

Directed chemical dimerisation enhances the antibacterial activity of the antimicrobial peptide MSI-78(4–20)
Rong Li A B # , Thomas N.G. Handley A # , Wenyi Li C * , Neil M. O’Brien-Simpson D , Mohammed Akhter Hossain A E and John D. Wade A E *
C * , Neil M. O’Brien-Simpson D , Mohammed Akhter Hossain A E and John D. Wade A E *
A Florey Institute of Neuroscience and Mental Health, University of Melbourne, Vic. 3010, Australia.
B Department of Biochemistry and Pharmacology, University of Melbourne, Vic. 3010, Australia.
C Department of Biochemistry and Chemistry, La Trobe Institute for Molecular Science, La Trobe University, Bundoora, Vic. 3086, Australia.
D ATCV (Antimicrobial and Cancer Therapeutics and Vaccines) Research Group, Division of Basic and Clinical Oral Sciences, The Melbourne Dental School, Royal Dental Hospital, University of Melbourne, Carlton, Vic. 3053, Australia.
E School of Chemistry, University of Melbourne, Vic. 3010, Australia.
Handling Editor: Mibel Aguilar
Australian Journal of Chemistry 76(8) 455-464 https://doi.org/10.1071/CH23022
Submitted: 31 January 2023 Accepted: 20 March 2023 Published: 31 May 2023
© 2023 The Author(s) (or their employer(s)). Published by CSIRO Publishing. This is an open access article distributed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License (CC BY-NC-ND)
Abstract
Antimicrobial resistance (AMR) is on the rise, leading to 700 000 deaths worldwide in 2020. Antimicrobial peptides (AMPs) are antibiotic agents that are active against multi-drug resistant pathogens and also have a reduced risk of AMR development. Previous studies have shown that dimerisation of the proline-rich antibacterial peptide (PrAMP) Chex1–Arg20 can enhance its antimicrobial activity while also reducing its toxicity. To determine if dimerisation via a simple disulfide bond can similarly improve other classes of AMPs, the α-helical cationic peptide MSI-78(4–20) was used as a model. The monomer alone, an S-carboxamidomethyl-capped N-terminal Cys–MSI-78(4–20) analogue and the disulfide-linked dimer were successfully synthesised and their antimicrobial activity and toxicity were determined. It was shown that dimerisation enhanced antimicrobial activity against the Gram-positive opportunistic pathogen Staphylococcus aureus ATCC 29213, the Gram-negative bacteria Escherichia coli ATCC 25922 and Pseudomonas aeruginosa ATCC 47615. The peptides showed no significant haemolytic activity with red blood cells and only induced 50% lactate dehydrogenase (LDH) release in mammalian cells at the highest tested concentration, 15 µM. The MSI-78(4–20) dimer was less cytotoxic than the monomer and S-alkyl monomer. Together, the data support the strategy of AMP chemically directed dimerisation as a means of producing potentially more therapeutically useful antimicrobial agents.
Keywords: antimicrobial peptide, antimicrobial resistance, chemical modification, dimer, disulfide dimerisation, infections, membrane active, solid-phase peptide synthesis.
Introduction
Owing to uncontrolled widespread use of antibiotics, antimicrobial-resistant (AMR) pathogens are rapidly evolving.[1,2,3] They caused ~700 000 global deaths in 2020 alone.[4] It is estimated that by 2050, the annual death rate will exceed 10 million.[5] To prevent this, there is an urgent need to develop new antibiotics to combat AMR pathogens. One class of antibiotic molecule that has gained significant attention in recent years is antimicrobial peptides (AMPs). AMPs are potent, specifically active molecules that, owing to their multiple modes of action, are active against pathogens that are resistant to traditional small-molecule antibiotics.[6–9] Many AMPs come from natural sources, where they act as host defence peptides, produced by various organisms against microbial pathogens including bacteria.[8–10]Many AMPs have been identified to date and are summarised in several reviews by their different antimicrobial activities.[7,8,11] As of January 2020, the US Food and Drug Administration (FDA) had approved seven AMPs for clinical application.[12]
There are several challenges that prevent widespread clinical use of AMPs: they are often toxic to mammalian cells at high concentrations, are sensitive to high salt concentrations and have poor in vivo stability in the presence of intrinsic proteases.[8,13,14] To overcome these limitations, and simultaneously enhance AMP antimicrobial activity, chemical modification strategies have been used such as: cyclisation to enhance chemical stability and decrease protease vulnerability, polyethylene glycol polymer conjugation (PEGylation) to reduce AMPs proteolytic degradation and enhance stability and solubility of AMPs, acetylation to improve resistance to proteolysis, and d-amino acid substitutions to improve stability and membrane incorporation potential.[8,14–17]
Bioconjugation, the chemical conjugation of one biomolecule to another molecule, is an appealing strategy to increase AMP potency while limiting unwanted effects.[18] Various bioconjugates of AMPs are possible including: AMP–polymer,[19] AMP–AMP,[20] AMP–nanoparticle,[21] AMP–photosensitiser[22] and AMP–antibiotics,[23] each having specialised applications such as drug delivery or as antimicrobial agents.[24–26] AMP multimerisation (a specific application of bio-conjugation) is an emerging and effective strategy to enhance the antimicrobial activity of AMPs by improving the target binding affinity and capacity to generate pores in bacterial membranes.[20,27,28] Multimeric AMPs can also act as ‘prodrugs’ that degrade into monomeric AMPs after administration, resulting in a rapid increase in local peptide concentration, which can surpass the bactericidal minimum concentration.[20,29,30]
In previous structure–activity studies on native insect proline-rich AMPs (PrAMPs) (pyrrhocoricin, drosocin and apidaecin), Otvos et al. designed a hybrid PrAMP, A3-APO, consisting of a covalent discontinuous dimer of two monomeric copies of the peptide Chex1–Arg20.[30–34] Chex1–Arg20 consists of 20 amino acids with the sequence Chex–Arg–Pro–Asp–Lys–Pro–Arg–Pro–Tyr–Leu–Pro–Arg–Pro–Arg–Pro–Pro–Arg–Pro–Val–Arg, where Chex represents cyclohexanecarboxylic acid.[35,30] To improve the pharmacological characteristics and understand the mechanism of action of Chex1–Arg20, we previously examined multimerisation of Chex1–Arg20, including A3-APO (dimeric Chex1–Arg20) and a disulfide-linked dimeric A3-APO (tetrameric Chex1–Arg20).[20,27] By increasing the valency from monomer to tetramer, the action on Escherichia coli changed from non-lytic to lytic, indicating that the tetramer acts against the membrane of E. coli.[20,28] Furthermore, the tetramer also showed lower cytotoxicity than the monomer and dimer against the mammalian cell lines H-4-II-E and HEK.[20,28] To further explore these findings based on the PrAMP Chex1–Arg20, we undertook to examine the effect of dimerisation on another class of AMPs using the cationic AMP MSI-78(4–20),[36] which was derived from MSI-78, an improved version of its parental peptide, magainin II.[37,38]
Magainin II is an amphipathic and cationic AMP, one of two peptides (magainin-1 and magainin-2) isolated from the skin of the African clawed frog, Xenopus laevis.[37,38] Magainin II is known to disrupt the bacterial membrane via toroidal pore formation.[38,39] In 1988, Zasloff et al. determined that the N-terminal and C-terminal amino acids are important for the activity of magainin II through generating several analogues of magainin II.[40] In the same year, Chen et al. determined that the helical structure enhances the antimicrobial activity of magainin II by substituting Gly for Ala at the eighth, thirteenth or eighteenth position.[39] Magainin II was further developed to generate MSI-78, through poly-lysine and poly-arginine substitution.[41–43] MSI-78 underwent a clinical trial in 1999 under the brand name Pexiganan to treat diabetic foot ulcers caused by Gram-positive organisms such as Staphylococcus aureus, and Gram-negative bacteria, such as Pseudomonas aeruginosa and E. coli.[44,45]
MSI-78 was recently developed into an improved analogue, MSI-78(4–20).[36,46] Compared with the original MSI-78 and other MSI-78 analogues, MSI-78(4–20) shows similar antimicrobial potency against Gram-positive Staphylococci strains and Gram-negative P. aeruginosa, and reduced toxicity to eukaryotic cells.[46] MSI-78(4–20) has a lower net charge of +8, higher hydrophobicity and a higher hydrophobic moment than its parent MSI-78, suggesting it has a greater capacity for membrane binding and disruption, ultimately killing the bacteria.[46] The higher hydrophobicity and hydrophobic moment of MSI-78(4–20) do not result in increased haemolytic activity.[46] MSI-78(4–20) was covalently bound to chitosan (a sugar found in crustaceans) and has been applied in bone implants and wound dressings to reduce associated infection risk.[47] This peptide was an interesting target for our dimerisation investigation.
