

Structural reassignment of a dibenz[b,f][1,4]oxazepin-11(10H)-one with potent antigiardial activity
Andrew G. Riches A * , Christopher J. S. Hart B C , Matthieu Schmit A , Emmanuel A. Debele A , Snigdha Tiash B , Erin Clapper B , Tina S. Skinner-Adams B * and John H. Ryan A
A * , Christopher J. S. Hart B C , Matthieu Schmit A , Emmanuel A. Debele A , Snigdha Tiash B , Erin Clapper B , Tina S. Skinner-Adams B * and John H. Ryan A
A CSIRO Manufacturing, Ian Wark Laboratory, Bayview Avenue, Clayton, Vic. 3168, Australia.
B Griffith Institute for Drug Discovery, Griffith University, Nathan, Qld 4111, Australia.
C Present address: Department of Microbiology and Molecular Genetics, University of California Davis, CA 95616, USA.
Handling Editor: Craig Hutton
Australian Journal of Chemistry 75(10) 839-845 https://doi.org/10.1071/CH22184
Submitted: 23 August 2022 Accepted: 14 October 2022 Published: 10 November 2022
© 2022 The Author(s) (or their employer(s)). Published by CSIRO Publishing. This is an open access article distributed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License (CC BY-NC-ND)
Abstract
A screen for compounds with antigiardial activity in the Compounds Australia Scaffolds library identified SN00797640 (supplied structure being 8-acylaminodibenzoxazepinone 1) as a hit compound with potent anti-parasitic activity (concentration for 50% growth inhibition of Giardia duodenalis, IC50 0.18 μM). To further explore the structure–activity relationships in this series, compound 1 and analogues, including its 7-acylaminodibenzoxazepinone regioisomer (2), were synthesized and assessed for anti-Giardia activity. While regioisomer 2 demonstrated antigiardial activity, resynthesized 1 and other 8-acylaminodibenzoxazepinone analogues were inactive. Comparison of spectroscopic and physical properties demonstrated the correct structure of SN00797640 to be 7-acylamino regioisomer 2. These results highlight the importance of independent synthesis in verifying the structure and activity of screening hits.
Keywords: antigiardial, dibenzoxazepinones, Giardia, giardiasis, medicinal chemistry, organic synthesis, parasite, screening, structural reassignment.
Introduction
A range of analytical, synthetic and medicinal chemistry techniques are required in the triage of drug screening hits, particularly in deciding which hit compounds should progress into a medicinal chemistry hit-to-lead program.[1] Many issues can be identified on analysis of an initial set of hits, including that library compounds plated out for screening have degraded over time or have been misidentified, false positives can originate from unknown contaminants or assay interference, and certain types of compounds are promiscuous, so-called frequent hitters.[2] The purification or resynthesis of hits enables their activity to be confirmed, leading to prioritization of the hit or, conversely, can reveal issues with quality, identity or inconsistent activity between batches that can lead to de-prioritization of the hit or at least demand further enquiry prior to progression.[3] In previous work on fragment-based screening against HIV integrase, we noted the isomerization of a hit compound occurring during testing that, once clarified, enabled development of more strongly binding compounds.[4] We now describe how the resynthesis of an assay hit from a phenotypic screen of a library of commercial compounds for antigiardial activity enabled reassignment of the supplied structure of the active compound.
In the search for new compounds with activity against the important intestinal parasite Giardia duodenalis,[5] a series of acylamino-dibenz[b,f][1,4]oxazepin-11(10H)-ones (Compounds Australia Scaffold Series CL9406) were identified as potent hits in a growth inhibition assay (Hart CJS, Riches AG, Tiash S, Clapper E, Ramu S, Zuegg J, Ryan JH, Skinner-Adams TS. unpubl. data). The commercially sourced compound SN00797640, with assigned structure 1 (Fig. 1), was the most potent hit (G. duodenalis mean + s.d. 50% growth inhibition (inhibitory concentration, IC50) = 0.18 ± 0.05 µM) in this series. To access additional material for further evaluation and to investigate structure–activity relationships (SARs) within the Scaffold Series CL9406, we resynthesized compound 1. However, the newly synthesized material as well as several analogues of 1 were found to be inactive. Herein, we describe how analytical and synthetic chemistry were used to reassign the structure of the active compound as the regioisomeric 7-acylamino 2.
