines and related α-alkyl-substituted phenethylamines.
The homochiral MDMA synthesised as described in this paper was investigated in non-human primate models of levodopa-induced dyskinesia in PD.[37] This study shed some light on the mechanism by which racemic MDMA alleviates major side effects of levodopa therapy. Specifically, the enantiomers work synergistically, with R-MDMA reducing severity of dyskinesia, while S-MDMA extends the therapeutic duration of levodopa. These insights have informed ongoing research into non-psychoactive MDMA analogues that improve the quality of levodopa therapy in PD animal models.[87–89]
Experimental
General
All solvents were distilled prior to use and, where appropriate, dried according to standard methods. K2CO3 was dried overnight in an oven at 140°C. Anhydrous reactions were performed using glassware that was dried in an oven at 140°C, then cooled under argon. Sonication was achieved with a Soniclean 120T (50/60 Hz, 60 W).
Flash chromatography and rapid silica filtration (RSF) were conducted using Merck silica gel 60 (63 200 μm). Thin-layer chromatography was conducted on Merck aluminium-supported silica sheets (silica gel 60 F254). Plates were visualised using UV light (254 nm) and amines were stained with ninhydrin.
Melting points were determined using a Reichert hot-stage melting point apparatus. Specific rotations were measured with a PerkinElmer 141 polarimeter (1 mL, 10 cm pathlength). Infrared spectra were recorded on a PerkinElmer SpectrumOne FTIR spectrometer (4000–400 cm−1). 1H and 13C NMR spectra were recorded in CDCl3 using Bruker ARX500 (500.1 MHz for 1H and 125.8 MHz for 13C), AV600 (600.1 MHz for 1H and 150.9 MHz for 13C) or AM300 (300 MHz for 1H) spectrometers. Chemical shifts are expressed in parts per million (ppm) relative to residual CHCl3 (1H, δ 7.26) or CDCl3 (13C, δ 77.16). Routine assignments of 13C spectra were made with the assistance of DEPT 135 and DEPT 90 experiments (DEPT, distortionless enhancement by polarisation transfer). High-resolution mass spectra (HRMS) were recorded with a VG-Autospec spectrometer using electron impact (EI) or fast-atom bombardment (FAB) ionisation, as indicated.
Enantioselective analytical HPLC was carried out on an Agilent 1200 Series HPLC instrument equipped with G1311A quaternary pump, G1315B diode-array detector (DAD) and G1321B fluorescence detector (FLD). Separation of enantiomers was performed on a Regis® amylose tris(3,5-dimethylphenylcarbamate) column (Reflect I-Amylose A, 5 μM, 250 × 4.6 mm, Regis Technologies, Inc., IL, USA) with a flow rate of 1.0 mL/min and 20 μL injection volume. All samples were subjected to isocratic elution with a 95:4.95:0.05 (v/v/v) n-hexane/EtOH/diethylamine mobile phase. UV detection was performed at 236, 254 and 280 nm; fluorescence detection was performed at an excitation wavelength of 285 nm and an emission wavelength of 320 nm.[82]
Synthesis

A solution of Br2 (5.1 mL, 0.10 mol) in DCM (100 mL) was added dropwise over 6 h to a stirred solution of 1,3-benzodioxole (11 mL, 0.10 mol) in DCM (150 mL) at 0°C. The mixture was allowed to warm to room temperature and stirring was continued overnight, after which time GC-MS analysis showed the starting material to have been consumed. The reaction mixture was diluted with sat. aq. Na2S2O7 (100 mL) and stirred for 10 min, the phases were separated and the aqueous phase was extracted with DCM (3 × 100 mL). The extract was washed with water (100 mL) and brine (100 mL), dried and concentrated under vacuum. The residue was distilled under reduced pressure to afford the bromide 18 as a pale yellow oil (18 g, 89%); bp 98–100°C at 3 mm Hg (lit.[90] 85°C at 1 mm Hg). 1H NMR (600 MHz) δ 6.96–6.93 (m, 2H, H4/H7), 6.69 (m, 1H, H6), 5.97 (s, 2H, H2) ppm. The 1H NMR spectrum matched the reported data.[91]

Mg flakes (2.44 g, 100 mmol) were stirred under argon overnight. THF (40 mL) was added to the blackened Mg, followed by dropwise addition of a solution of bromide 18 (4.02 g, 20.0 mmol) in anhydrous THF (20 mL). Initiation of Grignard reagent formation was indicated by warming of the reaction vessel and darkening of the solution. The reaction mixture was stirred for 4 h at rt, then 1 h at 45°C. The suspension was allowed to settle; the supernatant was cannulated from the remaining Mg, and was then cooled to −30°C. CuI (0.58 g, 3.0 mmol) was added and the pale yellow mixture was stirred for 45 min, then cooled to −78°C, and treated dropwise with a solution of aziridine R19[60,61] (2.113, 10.00 mmol) in THF (40 mL). The reaction mixture was allowed to warm gradually to rt and stirring was continued overnight. The reaction mixture was diluted with sat. aq. NH4Cl (120 mL) and concentrated under reduced pressure. The aqueous residue was extracted with Et2O (3 × 40 mL) and the extract was washed with brine (40 mL), dried and evaporated. The residue was subjected to flash chromatography. Elution with 1:7 EtOAc/petrol afforded the sulfonamide R20 as a colourless gum (3.10 g, 93%), which solidified to give an amorphous, colourless solid over a period of months; mp 59–63°C. +6.1° (c 1.2, CHCl3). IR νmax (film, DCM) 3286 (N–H), 1490, 1248, 1159, 1039 cm−1. 1H NMR (600 MHz) δ 7.62–7.59 (m (pseudo d), 2H, H2′/6′), 7.25–7.22 (m (pseudo d), 2H, H3′/5′), 6.65 (d, J = 7.8 Hz, 1H, H5′), 6.46 (dd, J = 7.9, 1.7 Hz, 1H, H6′), 6.42 (d, J = 1.7 Hz, 1H, H2′), 5.92 (d, J = 1.4 Hz, 1H, O2CH2a), 5.91 (d, J = 1.4 Hz, 1H, O2CH2b), 4.24 (br d, J = 7.1 Hz, 1H, NH), 3.47–3.40 (m, 1H, H2), 2.59 (dd, J = 13.8, 6.3 Hz, 1H, H1a), 2.55 (dd, J = 13.8, 6.9 Hz, 1H, H1b), 2.42 (s, 3H, 4″–CH3), 1.11 (d, J = 6.5 Hz, 3H, H3). 13C NMR (150.9 MHz) δ 147.8 (ArO), 146.5 (ArO), 143.3 (Ar), 137.6 (Ar), 130.9 (C1′), 129.7 (2 × ArH), 127.1 (2 × ArH), 122.5 (ArH), 109.6 (ArH), 108.4 (ArH), 101.1 (CH2O2), 51.1 (C2), 43.2 (C1), 21.64 (CH3), 21.63 (CH3). HRMS (EI): m/z calcd for C17H19O4S˙+ [M]˙+ 333.1029; observed 333.1028.