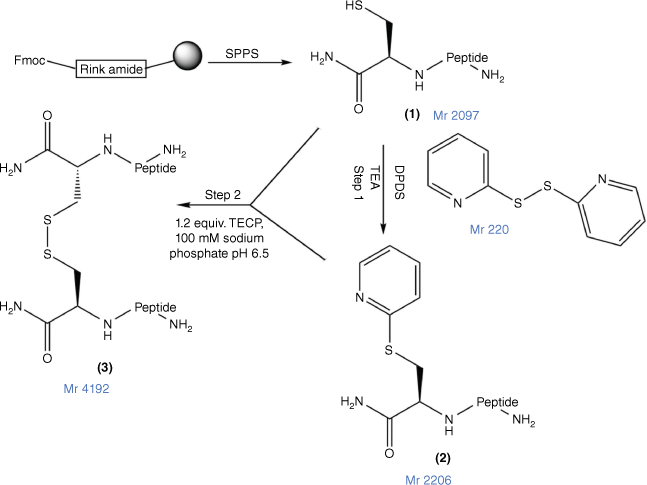
In this study, to confirm a correlation between PrAMP and other classes of AMPs for their capacity for peptide multimerisation to enhance their antimicrobial potency and pharmaceutical characteristics, we prepared a disulfide dimeric analogue of MSI-78(4–20), assessed its antimicrobial and cytotoxicity potency and compared it against both MSI-78(4–20) and an N-terminal Cys analogue in which the thiol group was capped by a carboxamidomethyl (alkyl) group (Fig. 1) to prevent its dimerisation.
The disulfide bond formation strategy for MSI-78(4–20) dimer. 1, Cys–MSI-78(4–20), with Cys at the N-terminus of the peptide; 2, DPDS-activated peptide (Cys(S-pyridyl)–MSI-78(4–20); the MSI-78(4–20) dimer 3 was prepared by combining 1 and 2 at pH 6.5 in the presence of tris-(2-carboxyethyl)phosphine (TCEP) (Fmoc, 9-fluorenylmethoxycarbonyl; SPPS, solid-phase peptide synthesis; DPDS, 2,2′-dipyridyl disulfide).
Materials and methods
Materials
O-(1H-6-Chlorobenzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HCTU) and 9-fluorenylmethoxycarbonyl (Fmoc)-l-amino acids were purchased from GL Biochem (China). 2,2′-Dithiodipyridine (DPDS), triisopropylsilane (TIPS), iodoacetamide and 3,6-dioxa-1,8-octanedithiol (DODT) were purchased from Sigma–Aldrich (Australia). Piperidine, acetonitrile (ACN), methanol (MeOH), N,N-diisopropylethylamine (DIPEA), dimethylformamide (DMF), diethyl ether and Rink amide resins (0.35 mmol/g) were purchased from Merck Millipore (Australia). Trifluoroacetic acid (TFA) was purchased from AusPep (Australia).
Solid-phase peptide synthesis
All peptides were synthesised using Fmoc solid-phase peptide synthesis[48,49] on a Biotage Initiator + Alstra microwave-assisted synthesiser using Rink amide resins (0.35 mmol/g loading). Peptides were coupled throughout with a 4-fold molar excess of the Fmoc-protected amino acids in the presence of 3.9-fold HCTU and 8-fold DIPEA. Resin-bond peptides were cleaved from the solid phase with a 5 mL cleavage cocktail (125 µL DODT:250 µL TIPS:4625 µL TFA) for 1.5 h. After cleavage, filtration was used to separate the resin solid and peptide in TFA. The peptide in TFA was concentrated under nitrogen gas. Ice-cold diethyl ether then was added to the concentrated solution and the peptide precipitated. The peptide was collected by centrifugation (1000g, 5 min) and washed twice using ice-cold diethyl ether. The precipitate was dried in a fume hood for 1 h. The crude peptides were purified by reverse-phase high performance liquid chromatography (RP-HPLC) on a Waters (USA) 600 semi-preparative RP-HPLC that incorporated a Waters 996 UV detector on a Phenomenex Gemini® C-18 column (particle size 5 µm, 150 × 21.2 mm, pore size 110 Å), with a gradient of 10–70% of 0.1% TFA in ACN and 0.1% TFA in H2O for 70 min, and then freeze-dried and stored at −20°C. The final peptides were analysed for purity and molecular weight by Shimadzu Nexera analytical HPLC incorporating an SPD-M40 UV detector using a C18 column (particle size 5 µm, 4.6 × 250 mm) and a Shimadzu MALDI-8020 matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometer (MALDI-TOF MS) using sinapinic acid as the matrix, independently.