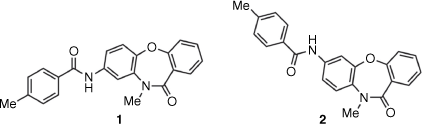
|
Results and discussion
Synthesis
Despite compound 1 being commercially available, a literature search found no reported methods for its preparation. We therefore developed our own synthesis of 1 (Scheme 1). Reaction of 2,4-dinitrofluorobenzene with the anion of methyl salicylate 3 gave biaryl ether 4, previously obtained from reaction of 3 with 2,4-dinitrochlorobenzene.[6,7] Catalytic hydrogenation/hydrogenolysis reduced both nitro groups of 4 to give diamine 5, which appears in a 2018 patent without any characterization data.[8]Acid-catalyzed lactamization of 5 then gave dibenzoxazepinone 6, which was reported in a 1974 paper as the product of thermally mediated cyclization of the ethyl ester analogue of 5, also without accompanying physical or spectroscopic data.[9] The amino group of 6 was protected as the novel phthalimide 7, which enabled selective N-methylation of the lactam, affording the previously unreported 8. Subsequent hydrazinolysis of the phthalimide 8 gave aniline 9. A literature search found no previous reports of compound 9. The coupling of 9 with p-toluic acid gave 8-acylaminodibenzoxazepinone 1. Additional analogues 10–14 were prepared by coupling amine 9 with other carboxylic acids or acid chlorides, and are characterized here for the first time.

|
The synthesis of the regioisomeric 7-acylaminodibenzoxazepinone 2 (Scheme 2) employed a tandem SNAr–Smiles rearrangement sequence to give tricyclic ring system 16 in one operation from 15 and 3,4-difluoronitrobenzene.[10] Hydrogenation of 16 then gave the 7-aminodibenzoxazepinone 17.[11] Amide coupling of 17 with p-toluic acid gave the 7-acylaminodibenzoxazepinone 2. A literature search found no reports describing either the preparation or physical or spectroscopic characterization of compound 2.

|
Biological activity and structural characterization
In contrast to the original commercially sourced compound, SN00797640 (IC50 = 0.18 ± 0.05 µM), compound 1 prepared according to Scheme 1 was inactive against Giardia (IC50 > 10 µM). As additional 8-acylaminodibenzoxazepinones 10–14, prepared to examine SARs, were also inactive, additional SN00797640 was purchased from ChemDiv for spectroscopic and physical property assessment. Significant differences were observed between the 1H NMR spectrum of the commercially sourced sample (SN00797640) and that of the in-house synthesized sample of 1 (Fig. 2). A large difference was also observed between the melting points of the commercially sourced sample of SN00797640 (mp 211–212°C) and in-house synthesized sample of 1 (mp 255–257°C). Conversely, compound 2 exhibited a 1H NMR spectrum and melting point (mp 211–213°C) in accord with SN00797640 (Fig. 2). Compound 2 also demonstrated potent in vitro activity against Giardia parasites (IC50 = 0.28 ± 0.04 µM) similar to that observed for SN00797640. When notified of this discrepancy, ChemDiv did not have any SN00797640 (ChemDiv code: MO13-0086) in stock with which they could independently verify its regiosomeric identity. However, they provided confirmation (by 2D NOESY spectroscopy) that other available members of the CL9406 series were 7-acylaminodibenzoxazepinones, rather than the corresponding 8-acylamino isomers, as originally specified in ChemDiv documentation (pers. comm.). The initial misassignment of the structure of SN00797640 serves as a reminder of the importance of using independent synthesis to verify the structure and activity of screening hits prior to undertaking more extensive SAR studies of a hit series.[1,2]

|
Conclusion
Synthesis of compounds 1 and 2 enabled reassignment of the structure of the antigiardial screening hit SN00797640 from the commercially supplied structure 1 to structure 2. In contrast to the 7-benzamido regioisomer 2, the 8-benzamido regioisomer 1 was inactive against Giardia duodenalis in vitro. The revision of this structure led to review of the CL9406 series by ChemDiv and confirmation that this series comprise all 7-acylamino isomers. With the structure of the hit compound confirmed, analogues of 2 can be synthesized and evaluated for antigiardial activity, enabling development of robust SARs.