K2CO3 (3.32 g, 24.0 mmol) and MeI (1.0 mL, 16 mmol) were added to a solution of sulfonamide R20 (2.66 g, 8.00 mmol) in anhydrous DMF (25 mL) under argon. The mixture was stirred for 20 h, after which time TLC indicated the reaction was complete. The reaction mixture was diluted with water (250 mL) and extracted with Et2O (3 × 80 mL). The extract was washed with water (2 × 80 mL) and brine (80 mL), dried and evaporated, and the residue was subjected to flash chromatography. Elution with DCM gave R21 as a colourless gum (2.77 g, quant.). −36.8° (c 1.5, CHCl3). IR νmax (film, DCM) 3422, 1490, 1335, cm−1. 1H NMR (600 MHz) δ 7.60–7.56 (m, 2H, H2″/6″), 7.25–7.21 (m, 2H, H3″/5″), 6.68 (d, J = 7.9 Hz, H5′), 6.58 (d, J = 1.7 Hz, 1H, 2′), 6.55 (dd, J = 7.9, 1.7 Hz, 1H, H6′), 5.93 (AB, (pseudo d ‘J’ = 1.5 Hz), 1H, CH2O2a), 5.92 (AB, (pseudo d ‘J’ = 1.5 Hz), 1H, CH2O2b), 4.24–4.17 (m, 1H, H2), 2.74 (s, 3H, NCH3), 2.61 (dd, J = 13.6, 6.5 Hz, 1H, H1′a), 2.49 (dd, J = 13.6, 8.3 Hz, 1H, H1′b), 2.40 (s, 3H, 4″-CH3), 0.95 (d, J = 6.7 Hz, 3H, H3). 13C NMR (150.9 MHz) δ 147.7 (ArO), 146.3 (ArO), 143.0 (Ar), 137.2 (Ar), 132.1 (C1′), 129.6 (2 × ArH), 127.2 (2 × ArH), 122.2 (ArH), 109.5 (ArH), 108.3 (ArH), 101.0 (CH2O2), 54.6 (C2), 40.8 (C1), 28.0 (NCH3), 21.6 (CH3), 17.0 (CH3). HRMS (EI): m/z calcd for C18H21O4S˙+ [M]˙+ 347.1186; observed 347.1186.

A solution of sulfonamide R21 (2.72 g, 7.80 mmol) in anhydrous THF (50 mL) was added to a suspension of Mg powder (1.90 g, 78.0 mmol) in dry MeOH (100 mL) under argon. The suspension was sonicated for 1 h, during which time a colourless precipitate formed and the Mg was consumed. Additional Mg powder (0.95 g, 39 mmol) was added, and sonication was continued for 1 h. Most of the solvent was evaporated, and the residue was diluted with saturated NaHCO3 (100 mL) and extracted with Et2O (3 × 30 mL). The extract was washed with brine, dried and evaporated, and the reside subjected to RSF. Elution with 48:1:1 Et2O/MeOH/NEt3 gave unreacted R21 as a colourless oil (1.42 g, 52%). Further elution afforded R-MDMA as a colourless oil (690 mg, 46%), −34.5° (c 1.1, CHCl3). 1H NMR (600 MHz) δ 6.74 (d, J = 7.9 Hz, 1H, H5′), 6.67 (d, J = 1.7 Hz, 1H, H2′), 6.63 (dd, J = 7.9, 1.7 Hz, 1H, H6′), 5.93 (s, 2H, CH2O2), 2.75–2.69 (m, 1H, H2), 2.61 (dd, J = 13.5, 7.2 Hz, 1H, H1a), 2.54 (dd, J = 13.5, 6.2 Hz, 1H, H1b), 2.39 (s, 3H, NCH3), 1.58 (br s, 1H, NH + H2O), 1.05 (d, J = 6.2 Hz, 3H, H3). 13C NMR (125.8 MHz) δ 147.8 (ArO), 146.1 (ArO), 133.3 (C1′), 122.3 (ArH), 109.6 (ArH), 108.3 (ArH), 101.0 (CH2O2), 56.6 (C2), 43.3 (C1), 34.1 (NCH3), 19.8 (C3). HRMS (EI): m/z calcd for C18H21O4S˙+ [M]˙+ 193.1097; observed 193.1102. The 1H spectrum matched the data previously reported for the racemate.[49]
A solution of the sulfonamide R21 (0.17 g, 0.50 mmol), NaBH4 (0.19 g, 5.0 mmol) and 1,4-dimethoxybenzene (0.28 g, 2.0 mmol) in 1:2 MeCN/EtOH (15 mL) in a quartz tube was irradiated at 254 nm in a Rayonet reactor for 5 h, after which time the reaction had stalled based on TLC analysis. The volatiles were evaporated and the residue was subjected to RSF. Elution with 25:73:2 EtOAc/petrol/NEt3 yielded R-MDMA as a colourless oil (32 mg, 33%), −33.7° (c 1.0, CHCl3), identical in all other respects with the material described above.