Preparation of MSI-78(4–20) dimer via disulfide chemistry
Cys–MSI-78(4–20) (Fig. 2; 1) was mixed with 10-fold molar excess of DPDS in 1 mL TFA (Fig. 1; Step 1). The reaction mixture was stirred for 60 min and then concentrated under nitrogen gas to 0.2 mL. Ice-cold diethyl ether was added to the concentrated solution and the peptide (Fig. 1; 2) was precipitated. The precipitate was dried in a fume hood for 1 h. Peptide 2 was analysed and purified by RP-HPLC with a gradient of 10–70% of 0.1% TFA in ACN and 0.1% TFA in H2O for 70 min, and then freeze-dried and stored at −20°C. The final purity (>95%) and molecular weight were confirmed by Nexera analytical HPLC (Supplementary Fig. S1) and MALDI-TOF MS (Supplementary Fig. S2).
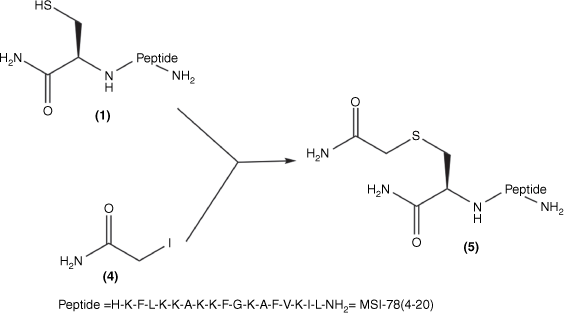
Synthesis of Cys(S-carboxamidomethyl)–MSI-78(4–20): Cys–MSI-78(4–20) 1; iodoacetamide 4; Cys(S-carboxamidomethyl)–MSI-78(4–20) 5 was obtained by combining 1 and 4 in MilliQ water.
Peptides 1 and 2 were separately dissolved in 100 mM sodium phosphate at pH 6.5. Basic 6 M Guanidine hydrochloride (GnHCl) was added dropwise into the solution until the solution became clear. Peptides 1 and 2 were then mixed for 30 min at room temperature. The final product, MSI-78(4–20) dimer (Fig. 1; 3), was analysed and purified by RP-HPLC and then freeze-dried and stored at −20°C.
Preparation of Cys(S-carboxamidomethyl)–MSI-78(4–20)
Cys–MSI-78(4–20) (1) was mixed with a 1.3-fold molar excess of TCEP solution in MilliQ-water for 10 min. A 10-fold mole excess (20 mg/mL) iodoacetamide solution (Fig. 1; 4) was then added to the solution of (1) at 37°C for the overnight reaction. The product (Fig. 2; 5) was purified and analysed (Supplementary Figs S1, S2) by RP-HPLC and then freeze-dried and stored at −20°C.
Antimicrobial assays
Minimal inhibitory concentrations (MICs) and minimal bactericidal concentrations (MBCs) of gentamicin, MSI-78(4–20) monomer, MSI-78(4–20) dimer and Cys(S-alkyl)–MSI-78(4–20) were determined. All peptide content was determined using an amino acid analyser, dissolved in 5% DMSO in Dulbecco’s phosphate buffered saline (DPBS) buffer to a final concentration of 1 mg/mL. E. coli ATCC 25922, S. aureus ATCC 29213 and P. aeruginosa ATCC 47615 were grown and maintained at 37°C on lysogeny broth (LB) agar plates.[20,50] Single colonies taken from the agar plates were used to inoculate Mueller Hinton Broth (MHB), which was incubated at 37°C overnight.The overnight culture (1 mL) was diluted into 20 mL of fresh MHB and placed for 1.5 h at 37°C in a shaking incubator until optical density at 600 nm (OD600) = 0.4–0.8, determined with a UV-visible spectrophotometer (Model EL05113296, Varian, Australia) with MHB as control.
Cells were then diluted to 2.5 × 106 cells/mL in MHB at 37°C immediately before the determination of MIC. Peptides and gentamicin were serially diluted in MHB to generate concentrations 2× final concentration in 96-well flat-bottom microtitre plates (Interpath Service, Melbourne, Vic). Peptides and gentamicin were diluted 1:1 (100 µL of peptide or gentamicin:100 µL of bacteria in MHB) and incubated at 37°C for 24 h. Bacterial growth was monitored for the determination of MIC at an optical density of 620 nm (OD620) using a Victor microplate reader, where no growth was where the OD620 was similar to the media control samples.
After E. coli and S. aureus MIC were incubated for 90 min, they were ready for MBC assays. A sample (20 µL) from each well was added to a fresh 96-well plate containing 180 µL DPBS and serially diluted 4 times. A sample (10 µL) of each serial dilution was dropped onto an LB agar plate that was divided into four segments, one for each of the dilutions, three times and dried in a laminar flow hood. After drying, E. coli plates were incubated at room temperature for 18 h and S. aureus plates were incubated at 37°C for 18 h. The MBC value was determined by counting the number of colonies in each dilution to observe the minimum concentration of peptide or gentamicin where no colonies are observed.
Cell proliferation assay
The lactate dehydrogenase (LDH) cytotoxicity assay was used to determine the cytotoxicity of peptides MSI-78(4–20) monomer, MSI-78(4–20) dimer and Cys(S-alkyl)–MSI-78(4–20) to mammalian HEK-293 cell lines. Gentamicin was used as control antibiotic. Briefly, 200 μL HEK-293 cells was added into flat-bottomed 96-well plates and incubated and cultured at 37°C, 5% CO2 for 28 h. After incubation, 100 µL of media was removed from each well of the plate containing HEK-293 cells. 50 µL of prepared tested peptides concentration were added into a new flat-bottomed 96-well plate that contained 100 µL of seeded cells, and then 0.5× dilutions of the peptides and control to a final volume of 100 µL. After that, 50 µL of each well in the plate that contained peptides and control was pipetted into the corresponding well of the plate that contained HEK-293 cells. The plate was incubated for 90 min at 37°C. After incubation, 50 µL from each well of the plate that was just incubated was transferred into a new 96-well flat-bottom plate.
LDH solution (50 µL) was added to each sample of the incubated plate. Then, the plate was covered with foil and was incubated for 30 min at room temperature. After incubation, 50 µL stop solution was added to each sample and any bubbles popped with a needle. Finally, the absorbance was recorded at 490 nm using a plate reader and percentage cytotoxicity was calculated with the following equation, where LDH release maximum = Absorbance of the wells with lysis solution.