Experimental
General chemistry experimental
Melting points were determined on a Büchi B-545 digital melting point apparatus and are uncorrected. NMR spectra were recorded on Bruker Avance 400 and 500 MHz NMR spectrometers at 25°C. 1H spectra in CDCl3 and d6-DMSO are referenced to residual solvent (δH 7.24 and 2.50 ppm respectively). 13C NMR spectra in CDCl3 and d6-DMSO are referenced to the central resonance of the CDCl3 ‘triplet’ (δC 77.23 ppm) and the d6-DMSO septet (δC 39.51 ppm) respectively. Thin layer chromatography (TLC) was performed on Merck pre-coated 0.25 mm silica F254 aluminium-backed plates (no. 5554). Column chromatography was performed using Merck (no. 9385, 230–400 mesh) silica gel 60. Analytical LCMS was performed on a Waters Acquity I Class with an Acquity UPLC BEH C18 1.7 mm 2.1 × 50 mm column with Photodiode Array (PDA) (254 nm) and Quadrupole Dalton (QDa) detection. Mobile phase: H2O (0.1% formic acid)/ACN (0.1% formic acid) 95:5 → 100:0 over 7 min and a flow rate of 0.4 mL/min with an injection volume of 1.00 μL. Mass spectrometric analyses were performed on a Thermo Scientific Q Exactive mass spectrometer fitted with an Atmospheric-Pressure Chemical Ionization (APCI) ion source or an Atmospheric Solids Analysis Source (ASAP) ion source. Positive and/or negative ions were recorded in an appropriate mass range at 140 000 mass resolution. The APCI probe was used without flow of solvent. The nitrogen nebulizing/desolvation gas used for vaporization was heated to 450°C in these experiments. The sheath gas flow rate was set to 25 and the auxiliary gas flow rate to 2 (all arbitrary units). The discharge current was 4 mA and the capillary temperature was 320°C. All anhydrous reactions were performed under a dry nitrogen atmosphere. All inorganic solutions are aqueous unless otherwise specified.
Synthesis
Methyl 2-(2,4-dinitrophenoxy)benzoate 4: A stirred mixture of 2,4-dinitrofluorobenzene (4.08 g, 21.9 mmol), methyl salicylate 3 (3.33 g, 21.9 mmol), caesium carbonate (7.14 g, 21.9 mmol) and DMF (15 mL) was heated to 60°C until TLC analysis (EtOAc/heptane, 20:80) showed complete reaction (30 min). The mixture was filtered, evaporated under reduced pressure and the residue partitioned between 1:1 heptane/EtOAc and 2 M Na2CO3. The organic phase was washed with water (×2), then brine, dried (Na2SO4) and evaporated under reduced pressure. The crude product was recrystallized from EtOAc/heptane to give 4 (6.11 g, 88%) as an off-white solid. Mp 90–91°C (lit.[6] 88°C, lit.[7] 85°C). NMR data have not been previously reported: 1H NMR (400 MHz, CDCl3) δ 8.86 (d, J = 2.8 Hz, 1H), 8.25 (dd, J = 9.3, 2.8 Hz, 1H), 8.09 (dd, J = 7.8, 1.7 Hz, 1H), 7.70–7.65 (m, 1H), 7.44 (apparent dt, J = 7.7, 1.1 Hz, 1H), 7.25 (dd, J = 8.1, 1.1 Hz, 1H), 6.79, d, J = 9.3 Hz, 1H), 3.73 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 164.4, 157.0, 152.5, 141.3, 139.0, 135.0, 133.2, 128.9, 127.4, 123.8, 123.7, 122.3, 117.3, 52.7.