R-MDMA was dissolved in excess methanolic HCl. The solution was evaporated under a stream of N2, and the solid residue was crystallised from iPrOH to afford R-MDMA.HCl as colourless shards, mp 185–186°C (lit.[51] 192–193°C (EtOH/Et2O)). −19.3° (c 1.1, H2O) (lit.[51] −17.5° (c 1, H2O)). 1H NMR (600 MHz, CDCl3) δ 9.64 (v br s, 2H, NH2), 6.75 (d, J = 7.9 Hz, 1H, H5′), 6.70 (d, J = 1.6 Hz, 1H, H2′), 6.68 (dd, J = 7.9, 1.7 Hz, 1H, H6′), 5.94 (AB (pseudo d ‘J’ = 1.5 Hz), 1H, CH2O2a), 5.94 (AB (pseudo d ‘J’ = 1.5 Hz), 1H, CH2O2b), 3.37 (dd, J = 13.2, 4.2 Hz, 1H, H1a), 3.30–3.22 (m, 1H, H2), 2.77 (dd, J = 13.2, 10.4 Hz, 1H, H1b), 2.69 (s, 3H, NCH3), 1.34 (d, J = 6.5 Hz, 3H, H3). The 1H NMR spectrum matched the previously reported data.[51]

Ring-opening as described for R20 from aziridine S19[60,61] (1.06 g, 5.02 mmol) afforded sulfonamide S20 as a colourless gum (1.62 g, 97%), −6.3° (c 1.3, CHCl3). HRMS (EI): m/z calcd for C17H19O4S˙+ [M]˙+ 333.1029; observed 333.1036. The IR, 1H and 13C NMR spectra were identical to those for the enantiomer R20.

Methylation of sulfonamide S20 (1.50 g, 4.50 mmol) as described above for R21 afforded S21 as a colourless gum (1.54 g, 99%). +38.4° (c 1.2, CHCl3). HRMS (EI): m/z calcd for C18H21O4S˙+ [M]˙+ 347.1186; observed 347.1189. The IR, 1H and 13C NMR spectra were identical to those for the enantiomer R21.

Mg powder (485 mg, 20.0 mmol) was added to a solution of S21 (695 mg, 2.00 mmol) in dry MeOH (20 mL) under argon, and the suspension was sonicated for 1 h, during which time a colourless precipitate formed, and the Mg was consumed. Additional Mg powder (485 mg, 20.0 mmol) was added and sonication was continued for 1 h. The reaction mixture was diluted with sat. aq. NaHCO3 (200 mL) and extracted with Et2O (3 × 60 mL). The extract was washed with brine, dried and evaporated, and the residue was subjected to RSF. Elution with 25:73:2 EtOAc/petrol/NEt3 afforded unreacted S21 as a colourless oil (217 mg, 56%). Further elution gave S-MDMA as a colourless oil (169 mg, 44%), +33.1° (c 1.2, CHCl3). HRMS (EI): m/z calcd for C18H21O4S˙+ [M]˙+ 193.1097; observed 193.1096. The 1H and 13C NMR spectra were identical to those for R-MDMA.
A solution of S21 (0.87 g, 2.5 mmol) in THF (10 mL) was added to a suspension of Mg powder (0.61 g, 25 mmol) in dry MeOH (15 mL) under argon, and the suspension was sonicated for 1 h. Additional Mg powder (0.61 g, 25 mmol, 2 h) was added and sonication was continued for 2 h. Workup as described for Method A was followed by flash chromatography. Elution with Et2O then 98:2 Et2O/NEt3 afforded unreacted S21 as a colourless oil (0.27 g, 31%). Further elution gave S-MDMA as a colourless oil (0.26 g, 54%), +32.7° (c 1.0, CHCl3), identical in all other respects to its enantiomer (R)-MDMA.
DME (50 mL) was added to a mixture of naphthalene (1.28 g, 10.0 mmol) and Na pieces (230 mg, 10.0 mmol) under argon. The surface of the Na immediately turned dark green. The reaction mixture was stirred for 2 h during which time the green colour darkened and persisted. The sodium naphthalide solution so formed was added dropwise to a solution of S21 (1.40 g, 4.03 mmol) in DME (30 mL) at −78°C. A persistent green endpoint was not discernible. The reaction mixture was stirred at −78°C for 1 h and at rt for 2 h, before being quenched with EtOH (10 mL) and evaporated. The solid residue was dissolved in Et2O, and the solution was washed with brine, dried and evaporated. The residue was subjected to RSF. Elution with petrol afforded naphthalene. Further elution with 1:3 EtOAc/petrol gave unreacted S21 as a colourless oil. Further elution with 25:73:2 EtOAc/petrol/NEt3 afforded S-MDMA as a colourless oil (392 mg, 50%), +33.0° (c 1.0, CHCl3) identical in all other respects to its enantiomer R-MDMA.

The hydrochloride was prepared as described above for R-MDMA, affording S-MDMA.HCl as colourless crystals; mp 184–186.0°C (MeOH/Et2O) (lit.[51] 192–193°C (EtOH/Et2O)). +18.3° (c 1.0, H2O) (lit.[51] +17.43° (c 1, H2O)), identical in all respects with its enantiomer R-MDMA.HCl.