Haemolysis assay
Sheep red blood cells (RBCs) were diluted at 1:20 ratio in PBS buffer at pH 7.4, after washing with 10 mL PBS 2 times (1000 g, 10 min). The number of RBCs was counted using a cell counter (Coulter particle counter Z series, Beckman Coulter), and they were then diluted to 2 × 107 cells/mL. A stock solution of peptides (100 μL) were serially diluted in a V-bottomed 96-well plate from 16 to 0.031 μM. Diluted RBCs (100 μL, 2 × 107 cells/mL in PBS) then were added to the V-bottomed 96-well plate. The cells were incubated at 37°C, 5% CO2 for 2 h before centrifugation at 1000 g for 10 min. Aliquots (100 μL) of the supernatant were transferred into a new flat-bottomed 96-well plate. The amount of haemoglobin released from RBCs was determined from absorbance values, which were measured at 405 nm with a PerkinElmer1420 Multilabel Counter VICTOR plate reader and Wallac software. Percentage haemolysis was determined with the following equation as previously described.[51] Wells with only RBCs in PBS buffer were used as negative control, while cells that contained 0.5% v/v Triton-X 100 were used as positive control.
Results
Synthesis of MSI-78(4–20) analogues
The MSI-78(4–20) monomer was synthesised via SPPS with a good overall yield of 18.5% (Table 1). An MSI-78(4–20) analogue was also generated with an additional cysteine at the N-terminus (Fig. 1; 1). Cys(S-alkyl)–MSI-78(4–20) was prepared by S-carboxyamidomethylating Cys–MSI-78(4–20) with iodoacetamide (Fig. 1; 4) in the presence of TCEP in a final yield of 43.8%.
| AMP | Sequence | Yield (%) | Purity (%) | Mw (g/mol) |
|---|---|---|---|---|
| MSI-78(4–20) monomer | 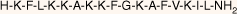 | 18.5 | >95 | 1994.6 |
| MSI-78(4–20) dimer | 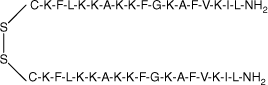 | 38.4 | >95 | 4193.4 |
| Cys(S-alkyl)–MSI-78(4–20) |  | 43.8 | >95 | 2153.7 |
Disulfide bonds can be formed through air oxidation (disulfide direct dimerisation). However, despite much effort, conditions were not found where complete dimerisation of Cys–MSI-78(4–20) was obtained within 48 h (data not shown). Instead, a chemically directed disulfide-mediated dimerisation strategy with DPDS was used.[52] After thiol activation by DPDS in TFA to yield Cys(S-pyridinyl)–MSI-78(4–20) (Supplementary Fig. S3; B and Fig. 1, step 1), this peptide was then combined with starting Cys–MSI-78(4–20) in sodium phosphate buffer (pH 6.5) in the presence of TCEP to yield the dimer (Supplementary Fig. S3; C) in 38.4% yield (Supplementary Fig. S3, Table 1). Comprehensive chemical characterisation of MSI-78(4–20) monomer, MSI-78(4–20) dimer and Cys(S-alkyl)–MSI-78(4–20) with Nexera analytical HPLC and MALDI-TOF MS confirmed their high purity and molecular identity (Supplementary Figs S1, S2).
Antimicrobial assays
Gentamicin and all MSI-78(4–20) analogues (MSI-78(4–20) monomer, dimer and Cys(S-alkyl)–MSI-78(4–20) were tested for antibacterial activity against Gram-positive bacteria S. aureus ATCC 29213, Gram-negative bacteria E. coli ATCC 25922 and P. aeruginosa ATCC 47615. Gentamicin was used as a control to compare with MSI-78(4–20) analogues. The MIC and MBC results are described in Supplementary Fig. S4 and summarised in Table 2.
| AMP | E. coli 25922 | S. aureus 29213 | P. aeruginosa 47615 | ||
|---|---|---|---|---|---|
| MBC (μM) | MIC (μM) | MBC (μM) | MIC (μM) | MIC (μM) | |
| Gentamicin | 1.26 | 5.0 | 0.62 | 0.62 | 2.5 |
| MSI-78(4–20) monomer | 7.82 | 3.91 | 15.63 | 15.63 | 1.96 |
| MSI-78(4–20) dimer | 3.81 | 1.95 | 3.81 | 3.81 | 1.90) |
| Cys(S-alkyl)–MSI-78(4–20) | 3.55 | 3.55 | 28.41 | 14.20 | 1.76 |
With the Gram-negative bacteria P. aeruginosa ATCC 47615, the MSI-78(4–20) analogues have similar antimicrobial activity, with MIC values of 1.96, 1.90 and 1.76 μM (Table 2). In comparison with the MIC results against E. coli ATCC 25922, MSI-78(4–20) dimer showed higher antimicrobial activity than the other two peptides, with a MIC value of 1.95 μM, whereas Cys(S-alkyl)–MSI-78(4–20) and MSI-78(4–20) monomer had MIC values of 3.91 and 3.55 μM (Table 2). Similarly, the Gram-positive bacterium S. aureus ATCC 29213 was more susceptible to MSI-78(4–20) dimer than the other two peptides, with MIC values of 3.81 μM while both of MSI-78(4–20) monomer and Cys(S-alkyl)–MSI-78(4–20) monomer were ~15 μM (Table 2). Also, in comparison with gentamicin, the MSI-78(4–20) analogues in Table 2 all showed higher antimicrobial activity (MIC 5 μM for P. aeruginosa ATCC 47615; MIC 2.5 μM for E. coli ATCC 25922) against the Gram-negative bacteria P. aeruginosa ATCC 47615 and E. coli ATCC 25922, while all MSI-78(4–20) analogues showed lower antimicrobial activity than gentamicin (MIC 0.62 μM for S. aureus ATCC 29213) against Gram-positive bacteria S. aureus ATCC 29213 (Table 2).
The MBC results followed a similar trend to the MIC results. According to Table 2, MSI-78(4–20) dimer was the most effective analogue against S. aureus ATCC 29213, with an MBC value of 3.81 μM, while Cys(S-alkyl)–MSI-78(4–20) and MSI-78(4–20) monomer had higher MBC values of 15.63 and 28.41 μM (Table 2). The MSI-78(4–20) dimer (MIC 3.81 μM) showed higher antimicrobial activity against E. coli ATCC 25922 than the MSI-78(4–20) monomer (MIC 7.82 μM), whereas the Cys(S-alkyl)-capped analogue maintained a similar potency with an MBC of 3.55 μM, similar to the MIC (Table 2). A comparison of MBC results and MIC results in general showed the MBC results were 2× higher than the MIC results.
Overall, based on the results of MBC and MIC values for the tested bacteria, several conclusions can be made: dimerisation of MSI-78(4–20) enhanced the antimicrobial activity against S. aureus ATCC 29213 and E. coli ATCC 25922, but maintained the antimicrobial activity against P. aeruginosa ATCC 47615. The MSI-78(4–20) monomer maintained similar antimicrobial potency against all three bacteria to Cys(S-alkyl)–MSI-78(4–20); MSI-78(4–20) analogues showed higher antimicrobial potency against Gram-negative bacteria than gentamicin, but lower antimicrobial potency against Gram-positive bacteria than gentamicin. Furthermore, the MBC values of all MSI-78(4–20) analogues and gentamicin were 2× higher than their MIC results.