Methyl 2-(2,4-diaminophenoxy)benzoate 5: Methyl 2-(2,4-dinitrophenoxy)benzoate 4 (3.25 g, 10.2 mmol) was hydrogenated (1 atm H2, balloon) over 10% Pd/C (50% H2O, 1.09 g, 0.511 mmol) in ethanol (100 mL) and the mixture stirred vigorously at room temperature for 3 h (reaction complete by TLC analysis), filtered through a pad of Celite and evaporated under reduced pressure to give a brown solid. The crude product was dissolved in EtOAc and filtered through a short plug of silica to remove baseline impurities to give diamine 5 (2.53 g, 96%) as a tan solid. Mp 128–131°C. HRMS (high-resolution mass spectrometry, APCI, +ve ion) m/z calcd for C₁₄H₁₅O₃N₂ [M + H+] 259.1077, found 259.1077. 1H NMR (400 MHz, CDCl3) δ 7.77 (dd, J = 7.7, 1.8 Hz, 1H), 7.36–7.30 (m, 1H), 7.03–6.98 (m, 1H), 6.87 (dd, J = 8.4, 0.8 Hz, 1H), 6.76 (d, J = 8.4 Hz, 1H), 6.14 (d, J = 2.6 Hz, 1H), 6.06 (dd, J = 8.4, 2.6 Hz, 1H), 3.92 (broad s, 2H), 3.89 (s, 3H), 3.49 (br. s, 2H). 13C NMR (125 MHz, CDCl3) δ 167.2, 158.2, 144.6, 140.2, 135.0, 133.5, 131.5, 122.9, 121.8, 120.9, 116.1, 105.4, 103.3, 52.4.
8-Aminodibenz[b,f][1,4]oxazepin-11(10H)-one 6: A mixture of methyl 2-(2,4-diaminophenoxy)benzoate 5 (2.39 g, 9.25 mmol), methanesulfonic acid (50 μL) and xylenes (50 mL) was refluxed for 20 h. After cooling, the solvent was evaporated under reduced pressure and the residue was triturated with EtOAc to give the 8-aminodibenzooxazepinone 6 (1.78 g, 85%) as a yellow powder. Mp 236–240°C (dec.). HRMS (APCI, +ve ion) m/z calcd for C13H11N2O2+ [M + H+] 227.0814, found 227.0815. 1H NMR (400 MHz, d6-DMSO) δ 10.34 (broad s, 1H), 7.73 (dd, J = 7.7, 1.7 Hz, 1H), 7.59–7.54 (m, 1H), 7.30–7.23 (m, 2H), 7.00 (d, J = 8.5 Hz, 1H), 6.41 (d, J = 2.6 Hz, 1H), 6.36 (dd, J = 8.5, 2.6 Hz, 1H) (NH2 signal not observed). 13C NMR (125 MHz, d6-DMSO) δ 166.2, 159.6, 146.6, 141.2, 134.0, 131.3, 131.1, 126.1, 124.8, 121.2, 120.3, 110.2, 105.8.