CuI (57 mg, 0.30 mmol) was added to a stirred solution of Grignard reagent (prepared as described above for R20 from 5-bromobenzodioxole 18 (402 mg, 2.00 mmol)) in anhydrous THF (3 mL) at −78°C under argon. After 2 h, a solution of aziridine S22 (287 mg, 1.05 mmol) in anhydrous THF (5 mL) was added dropwise to the reaction mixture and stirring was continued for 2 h, whereupon a brown solution formed. The reaction mixture was gradually warmed to −10°C over a period of 4 h, held at this temperature for 30 min, then quenched with saturated aqueous NH4Cl (30 mL) and extracted with Et2O (3 × 30 mL). The extract was washed with water (30 mL) and brine (30 mL), dried and evaporated. The brown residue was subjected to flash chromatography. Elution with 1:9 EtOAc/petrol afforded slightly impure 24 as a colourless gum that crystallised from MeOH/H2O to give 24 as fine colourless flakes (189 mg, 46%); mp 115–116°C. Rf (1:9 EtOAc/petrol) 0.15. IR (KBr) νmax cm−1: 3202, 1248, 1159, 1038. [α]D 4.3° (c 1.50, CHCl3). 1H NMR (500 MHz) δ 7.70–7.66 (m, 2H, H2‴/H6‴), 7.33–7.29 (m, 2H, H3‴/H5‴), 7.29–7.24 (m, 2H, H3″/H5″ + CHCl3), 7.23–7.19 (m, 1H, H4″), 7.09–7.06 (m, 2H, H2″/H6″), 6.71 (d, J = 8.0 Hz, 1H, H5′), 6.57 (ddd, J = 8.0, 1.8, 0.4 Hz, 1H, H6′), 6.53 (d, J = 1.8 Hz, 1H, H2′), 5.91 (q [AB], J = 1.4 Hz, 2H, CH2O2), 4.29 (dd (app. t), J = 6.2 Hz, 1H, NH), 3.97 (dd (app. t), J = 7.9 Hz, 1H, H2), 3.54–3.43 (m, 2H, H1), 2.45 (s, 3H, CH3) ppm. 13C NMR (125 MHz) δ 148.2 (ArO), 146.8 (ArO), 143.7 (Ar), 140.9 (Ar), 136.9 (Ar), 134.7 (Ar), 129.9 (2 × ArH), 129.0 (2 × ArH), 127.9 (2 × ArH), 127.31 (2 × ArH), 127.30, 121.2 (ArH), 108.6 (ArH), 108.4 (ArH), 101.2 (CH2O2), 50.4 (C1 or C2), 47.4 (C1 or C2), 21.7 (CH3). HRMS (FAB): m/z calcd for C22H21NO4S˙+ [M]˙+ 395.1186; observed 395.1173. 1H NMR spectroscopic assignments were made with the assistance of D2O exchange and COSY (correlation spectroscopy) experiments.
Acknowledgements
KDL was the recipient of an Australian Postgraduate Award. We thank Drs Lindsay Burn and Tony Reeder for assistance with NMR spectroscopy and mass spectrometry, respectively. Hung Nguyen qualifies for authorship but was unable to be contacted to approve being included as an author of this paper. We acknowledge the traditional custodians of the land on which this work was undertaken, the Whadjuk Noongar people. We respect their continuing culture and the contribution they make to UWA, Perth and the region.
References
1 Shulgin AT. The background and chemistry of MDMA. J Psychoactive Drugs 1986; 18: 291-304.
| Crossref | Google Scholar |
2 Pentney AR. An exploration of the history and controversies surrounding MDMA and MDA. J Psychoactive Drugs 2001; 33: 213-21.
| Crossref | Google Scholar |
3 Freudenmann RW, Öxler F, Bernschneider-Reif S. The origin of MDMA (ecstasy) revisited: the true story reconstructed from the original documents. Addiction 2006; 101: 1241-5.
| Crossref | Google Scholar |
4 Karch SB. A historical review of MDMA. Open Forensic Sci J 2011; 4: 20-4.
| Crossref | Google Scholar |
5 Dunlap LE, Andrews AM, Olson DE. Dark classics in chemical neuroscience: 3,4-methylenedioxymethamphetamine. ACS Chem Neurosci 2018; 9: 2408-27.
| Crossref | Google Scholar |
6 Walker S, Pullella G, Piggott M, Duggan P. Introduction to the chemistry and pharmacology of psychedelic drugs. Aust J Chem 2023; 76:.
| Crossref | Google Scholar |
7 Johnson MP, Hoffman AJ, Nichols DE. Effects of enantiomers of MDA, MDMA and related analogues on [3H]serotonin and [3H]dopamine release from superfused rat brain slices. Eur J Pharmacol 1986; 132: 269-76.
| Crossref | Google Scholar |
8 Young MB, Andero R, Ressler KJ, Howell LL. 3,4-Methylenedioxymethamphetamine facilitates fear extinction learning. Transl Psychiatry 2015; 5: e634.
| Crossref | Google Scholar |
9 Sessa B, Higbed L, Nutt D. A review of 3,4-methylenedioxymethamphetamine (MDMA)-assisted psychotherapy. Front Psychiatry 2019; 10: 138.
| Crossref | Google Scholar |
10 Mitchell JM, Bogenschutz M, Lilienstein A, Harrison C, Kleiman S, Parker-Guilbert K, Ot’alora G M, Garas W, Paleos C, Gorman I, Nicholas C, Mithoefer M, Carlin S, Poulter B, Mithoefer A, Quevedo S, Wells G, Klaire SS, van der Kolk B, Tzarfaty K, Amiaz R, Worthy R, Shannon S, Woolley JD, Marta C, Gelfand Y, Hapke E, Amar S, Wallach Y, Brown R, Hamilton S, Wang JB, Coker A, Matthews R, de Boer A, Yazar-Klosinski B, Emerson A, Doblin R. MDMA-assisted therapy for severe PTSD: a randomized, double-blind, placebo-controlled phase 3 study. Nat Med 2021; 27: 1025-33.
| Crossref | Google Scholar |
11 Prior positive results confirmed in MAPS-sponsored, philanthropy-funded Phase 3 Trial: MAPS: Multidisciplinary Association for Psychedelic Studies. 2023. Available at https://maps.org/2023/01/05/prior-positive-results-confirmed/ [accessed 25 March 2023].