Haemolysis assays
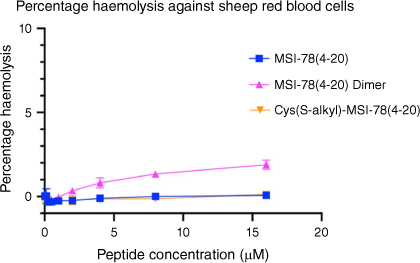
Following the antimicrobial assays, the MSI-78(4–20) analogues were tested to assay their haemolytic properties against sheep blood cells (Fig. 3) to evaluate their toxicity in mammalian cells at the range of their MIC. Both MSI-78(4–20) and the S-alkyl analogue presented no measurable haemolytic activity, whereas the MSI-78(4–20) dimer showed very low toxicity at 16 μM (only 3% haemolysis).
Haemolytic activity of MSI-78(4–20) analogues on sheep red blood cells. Peptide concentrations were tested up to 16 μM. Cells treated with DPBS buffer only were used as negative control while cells treated with 0.5% v/v Triton X-100 were used as positive control. All data are expressed as mean ± s.d. indicated by the error bars. Assays were performed three times in duplicate.
Cell proliferation assays
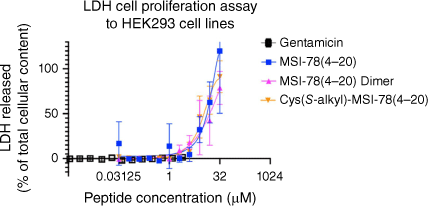
The MSI-78(4–20) analogues were examined for toxicity in the LDH cell proliferation assay using HEK293 cells (Fig. 4, Table 3). LDH release into the media is measured as an indicator of plasma membrane damage in cells.[53] As shown in Fig. 4, from 0 to 1 μM, the three peptides showed no cytotoxicity to HEK293 cell lines. However, from 1 to 16 μM, LDH release activity immediately started to grow for the three analogues, with the MSI-78(4–20) dimer increasing more rapidly than the other two monomers. Up to the highest concentration of 16 μM, they stayed at a similar 50% LDH cell release. Thus, we showed that in the LDH cell proliferation assay, the MSI-78(4–20) dimer maintained the same cytotoxicity level as MSI-78(4–20) monomer and Cys(S-alkyl)–MSI-78(4–20).
Summary of LDH cell proliferation assay to HEK293 cell lines. All peptides were analysed up to 32 μM for MSI-78(4–20) monomer and Cys(S-alkyl)–MSI-78(4–20) and 16 μM for MSI-78(4–20) dimer. All data are expressed as mean ± s.d. indicated by the error bars. Assays were performed three times in duplicate.
Discussion
The recognition that AMPs represent an exciting opportunity to potentially address the major issue of AMR has led to a substantial level of academic research over the past decade, mainly focused on identifying new peptides, their modes of action and development of analogues with greater potency and bacterial selectivity. This is highlighted by the fact that nearly 2000 vertebrate peptides have thus far been reported to possess antimicrobial activity.[54] AMPs generally are short sequences (10–20 amino acids), which makes their chemical synthesis convenient, enabling structure–function study and site-selective modifications. SPPS is predominant as it is straightforward and enables the production of large and complex peptides such as insulin-like peptides, as well as of modified analogues that would not generally be possible through recombinant DNA techniques.[55] Example peptide modifications include O-phosphorylation, glycosylation, lipidation and multimerisation.[8,27,56,57]
MSI-78(4–20) monomer, MSI-78(4–20) disulfide dimer and Cys(S-alkyl)–MSI-78(4–20) were each successfully chemically synthesised and purified with a >95% purity for bioassay (Supplementary Figs S1, S2). The free thiol of Cys–MSI-78(4–20) was capped by S-akylation on reaction with iodoacetamide to prevent its oxidation to a dimer. Although disulfide dimerisation can be achieved in several ways, in this case, air oxidation was shown to be inefficient, requiring a very long reaction time (>48 h) and producing a poor yield.[58,59] In contrast, the DPPS-directed disulfide-mediated dimerisation strategy was straightforward for preparing the dimer by nucleophilic substitution.
Antimicrobial assays
As gentamicin is a widely used oxygen-dependent bactericidal antibiotic, which belongs to the aminoglycoside family and has high antimicrobial activity against common aerobic Gram-negative bacteria, it was used as control to compare with the MSI-78(4–20) analogue antimicrobial activity.[60] Control gentamicin and each MSI-78(4–20) analogue were tested in MIC and MBC antibacterial assays against Gram-positive S. aureus strain ATCC 29213, Gram-negative straind E. coli ATCC 25922 and P. aeruginosa ATCC 47615. The MIC is calculated by the lowest concentration of the antimicrobial agent that inhibits bacterial growth completely,[50] while MBC is the lowest concentration level of antimicrobial agent that leads to bacterial death, determined from the number of colony-forming unit plotted versus peptide concentration.[61] The MBC results of all MSI-78(4–20) analogues and gentamicin were 2 times higher than the MIC results. This was due to the MIC only inhibiting the growth bacteria, but not completely kill the bacteria at the same MIC concentration. Thus, MBC results of MSI-78(4–20) analogues and gentamicin were shown 1 or 2 times higher than their MIC values.
Regarding the comparative antimicrobial activity between Cys(S-alkyl)–MSI-78(4–20) and MSI-78(4–20) monomer against the three bacteria (Gram-negative P. aeruginosa ATCC 47615 strain and E. coli ATCC 25922, Gram-positive S. aureus ATCC 29213), the consistency between their MIC and MBC values confirmed that the addition of cysteine on MSI-78(4–20) at the N-terminus did not significantly alter antimicrobial activity (Table 2). This was because the same total charge is maintained between the MSI-78(4–20) monomer, Cys–MSI-78(4–20), and Cys(S-alkyl)–MSI-78(4–20). Thus, Cys–MSI-78(4–20) and Cys(S-alkyl)-MSI-78(4–20) both had similar antimicrobial activity against different bacteria. By comparing MIC and MBC results of the MSI-78(4–20) analogues, dimerisation of MSI-78(4–20) did not improve antimicrobial activity against P. aeruginosa ATCC 47615, but it significantly enhanced the activity against S. aureus ATCC 29213 and E. coli ATCC 25922.