2-(11-Oxo-10,11-dihydrodibenzo[b,f][1,4]oxazepin-8-yl)isoindoline-1,3-dione 7: The 8-aminodibenzoxazepinone 6 (1.45 g, 6.41 mmol) was dissolved in dry DMF (20 mL) and phthalic anhydride (949 mg, 6.41 mmol) was added. After stirring at room temperature for 2 h, no starting amine was detected by TLC. The solvent was evaporated under reduced pressure, acetic acid (30 mL) was added, and the mixture was heated at reflux for 3 h, then evaporated. Trituration of the residue with EtOAc gave 7 (1.84 g, 81%) as a colourless solid. Mp 298–301°C. HRMS (APCI, +ve ion) m/z calcd for C21H13N2O4+ [M + H+] 357.0870, found 357.0871. 1H NMR (400 MHz, d6-DMSO) δ 10.71 (broad s, 1H), 7.98–7.87 (m, 4H), 7.79 (dd, J = 7.8, 1.7 Hz, 1H), 7.67–7.62 (m, 1H), 7.49 (d, J = 8.4 Hz, 1H), 7.41–7.32 (m, 2H), 7.26–7.21 (m, 2H). 13C NMR (125 MHz, d6-DMSO) δ 166.8, 165.7, 158.6, 149.8, 134.7, 134.6, 131.6, 131.5, 131.5, 129.3, 125.7, 125.6, 124.3, 123.5, 121.7, 120.6, 120.5.
2-(10-Methyl-11-oxo-10,11-dihydrodibenzo[b,f][1,4]oxazepin-8-yl)isoindoline-1,3-dione 8: Sodium hydride (60% in oil, 104 mg, 2.60 mmol) was added to a stirred suspension of the lactam 7 (841 mg, 2.36 mmol) in DMF (10 mL). After 15 min, the bubbling had subsided to give a clear solution, and methyl iodide (1.67 g, 11.8 mmol) was added and the mixture was stirred at room temperature for 2 h to give a thick slurry. The DMF was evaporated under reduced pressure and the residue was mixed with 1 M HCl and the solid collected by filtration, dried at the pump, and washed with methyl tert-butyl ether to give the title compound 8 (760 mg, 87%) as a tan powder. Mp 241–242°C. HRMS (APCI, +ve ion) m/z calcd for C22H15N2O4+ [M + H+] 371.1026, found 371.1024. 1H NMR (400 MHz, d6-DMSO) δ 8.01–7.88 (m, 4H), 7.78 (dd, J = 7.8, 1.7 Hz; 1H), 7.65–7.60 (m, 2H), 7.55 (d, J = 8.5 Hz, 1H), 7.41 (broad d, J = 8.0 Hz, 1H), 7.37–7.30 (m, 2H), 3.48 (s, 3H). 13C NMR (125 MHz, d6-DMSO) δ 166.8, 165.1, 159.7, 152.3, 135.7, 134.8, 134.2, 131.9, 131.5, 129.6, 125.7, 125.7, 125.6, 123.5, 122.5, 121.5, 120.0, 36.3.
10-Methyl-8-aminodibenz[b,f][1,4]oxazepin-11(10H)-one 9: Hydrazine hydrate (1.03 g, 20.5 mmol) was added to a suspension of phthalimide 8 (759 mg, 2.05 mmol) in acetonitrile. The mixture was refluxed for 1 h, during which time it became homogeneous, before a voluminous precipitate appeared. After cooling to room temperature, the solid was removed by filtration and the filtrate was evaporated to give a tan solid. Column chromatography (EtOAc/CHCl3, 0:100 → 10:90) gave the amine 9 (445 mg, 90%) as a colourless oil that solidified on standing. Mp 230–238°C (dec.). HRMS (ESI, +ve ion) m/z calcd for C₁₄H₁₃O₂N₂+ [M + H+] 241.0972, found 241.0970. 1H NMR (400 MHz, CDCl3) δ 7.84 (dd, J = 7.8, 1.7 Hz, 1H), 7.43–7.38 (m, 1H), 7.19–7.11 (m, 2H), 7.00 (d, J = 8.6 Hz, 1H), 6.49 (d, J = 2.7 Hz, 1H), 6.40 (dd, J = 8.6, 2.7 Hz, 1H), 3.62 (broad s, 2H), 3.52 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 166.7, 161.0, 146.1, 144.2, 136.3, 133.4, 132.2, 126.4, 124.9, 121.8, 119.6, 112.4, 108.5, 36.5.