12 Change to classification of psilocybin and MDMA to enable prescribing by authorised psychiatrists: Therapeutic Goods Administration. Department of Health and Aged Care, Australian Government; 2023. Available at https://www.tga.gov.au/news/media-releases/change-classification-psilocybin-and-mdma-enable-prescribing-authorised-psychiatrists [updated 3 February 2023; accessed 25 March 2023].
13 Brewerton TD, Wang JB, Lafrance A, Pamplin C, Mithoefer M, Yazar-Klosinki B, Emerson A, Doblin R. MDMA-assisted therapy significantly reduces eating disorder symptoms in a randomized placebo-controlled trial of adults with severe PTSD. J Psychiatr Res 2022; 149: 128-35.
| Crossref | Google Scholar |
14 Brewerton TD, Lafrance A, Mithoefer MC. The potential use of N-methyl-3,4-methylenedioxyamphetamine (MDMA) assisted psychotherapy in the treatment of eating disorders comorbid with PTSD. Med Hypotheses 2021; 146: 110367.
| Crossref | Google Scholar |
15 Sessa B, Aday JS, O’Brien S, Curran HV, Measham F, Higbed L, Nutt DJ. Debunking the myth of ‘Blue Mondays’: no evidence of affect drop after taking clinical MDMA. J Psychopharmacol 2022; 36: 360-7.
| Crossref | Google Scholar |
16 Nicholas CR, Wang JB, Coker A, Mitchell JM, Klaire SS, Yazar-Klosinski B, Emerson A, Brown RT, Doblin R. The effects of MDMA-assisted therapy on alcohol and substance use in a phase 3 trial for treatment of severe PTSD. Drug Alcohol Depend 2022; 233: 109356.
| Crossref | Google Scholar |
17 Sessa B, Higbed L, O’Brien S, Durant C, Sakal C, Titheradge D, Williams TM, Rose-Morris A, Brew-Girard E, Burrows S, Wiseman C, Wilson S, Rickard J, Nutt DJ. First study of safety and tolerability of 3,4-methylenedioxymethamphetamine-assisted psychotherapy in patients with alcohol use disorder. J Psychopharmacol 2021; 35: 375-83.
| Crossref | Google Scholar |
18 Sessa B, Sakal C, O’Brien S, Nutt D. First study of safety and tolerability of 3,4-methylenedioxymethamphetamine (MDMA)-assisted psychotherapy in patients with alcohol use disorder: preliminary data on the first four participants. BMJ Case Rep 2019; 12: e230109.
| Crossref | Google Scholar |
19 Sessa B. Why MDMA therapy for alcohol use disorder? And why now? Neuropharmacology 2018; 142: 83-8.
| Crossref | Google Scholar |
20 Rezvani AH, Garges PL, Miller DB, Gordon CJ. Attenuation of alcohol consumption by MDMA (ecstasy) in two strains of alcohol-preferring rats. Pharmacol Biochem Behav 1992; 43: 103-10.
| Crossref | Google Scholar |
21 Jerome L, Schuster S, Yazar-Klosinski BB. Can MDMA play a role in the treatment of substance abuse? Curr Drug Abuse Rev 2013; 6: 54-62.
| Crossref | Google Scholar |
22 Pitts EG, Curry DW, Hampshire KN, Young MB, Howell LL. (±)-MDMA and its enantiomers: potential therapeutic advantages of R(−)-MDMA. Psychopharmacology 2018; 235: 377-92.
| Crossref | Google Scholar |
23 Schimmers N, Breeksema JJ, Smith-Apeldoorn SY, Veraart J, van den Brink W, Schoevers RA. Psychedelics for the treatment of depression, anxiety, and existential distress in patients with a terminal illness: a systematic review. Psychopharmacology 2022; 239: 15-33.
| Crossref | Google Scholar |
24 Luoma JB, Shahar B, Kati Lear M, Pilecki B, Wagner A. Potential processes of change in MDMA-assisted therapy for social anxiety disorder: enhanced memory reconsolidation, self-transcendence, and therapeutic relationships. Hum Psychopharmacol 2022; 37: e2824.
| Crossref | Google Scholar |
25 Wolfson PE, Andries J, Feduccia AA, Jerome L, Wang JB, Williams E, Carlin SC, Sola E, Hamilton S, Yazar-Klosinski B, Emerson A, Mithoefer MC, Doblin R. MDMA-assisted psychotherapy for treatment of anxiety and other psychological distress related to life-threatening illnesses: a randomized pilot study. Sci Rep 2020; 10: 20442.
| Crossref | Google Scholar |
26 Danforth AL, Grob CS, Struble C, Feduccia AA, Walker N, Jerome L, Yazar-Klosinski B, Emerson A. Reduction in social anxiety after MDMA-assisted psychotherapy with autistic adults: a randomized, double-blind, placebo-controlled pilot study. Psychopharmacology 2018; 235: 3137-48.
| Crossref | Google Scholar |
27 Danforth AL, Struble CM, Yazar-Klosinski B, Grob CS. MDMA-assisted therapy: A new treatment model for social anxiety in autistic adults. Prog Neuropsychopharmacol Biol Psychiatry 2016; 64: 237-49.
| Crossref | Google Scholar |
28 Christie D, Yazar-Klosinski B, Nosova E, Kryskow P, Siu W, Lessor D, Argento E. MDMA-assisted therapy is associated with a reduction in chronic pain among people with post-traumatic stress disorder. Front Psychiatry 2022; 13: 939302.