Based on our previous results for multimeric PrAMPs, it was hypothesised that the dimerisation of MSI-78(4–20) would decrease antimicrobial potency against Gram-positive bacteria, but either increase or maintain antimicrobial potency against Gram-negative bacteria.[20,28] As MSI-78(4–20) monomer has high hydrophobicity and amphipathicity characteristics and an alpha-helical structure, MSI-78(4–20) can readily bind to the bacterial membrane via toroidal pore formation on disrupting the membrane.[46,62] Owing to the MSI-78(4–20) monomer’s mechanism of action, toroidal pore formation in bacterial membranes and its characteristics – high hydrophobicity, high amphipathicity and alpha helical structure – the dimerisation of MSI-78(4–20) was expected to increase membrane permeabilising activity like Chex1–Arg20 tetramer compared with its respective monomer, resulting in increasing or maintaining antimicrobial activity against Gram-negative bacteria.[20] However, it was found that MSI-78(4–20) dimerisation increased antimicrobial potency against both Gram-positive bacteria S. aureus strain ATCC 29213 and Gram-negative strain E. coli ATCC 25922 but not against P. aeruginosa ATCC 47615. This may be due to several reasons. First, as the disulfide bond for dimerising MSI-78(4–20) at the N-terminus is an S-reducible linkage, this could readily easily release the two monomers of Cys–MSI-78(4–20) from the prodrug.[63] Both MSI-78(4–20) and Cys-MSI-78(4–20) have the same charge of +8, which suggests they may share characteristics, such as α-helix secondary structure, high hydrophobicity and amphipathic activity. As the hydrophobicity and charge of peptides are key contributors to the ability of AMPs to insert into lipid bilayers, the high hydrophobicity and high charge of MSI-78(4–20) may enable disruption of both Gram-positive and Gram-negative bacteria membranes, contributing to bacterial cell death.[64] Second, based on the fact that the thick peptidoglycan in Gram-positive bacteria has a higher negative charge than Gram-negative bacteria at pH 8.0, a peptide with higher hydrophobicity and higher positive charge would more likely attach to the more negatively charged bacteria.[65] Thus, after MSI-78(4–20) dimer releases two monomers in the cell, both may more easily attach to the Gram-positive bacterial membrane, resulting in a disruption of the membrane and an increase in antimicrobial activity against Gram-positive bacteria.[65]
LDH cell proliferation assays and haemolysis assays
From the results of the haemolytic assay against sheep RBCs (Table 3, Fig. 3), it was found that up to 16 μM, no haemolysis was caused by the MSI-78(4–20) monomer and Cys(S-alkyl)–MSI-78(4–20) and no toxicity for all MSI-78(4–20) analogues up to highest tested concentration. We determined dimerisation of MSI-78(4–20) maintained a toxicity similar to the MSI-78(4–20) monomer up to 16 μM peptide concentration, which was equal to or higher than their MIC results. The further LDH assay showed indicated that dimerisation of MSI-78(4–20) maintained pharmacologic activity. The dose response data shown in Supplementary Fig. S4 for MSI-78(4–20) dimer, MSI-78(4–20) monomer and acetamide-Cys–MSI-78(4–20) showed only a minor differences in cell toxicity. At their highest concentration of 32 μM, each peptide showed nearly 50% LDH released in HEK293 cells.
Overall, MSI-78(4–20) dimer maintained the LDH cytotoxicity to the HEK293 cell line and also had similar haemolytic activity against sheep blood cells to the MSI-78(4–20) monomer, which is a significant achievement and step towards a potential therapeutic application.
Conclusions
A disulfide dimer of MSI-78(4–20) was successfully synthesised and its antibacterial activity against Gram-positive and Gram-negative bacteria was determined. Dimerisation of MSI-78(4–20) enhanced the antibacterial activity against Gram-positive bacteria S. aureus and Gram-negative E. coli but not against the Gram-negative bacteria P. aeruginosa. MSI-78(4–20) dimerisation maintained the pharmacological characteristics of MSI-78(4–20), showing consistency in toxicity assays with HEK293 cells and showing no toxicity. This preliminary finding confirms our hypothesis that, like the proline-rich peptide A3APO, dimerisation of the cationic peptide MSI-78(4–20) improves its antimicrobial activity against both Gram-negative and Gram-positive bacteria and also retains low cytotoxicity. Chemically directed dimerisation via a disulfide bond thus represents a generally useful means of producing antimicrobial agents with potentially improved bacterial selectivity.
Data availability
The data that support this study are available in the article and accompanying online supplementary material.
Declaration of funding
This work was supported by grants to W.L. (NHMRC Investigator grant APP2018256 and Australian Dental Research Foundation Grant), to J.D.W. (NHMRC Project grant APP1158841, NHMRC Principal Research Fellowship APP1117483); to N.M.O-S (NHMRC Project grants APP1142472, APP1158841, APP1185426, ARC funding DP210102781, DP160101312, LE200100163) and Australian Dental Research Funding in antimicrobial materials. Studies at the The Florey Institute of Neuroscience and Mental Health were supported by the Victorian Government’s Operational Infrastructure Support Program.
References
1 Prestinaci F, Pezzotti P, Pantosti A. Antimicrobial resistance: A global multifaceted phenomenon. Pathog Glob Health 2015; 109: 309-318.
| Crossref | Google Scholar |
2 Strachan CR, Davies J. The whys and wherefores of antibiotic resistance. Cold Spring Harb Perspect Med 2017; 7: a025171.
| Crossref | Google Scholar |
3 Llor C, Bjerrum L. Antimicrobial resistance: Risk associated with antibiotic overuse and initiatives to reduce the problem. Ther Adv Drug Saf 2014; 5: 229-241.
| Crossref | Google Scholar |
4 Hendriksen RS, Vieira AR, Karlsmose S, Wong DMALF, Jensen AB, Wegener HC, et al. Global monitoring of Salmonella serovar distribution from the world health organization global foodborne infections network country data bank: Results of quality assured laboratories from 2001 to 2007. Foodborne Pathog Dis 2011; 8: 887-900.
| Crossref | Google Scholar |
5 O’Neill J. Antimicrobial resistance: Tackling a crisis for the health and wealth of nations. London: Review on Antimicrobial Resistance; 2014. Available at https://amr-review.org/ [verified 5 January 2022].
6 Huan Y, Kong Q, Mou H, Yi H. Antimicrobial peptides: Classification, design, application and research progress in multiple fields. Front Microbiol 2020; 11: 582779.
| Crossref | Google Scholar |
7 Li W, Separovic F, O’Brien-Simpson NM, Wade JD. Chemically modified and conjugated antimicrobial peptides against superbugs. Chem Soc Rev 2021; 50: 4932-4973.
| Crossref | Google Scholar |
8 Park Y-K, Hahm K-S. Antimicrobial peptides (AMPs): Peptide structure and mode of action. BMB Rep 2005; 38: 507-516.
| Crossref | Google Scholar |
9 Shai Y. Mode of action of membrane active antimicrobial peptides. Biopolymers 2002; 66: 236-248.
| Crossref | Google Scholar |
10 Hancock REW, Haney EF, Gill EE. The immunology of host defence peptides: Beyond antimicrobial activity. Nat Rev Immunol 2016; 16: 321-334.
| Crossref | Google Scholar |
11 Lin B, Li R, Handley TNG, Wade JD, Li W, O’Brien-Simpson NM. Cationic antimicrobial peptides are leading the way to combat oropathogenic infections. ACS Infect Dis 2021; 7: 2959-2970.
| Crossref | Google Scholar |
12 Chen CH, Lu TK. Development and challenges of antimicrobial peptides for therapeutic applications. Antibiotics 2020; 9: 24.
| Crossref | Google Scholar |
13 Kumar P, Kizhakkedathu JN, Straus SK. Antimicrobial peptides: Diversity, mechanism of action and strategies to improve the activity and biocompatibility in vivo. Biomolecules 2018; 8: 4.
| Crossref | Google Scholar |
14 Rajchakit U, Sarojini V. Recent developments in antimicrobial-peptide-conjugated gold nanoparticles. Bioconjug Chem 2017; 28: 2673-2686.
| Crossref | Google Scholar |
15 Gao Y, Fang H, Fang L, Liu D, Liu J, Su M, et al. The modification and design of antimicrobial peptide. Curr Pharm Des 2018; 24: 904-910.
| Crossref | Google Scholar |
16 Amerikova M, Pencheva El-Tibi I, Maslarska V, Bozhanov S, Tachkov K. Antimicrobial activity, mechanism of action, and methods for stabilisation of defensins as new therapeutic agents. Biotechnol Biotechnol Equip 2019; 33: 671-682.
| Crossref | Google Scholar |
17 Manteghi R, Pallagi E, Olajos G, Csóka I. Pegylation and formulation strategy of anti-microbial peptide (AMP) according to the quality by design approach. Eur J Pharm Sci 2020; 144: 105197.
| Crossref | Google Scholar |
18 Reinhardt A, Neundorf I. Design and application of antimicrobial peptide conjugates. Int J Mol Sci 2016; 17: 701.
| Crossref | Google Scholar |
19 Sun Z, Ma L, Sun X, Sloan AJ, O’Brien-Simpson NM, Li W. The overview of antimicrobial peptide-coated implants against oral bacterial infections. Aggregate 2023; e309.
| Crossref | Google Scholar |
20 Li W, O’Brien-Simpson NM, Tailhades J, Pantarat N, Dawson RM, Otvos Jr L, et al. Multimerization of a proline-rich antimicrobial peptide, Chex–Arg20, alters its mechanism of interaction with the Escherichia coli membrane. Chem Biol 2015; 22: 1250-1258.
| Crossref | Google Scholar |
21 Rai A, Pinto S, Evangelista MB, Gil H, Kallip S, Ferreira MG, et al. High-density antimicrobial peptide coating with broad activity and low cytotoxicity against human cells. Acta Biomater 2016; 33: 64-77.
| Crossref | Google Scholar |
22 Liu F, Soh Yan Ni A, Lim Y, Mohanram H, Bhattacharjya S, Xing B. Lipopolysaccharide neutralizing peptide–porphyrin conjugates for effective photoinactivation and intracellular imaging of gram-negative bacteria strains. Bioconjug Chem 2012; 23: 1639-1647.
| Crossref | Google Scholar |
23 Li W, O’Brien‐Simpson NM, Holden JA, Otvos L, Reynolds EC, Separovic F, et al. Covalent conjugation of cationic antimicrobial peptides with a β‐lactam antibiotic core. Pept Sci 2018; 110: e24059.
| Crossref | Google Scholar |
24 Cao Y, Nguyen GK, Chuah S, Tam JP, Liu C-F. Butelase-mediated ligation as an efficient bioconjugation method for the synthesis of peptide dendrimers. Bioconjug Chem 2016; 27: 2592-2596.
| Crossref | Google Scholar |
25 Darbre T, Reymond J-L. Peptide dendrimers as artificial enzymes, receptors, and drug-delivery agents. Acc Chem Res 2006; 39: 925-934.
| Crossref | Google Scholar |
26 Sadler K, Tam JP. Peptide dendrimers: Applications and synthesis. Rev Mol Biotechnol 2002; 90: 195-229.
| Crossref | Google Scholar |
27 Li W, Lin F, Hung A, Barlow A, Sani M-A, Paolini R, et al. Enhancing proline-rich antimicrobial peptide action by homodimerization: Influence of bifunctional linker. Chem Sci 2022; 13: 2226-2237.
| Crossref | Google Scholar |
28 Li W, Sani M-A, Jamasbi E, Otvos Jr L, Hossain MA, Wade JD, et al. Membrane interactions of proline-rich antimicrobial peptide, Chex1–Arg20, multimers. Biochim Biophys Acta 2016; 1858: 1236-1243.
| Crossref | Google Scholar |
29 Cudic M, Condie BA, Weiner DJ, Lysenko ES, Xiang ZQ, Insug O, et al. Development of novel antibacterial peptides that kill resistant isolates. Peptides 2002; 23: 2071-2083.
| Crossref | Google Scholar |
30 Li W, Tailhades J, O’Brien-Simpson N, Separovic F, Otvos Jr L, Hossain MA, et al. Proline-rich antimicrobial peptides: Potential therapeutics against antibiotic-resistant bacteria. Amino Acids 2014; 46: 2287-2294.
| Crossref | Google Scholar |
31 Rozgonyi F, Szabo D, Kocsis B, Ostorhazi E, Abbadessa G, Cassone M, et al. The antibacterial effect of a proline-rich antibacterial peptide a3-apo. Curr Med Chem 2009; 16: 3996-4002.
| Crossref | Google Scholar |
32 Otvos Jr L, de Olivier Inacio V, Wade JD, Cudic P. Prior antibacterial peptide-mediated inhibition of protein folding in bacteria mutes resistance enzymes. Antimicrob Agents Chemother 2006; 50: 3146-3149.
| Crossref | Google Scholar |
33 Hoffmann R, Bulet P, Urge L, Otvos L. Range of activity and metabolic stability of synthetic antibacterial glycopeptides from insects. Biochim Biophys Acta Gen Subj 1999; 1426: 459-467.
| Crossref | Google Scholar |
34 Otvos Jr L, Wade JD, Lin F, Condie BA, Hanrieder J, Hoffmann R. Designer antibacterial peptides kill fluoroquinolone-resistant clinical isolates. J Med Chem 2005; 48: 5349-5359.
| Crossref | Google Scholar |
35 Li W, Tailhades J, Hossain MA, O’Brien-Simpson NM, Reynolds EC, Otvos L, et al. C-Terminal modifications broaden activity of the proline-rich antimicrobial peptide, Chex1–Arg20. Aust J Chem 2015; 68: 1373-1378.
| Crossref | Google Scholar |
36 Lin B, Hung A, Li R, Barlow A, Singleton W, Matthyssen T, et al. Systematic comparison of activity and mechanism of antimicrobial peptides against nosocomial pathogens. Eur J Med Chem 2022; 231: 114135.