| Crossref | Google Scholar |
29 Anderson III GM, Braun G, Braun U, Nichols DE, Shulgin AT. Absolute configuration and psychotomimetic activity. NIDA Res Monogr 1978; 22: 8-15.
| Google Scholar |
30 Young R, Glennon RA. MDMA (N-methyl-3,4-methylenedioxyamphetamine) and its stereoisomers: similarities and differences in behavioral effects in an automated activity apparatus in mice. Pharmacol Biochem Behav 2008; 88: 318-31.
| Crossref | Google Scholar |
31 Nichols DE, Lloyd DH, Hoffman AJ, Nichols MB, Yim GKW. Effects of certain hallucinogenic amphetamine analogs on the release of [3H]-serotonin from rat brain synaptosomes. J Med Chem 1982; 25: 530-5.
| Crossref | Google Scholar |
32 Verrico CD, Miller GM, Madras BK. MDMA (ecstasy) and human dopamine, norepinephrine, and serotonin transporters: implications for MDMA-induced neurotoxicity and treatment. Psychopharmacology 2006; 189: 489-503.
| Crossref | Google Scholar |
33 Murnane KS, Fantegrossi WE, Godfrey JR, Banks ML, Howell LL. Endocrine and neurochemical effects of 3,4-methylenedioxymethamphetamine and its stereoisomers in Rhesus monkeys. J Pharmacol Exp Ther 2010; 334: 642-50.
| Crossref | Google Scholar |
34 Lyon RA, Glennon RA, Titeler M. 3,4-Methylenedioxymethamphetamine (MDMA): stereoselective interactions at brain 5-HT1 and 5-HT2 receptors. Psychopharmacology 1986; 88: 525-6.
| Crossref | Google Scholar |
35 Toll L, Berzetei-Gurske IP, Polgar WE, Brandt SR, Adapa ID, Rodriguez L, Schwartz RW, Haggart D, O’Brien A, White A, Kennedy JM, Craymer K, Farrington L, Auh JS. Standard binding and functional assays related to medications development division testing for potential cocaine and opiate narcotic treatment medications. NIDA Res Monogr 1998; 178: 440-66.
| Google Scholar |
36 Nash JF, Roth BL, Brodkin JD, Nichols DE, Gudelsky GA. Effect of the R(−) and S(+) isomers of MDA and MDMA on phosphatidylinositol turnover in cultured cells expressing 5-HT2A or 5-HT2C receptors. Neurosci Lett 1994; 177: 111-5.
| Crossref | Google Scholar |
37 Huot P, Johnston TH, Lewis KD, Koprich JB, Reyes MG, Fox SH, Piggott MJ, Brotchie JM. Characterization of 3,4-methylenedioxymethamphetamine (MDMA) enantiomers in vitro and in the MPTP-lesioned primate: R-MDMA reduces severity of dyskinesia, whereas S-MDMA extends duration of ON-time. J Neurosci 2011; 31: 7190-8.
| Crossref | Google Scholar |
38 Murnane KS, Kimmel HL, Rice KC, Howell LL. The neuropharmacology of prolactin secretion elicited by 3,4-methylenedioxymethamphetamine (‘ecstasy’): a concurrent microdialysis and plasma analysis study. Horm Behav 2012; 61: 181-90.
| Crossref | Google Scholar |
39 Curry DW, Young MB, Tran AN, Daoud GE, Howell LL. Separating the agony from ecstasy: R(−)-3,4-methylenedioxymethamphetamine has prosocial and therapeutic-like effects without signs of neurotoxicity in mice. Neuropharmacology 2018; 128: 196-206.
| Crossref | Google Scholar |
40 Acute effects of R- and S-MDMA in healthy subjects (R-S-MDMA). Basel, Switzerland: University Hospital, ClinicalTrials.gov, National Institutes of Health; 2022. Available at https://clinicaltrials.gov/ct2/show/NCT05277636 [accessed 26 March 2023].
41 Ecstasy & agony, BBC Horizon Series, Harrison J. 2001. Available at https://www.youtube.com/watch?v=FfP-cUTER5c [accessed 26 February 2023].
42 Sgambato V, Tremblay L. Pathophysiology of dyskinesia and behavioral disorders in non-human primates: the role of serotonergic fibers. J Neural Transm 2018; 125: 1145-56.
| Crossref | Google Scholar |
43 Beaudoin-Gobert M, Epinat J, Météreau E, Duperrier S, Neumane S, Ballanger B, Lavenne F, Liger F, Tourvielle C, Bonnefoi F, Costes N, Bars DL, Broussolle E, Thobois S, Tremblay L, Sgambato-Faure V. Behavioural impact of a double dopaminergic and serotonergic lesion in the non-human primate. Brain 2015; 138: 2632-47.
| Crossref | Google Scholar |
44 Lettfuss NY, Fischer K, Sossi V, Pichler BJ, von Ameln-Mayerhofer A. Imaging DA release in a rat model of L-DOPA-induced dyskinesias: a longitudinal in vivo PET investigation of the antidyskinetic effect of MDMA. Neuroimage 2012; 63: 423-33.
| Crossref | Google Scholar |
45 Bishop C, Taylor JL, Kuhn DM, Eskow KL, Park JY, Walker PD. MDMA and fenfluramine reduce L-DOPA-induced dyskinesia via indirect 5-HT1A receptor stimulation. Eur J Neurosci 2006; 23: 2669-76.
| Crossref | Google Scholar |
46 Iravani MM, Jackson MJ, Kuoppamäki M, Smith LA, Jenner P. 3,4-Methylenedioxymethamphetamine (ecstasy) inhibits dyskinesia expression and normalizes motor activity in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated primates. J Neurosci 2003; 23: 9107-15.
| Crossref | Google Scholar |
47 Sgambato V. Breathing new life into neurotoxic-based monkey models of Parkinson’s disease to study the complex biological interplay between serotonin and dopamine. Prog Brain Res 2021; 261: 265-85.