| Crossref | Google Scholar |
37 Giovannini MG, Poulter L, Gibson BW, Williams DH. Biosynthesis and degradation of peptides derived from Xenopus laevis prohormones. Biochem J 1987; 243: 113-120.
| Crossref | Google Scholar |
38 Zasloff M. Magainins, a class of antimicrobial peptides from Xenopus skin: Isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc Natl Acad Sci 1987; 84: 5449-5453.
| Crossref | Google Scholar |
39 Chen H-C, Brown JH, Morell JL, Huang CM. Synthetic magainin analogues with improved antimicrobial activity. FEBS Letters 1988; 236: 462-466.
| Crossref | Google Scholar |
40 Zasloff M, Martin B, Chen H-C. Antimicrobial activity of synthetic magainin peptides and several analogues. Proc Natl Acad Sci 1988; 85: 910-913.
| Crossref | Google Scholar |
41 Cuervo JH, Rodriguez B, Houghten R. The magainins: Sequence factors relevant to increased antimicrobial activity and decreased hemolytic activity. Pept Res 1988; 1: 81-86.
| Google Scholar |
42 Bessalle R, Haas H, Goria A, Shalit I, Fridkin M. Augmentation of the antibacterial activity of magainin by positive-charge chain extension. Antimicrob Agents Chemother 1992; 36: 313-317.
| Crossref | Google Scholar |
43 Maloy WL, Kari UP. Structure–activity studies on magainins and other host defense peptides. Biopolymers 1995; 37: 105-122.
| Crossref | Google Scholar |
44 Ge Y, MacDonald DL, Holroyd KJ, Thornsberry C, Wexler H, Zasloff M. In vitro antibacterial properties of pexiganan, an analog of magainin. Antimicrob Agents Chemother 1999; 43: 782-788.
| Crossref | Google Scholar |
45 Lipsky BA, Holroyd KJ, Zasloff M. Topical versus systemic antimicrobial therapy for treating mildly infected diabetic foot ulcers: A randomized, controlled, double-blinded, multicenter trial of pexiganan cream. Clin Infect Dis 2008; 47: 1537-1545.
| Crossref | Google Scholar |
46 Monteiro C, Pinheiro M, Fernandes M, Maia S, Seabra CL, Ferreira-da-Silva F, et al. A 17-mer membrane-active MSI-78 derivative with improved selectivity toward bacterial cells. Mol Pharm 2015; 12: 2904-2911.
| Crossref | Google Scholar |
47 Monteiro C, Fernandes H, Oliveira D, Vale N, Barbosa M, Gomes P, et al. AMP–chitosan coating with bactericidal activity in the presence of human plasma proteins. Molecules. 2020; 25: 3046.
| Crossref | Google Scholar |
48 Li W, O’Brien-Simpson N, Hossain M, Wade J. The 9-fluorenylmethoxycarbonyl (Fmoc) group in chemical peptide synthesis – its past, present and future. Aust J Chem 2020; 73: 271-276.
| Crossref | Google Scholar |
49 Li W, Wade JD, Reynolds E, O’Brien-Simpson NM. Chemical modification of cellulose membranes for SPOT synthesis. Aust J Chem 2020; 73: 78-84.
| Crossref | Google Scholar |
50 Lambert RJW, Pearson J. Susceptibility testing: Accurate and reproducible minimum inhibitory concentration (MIC) and non-inhibitory concentration (NIC) values. J Appl Microbiol 2000; 88: 784-790.
| Crossref | Google Scholar |
51 Lin B, Hung A, Singleton W, Darmawan KK, Moses R, Yao B, et al. The effect of tailing lipidation on the bioactivity of antimicrobial peptides and their aggregation tendency: Special issue: Emerging investigators. Aggregate 2023; e329.
| Crossref | Google Scholar |
52 Maruyama K, Nagasawa H, Suzuki A. 2,2′-Bispyridyl disulfide rapidly induces intramolecular disulfide bonds in peptides. Peptides. 1999; 20: 881-884.
| Crossref | Google Scholar |
53 Smith SM, Wunder MB, Norris DA, Shellman YG. A simple protocol for using a ldh-based cytotoxicity assay to assess the effects of death and growth inhibition at the same time. PLoS One 2011; 6: e26908.
| Crossref | Google Scholar |
54 Zhang Q-Y, Yan Z-B, Meng Y-M, Hong X-Y, Shao G, Ma J-J, et al. Antimicrobial peptides: Mechanism of action, activity and clinical potential. Mil Med Res 2021; 8: 48.
| Crossref | Google Scholar |
55 Shabanpoor F, Hughes RA, Bathgate RAD, Zhang S, Scanlon DB, Lin F, et al. Solid-phase synthesis of europium-labeled human INSL3 as a novel probe for the study of ligand−receptor interactions. Bioconjug Chem 2008; 19: 1456-1463.
| Crossref | Google Scholar |
56 Chandrashekar C, Hossain MA, Wade JD. Chemical glycosylation and its application to glucose homeostasis-regulating peptides. Front Chem 2021; 9: 650025.
| Crossref | Google Scholar |
57 Kitas EA, Perich JW, Wade JD, Johns RB, Tregear GW. Fmoc-polyamide solid phase synthesis of an O-phosphotyrosine-containing tridecapeptide. Tetrahedron Lett 1989; 30: 6229-6232.
| Crossref | Google Scholar |
58 Ruano JLG, Parra A, Alemán J. Efficient synthesis of disulfides by air oxidation of thiols under sonication. Green Chem 2008; 10: 706-711.
| Crossref | Google Scholar |
61 Sani M-A, Whitwell TC, Gehman JD, Robins-Browne RM, Pantarat N, Attard T, et al. Maculatin 1.1 disrupts Staphylococcus aureus lipid membranes via a pore mechanism. Antimicrob Agents Chemother 2013; 57: 3593-3600.
| Crossref | Google Scholar |
62 Monteiro C, Fernandes M, Pinheiro M, Maia S, Seabra CL, Ferreira-da-Silva F, et al. Antimicrobial properties of membrane-active dodecapeptides derived from msi-78. Biochim Biophys Acta Biomembr 2015; 1848: 1139-1146.
| Crossref | Google Scholar |
63 Saito G, Swanson JA, Lee K-D. Drug delivery strategy utilizing conjugation via reversible disulfide linkages: Role and site of cellular reducing activities. Adv Drug Deliv Rev 2003; 55: 199-215.
| Crossref | Google Scholar |
64 Matsuzaki K. Control of cell selectivity of antimicrobial peptides. Biochim Biophys Acta - Biomembr 2009; 1788: 1687-1692.
| Crossref | Google Scholar |
65 Malanovic N, Lohner K. Antimicrobial peptides targeting gram-positive bacteria. Pharmaceuticals 2016; 9: 59.
| Crossref | Google Scholar |