| Crossref | Google Scholar |
48 Wasik AM, Gandy MN, McIldowie M, Holder MJ, Chamba A, Challa A, Lewis KD, Young SP, Scheel-Toellner D, Dyer MJ, Barnes NM, Piggott MJ, Gordon J. Enhancing the anti-lymphoma potential of 3,4-methylenedioxymethamphetamine (‘ecstasy’) through iterative chemical redesign: mechanisms and pathways to cell death. Invest New Drugs 2012; 30: 1471-83.
| Crossref | Google Scholar |
49 Gandy MN, McIldowie M, Lewis K, Wasik AM, Salomonczyk D, Wagg K, Millar ZA, Tindiglia D, Huot P, Johnston T, Thiele S, Nguyen B, Barnes NM, Brotchie JM, Martin-Iverson MT, Nash J, Gordon J, Piggott MJ. Redesigning the designer drug ecstasy: non-psychoactive MDMA analogues exhibiting Burkitt’s lymphoma cytotoxicity. MedChemComm 2010; 1: 287-93.
| Crossref | Google Scholar |
50 Meredith EJ, Holder MJ, Chamba A, Challa A, Lee AD, Bunce CM, Drayson MT, Pilkington G, Blakely RD, Dyer MJS, Barnes NM, Gordon J. The serotonin transporter (SLC6A4) is present in B-cell clones of diverse malignant origin: probing a potential antitumor target for psychotropics. FASEB J 2005; 19: 1187-9.
| Crossref | Google Scholar |
51 Nichols DE, Hoffman AJ, Oberlender RA, Jacob III P, Shulgin AT. Derivatives of 1-(1,3-benzodioxol-5-yl)-2-butanamine: representatives of a novel therapeutic class. J Med Chem 1986; 29: 2009-15.
| Crossref | Google Scholar |
52 Llabrés S, García-Ratés S, Cristóbal-Lecina E, Riera A, Borrell JI, Camarasa J, Pubill D, Luque FJ, Escubedo E. Molecular basis of the selective binding of MDMA enantiomers to the α4β2 nicotinic receptor subtype: synthesis, pharmacological evaluation and mechanistic studies. Eur J Med Chem 2014; 81: 35-46.
| Crossref | Google Scholar |
54 Effenberger F, Jäger J. Stereoselective synthesis of (S)-3,4-methylenedioxyamphetamines from (R)-cyanohydrins. Chem Eur J 1997; 3: 1370-4.
| Crossref | Google Scholar |
55 Pizarro N, de la Torre R, Farré M, Segura J, Llebaria A, Joglar J. Synthesis and capillary electrophoretic analysis of enantiomerically enriched reference standards of MDMA and its main metabolites. Bioorg Med Chem 2002; 10: 1085-92.
| Crossref | Google Scholar |
56 Lourenço TC, Bósio GC, Cassiano NM, Cass QB, Moreau RLM. Chiral separation of 3,4-methylenedioxymethamphetamine (MDMA) enantiomers using batch chromatography with peak shaving recycling and its effects on oxidative stress status in rat liver. J Pharm Biomed Anal 2013; 73: 13-7.
| Crossref | Google Scholar |
57 Hashimoto K, Hirai K, Goromaru T. Synthesis of racemic, S(+)- and R(−)-N-[methyl-3H]3,4-methylenedioxymethamphetamine. J Labelled Comp Radiopharm 1990; 28: 465-9.
| Crossref | Google Scholar |
59 Nenajdenko VG, Karpov AS, Balenkova ES. A new convenient approach to chiral β-aryl(heteroaryl)alkylamines. Tetrahedron Asymmetry 2001; 12: 2517-27.
| Crossref | Google Scholar |
60 Berry MB, Craig D. A convenient method for the preparation of enantiomerically pure 2-substituted N-tosylaziridines. Synlett 1992; 1992 41-4.
| Crossref | Google Scholar |
61 Pei Y, Brade K, Brulé E, Hagberg L, Lake F, Moberg C. A general method for the preparation of chiral TREN derivatives. Eur J Org Chem 2005; 2005: 2835-40.
| Crossref | Google Scholar |
62 Dallacker F, Zegers H. Derivatives of methylenedioxybenzene. XII. Introduction of sulfur into pyrocatechol methylene ether derivatives. Justus Liebigs Ann Chem 1965; 689: 156-62.
| Crossref | Google Scholar |
63 Tanner D. Chiral aziridines. Preparation and stereoselective transformations. Angew Chem 1994; 106: 625-46.
| Crossref | Google Scholar |
64 Onistschenko A, Buchholz B, Stamm H. Reactions with aziridines - 39: ring opening of activated 2,2-dimethylaziridines by Grignard reagents. A mechanistic study. Tetrahedron 1987; 43: 565-76.
| Crossref | Google Scholar |
65 Buchowlecki W, Grosman-Zjawiona Z, Zjawiony J. 1-Diphenylphosphinyl-2,2-dimethylaziridine – a new precursor of α,α-dimethylarylalkylamines. Tetrahedron Lett 1985; 26: 1245-8.
| Crossref | Google Scholar |
66 Kozikowski AP, Ishida H, Isobe K. Ring opening reactions of aziridines with organometallics. J Org Chem 1979; 44: 2788-90.
| Crossref | Google Scholar |
68 Alonso DA, Andersson PG. Deprotection of sulfonyl aziridines. J Org Chem 1998; 63: 9455-61.
| Crossref | Google Scholar |
69 Ji S, Gortler LB, Waring A, Battisti AJ, Bank S, Closson WD, Wriede PA. Cleavage of sulfonamides with sodium naphthalene. J Am Chem Soc 1967; 89: 5311-2.
| Crossref | Google Scholar |
70 Lautens M, Fagnou K, Zunic V. An expedient enantioselective route to diaminotetralins: application in the preparation of analgesic compounds. Org Lett 2002; 4: 3465-8.
| Crossref | Google Scholar |
71 Hamada T, Nishida A, Yonemitsu O. Selective removal of electron-accepting p-toluene- and naphthalenesulfonyl protecting groups for amino function via photoinduced donor–acceptor ion pairs with electron-donating aromatics. J Am Chem Soc 1986; 108: 140-5.
| Crossref | Google Scholar |
72 Art JF, Kestemont JP, Soumillion JP. Photodetosylation of sulfonamides initiated by electron transfer from an anionic sensitizer. Tetrahedron Lett 1991; 32: 1425-8.
| Crossref | Google Scholar |
73 Hu XE. Nucleophilic ring opening of aziridines. Tetrahedron 2004; 60: 2701-43.
| Crossref | Google Scholar |
74 Hu XE, Kim NK, Ledoussal B, Colson A-O. Regio- and stereo-controlled copper organometallic addition to a piperidinyl aziridine: synthesis of trans-3-amino-4-alkyl-piperidines. Tetrahedron Lett 2002; 43: 4289-93.
| Crossref | Google Scholar |
75 Bieber LW, De Araújo MCF. Short and efficient synthesis of optically active N-tosyl aziridines from 2-amino alcohols. Molecules 2002; 7: 902-6.
| Crossref | Google Scholar |
76 Sun H, Yang C, Lin R, Xia W. Regioselective ring-opening nucleophilic addition of aziridines through photoredox catalyst. Adv Synth Catal 2014; 356: 2775-80.
| Crossref | Google Scholar |
77 Zhang R, McIntyre PJ, Collins PM, Foley DJ, Arter C, von Delft F, Bayliss R, Warriner S, Nelson A. Construction of a shape-diverse fragment set: design, synthesis and screen against aurora-A kinase. Chem Eur J 2019; 25: 6831-9.
| Crossref | Google Scholar |
78 Toshimitsu A, Abe H, Hirosawa C, Tamao K. Conversion of chiral oxiranes into chiral aziridines with retention of configuration by way of chiral episulfonium ions and reactions of the aziridines with Grignard reagents. J Chem Soc Perkin Trans 1 1994; 3465-71.
| Crossref | Google Scholar |
79 Nichols DE, Barfknecht CF, Rusterholz DB, Benington F, Morin RD. Asymmetric synthesis of psychotomimetic phenylisopropylamines. J Med Chem 1973; 16: 480-3.
| Crossref | Google Scholar |
80 Morimoto BH, Lovell S, Kahr B. Ecstasy: 3,4-methylenedioxymethamphetamine (MDMA). Acta Crystallogr C 1998; 54: 229-31.
| Crossref | Google Scholar |
81 Schoenberger T. Guideline for qNMR Analysis. European Network of Forensic Science Institutes; 2019. Available at https://enfsi.eu/wp-content/uploads/2017/06/qNMR-Guideline_version001.pdf [accessed 28 March 2023].
82 Sadeghipour F, Veuthey JL. Enantiomeric separation of four methylenedioxylated amphetamines on β-cyclodextrin chiral stationary phases. Chromatographia 1998; 47: 285-90.
| Crossref | Google Scholar |
83 Perry JA, Rateike JD, Szczerba TJ. Eluting trace components before major constituents: I. Sensitivity enhancement in analytical determinations of optical purity. J Chromatogr A 1987; 389: 57-64.
| Crossref | Google Scholar |
84 Tok KC, Gumustas M, Jibuti G, Suzen HS, Ozkan SA, Chankvetadze B. The effect of enantiomer elution order on the determination of minor enantiomeric impurity in ketoprofen and enantiomeric purity evaluation of commercially available dexketoprofen formulations. Molecules 2020; 25: 5865.
| Crossref | Google Scholar |
86 Sherwood AM, Burkhartzmeyer EK, Williamson SE, Faley MT. Swim in the Chiral Pool: MDMA and MDA Enantiomers from Alanine-Derived Precursors. ACS Omega 2023; 8: 22132-7.
| Crossref | Google Scholar |
87 Huot P, Johnston TH, Gandy MN, Reyes MG, Fox SH, Piggott MJ, Brotchie JM. The monoamine re-uptake inhibitor UWA-101 improves motor fluctuations in the MPTP-lesioned common marmoset. PLoS One 2012; 7: e45587.
| Crossref | Google Scholar |
88 Johnston TH, Millar Z, Huot P, Wagg K, Thiele S, Salomonczyk D, Yong-Kee CJ, Gandy MN, McIldowie M, Lewis KD, Gomez-Ramirez J, Lee J, Fox SH, Martin-Iverson M, Nash JE, Piggott MJ, Brotchie JM. A novel MDMA analogue, UWA-101, that lacks psychoactivity and cytotoxicity, enhances L-DOPA benefit in parkinsonian primates. FASEB J 2012; 26: 2154-63.
| Crossref | Google Scholar |
89 Huot P, Johnston TH, Lewis KD, Koprich JB, Reyes MG, Fox SH, Piggott MJ, Brotchie JM. UWA-121, a mixed dopamine and serotonin re-uptake inhibitor, enhances l-DOPA anti-parkinsonian action without worsening dyskinesia or psychosis-like behaviours in the MPTP-lesioned common marmoset. Neuropharmacology 2014; 82: 76-87.
| Crossref | Google Scholar |
90 Gensler WJ, Stouffer JE. Compounds related to podophyllotoxin. IX. 3,4-Methylenedioxyphenyllithium. J Org Chem 1958; 23: 908-10.
| Crossref | Google Scholar |
91 Gavara L, Boisse T, Rigo B, Hénichart J-P. A new method of bromination of aromatic rings by an iso-amyl nitrite/HBr system. Tetrahedron 2008; 64: 4999-5004.
| Crossref | Google Scholar |