4-Methyl-N-(10-methyl-11-oxo-10,11-dihydrodibenzo[b,f][1,4]oxazepin-8-yl)benzamide 1: A vial was charged with amine 9 (30 mg, 0.125 mmol), 4-methylbenzoic acid (19 mg, 0.137 mmol) and pyridine (200 μL). After 10 min. 2,4,6-tripropyl-1,3,5,2,4,6-trioxatriphosphorinane-2,4,6-trioxide (T3P) (50% solution in EtOAc, 112 μL, 0.188 mmol) was added dropwise and the mixture was stirred at room temperature for 3 h. The mixture was partitioned between 1 M HCl and EtOAc. The organic phase was washed successively with sat. aq. NaHCO3 and brine, dried (MgSO4) and evaporated. Column chromatography (EtOAc/CHCl3, 10:90 → 30:70) gave amide 1 (20 mg, 45%) as a colourless oil, which crystallized on standing. Mp 255–257°C. LCMS: tR 2.99 min. (98.3% purity). HRMS (ESI, +ve ion) m/z calcd for C22H19N2O3+ [M + H+] 359.1390, found 359.1391. 1H NMR (500 MHz, CDCl3) δ 7.87 (dd, J = 7.8, 1.7 Hz, 1H), 7.81–7.78 (m, 1H), 7.75–7.71 (m, 2H), 7.74 (broad s, 1H), 7.46–7.41 (m, 1H), 7.30–7.25 (m, 2H), 7.24–7.15 (m, 4H), 3.60 (s, 3H), 2.41 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 166.4, 165.6, 160.5, 150.0, 142.8, 136.4, 135.7, 133.5, 132.4, 131.5, 129.5, 127.0, 126.2, 125.2, 121.6, 119.7, 117.6, 114.7, 36.8, 21.5.
4-Chloro-N-(10-methyl-11-oxo-10,11-dihydrodibenzo[b,f][1,4]oxazepin-8-yl)benzamide 10. Prepared as for 1 from aryl amine 9 (29 mg, 0.120 mmol) and 4-chlorobenzoic acid (21 mg, 0.132 mmol) using T3P. The crude product was purified by column chromatography (CHCl3) to give product 10 (32 mg, 96%) as a colourless solid. Mp 256–257°C. LCMS: tR 3.08 min. (99.9% purity). HRMS (ESI, +ve ion) m/z calcd for C₂₁H₁O₃N₂³⁵Cl+ [M + H+] 379.0844, found 379.0844. 1H NMR (400 MHz, CDCl3) δ 7.85 (dd, J = 7.7, 1.6 Hz, 1H), 7.83 (broad s, 1H), 7.80–7.75 (m, 2H), 7.75 (brd, J = 1.9 Hz, 1H), 7.46–7.41 (m, 3H), 7.22–7.15 (m, 4H), 3.61 (s, 3H). 13C NMR (125 MHz, d6-DMSO) δ 165.3, 164.4, 159.9, 148.8, 136.8, 136.6, 135.2, 134.0, 133.2, 131.9, 129.6, 128.5, 125.8, 125.5, 121.1, 119.9, 118.2, 114.9, 36.2.
4-Cyano-N-(10-methyl-11-oxo-10,11-dihydrodibenzo[b,f][1,4]oxazepin-8-yl)benzamide 11. Aryl amine 9 (50 mg, 0.208 mmol) was dissolved in pyridine (0.5 mL) and 4-cyanobenzoyl chloride (38 mg, 0.229 mmol) was added and the mixture stirred at room temperature for 18 h. The resulting suspension was diluted with methyl tert-butyl ether (MTBE) and the solid was collected by filtration to give amide 11 (42 mg, 55%) as a pale grey solid. Mp 267–269°C. LCMS: tR 2.73 min. (96.1% purity). HRMS (ESI, +ve ion) m/z calcd for C₂₂H₁O₃N₃+ [M + H+] 370.1186, found 370.1187. 1H NMR (400 MHz, d6-DMSO) δ 10.60 (broad s, 1H), 8.11–8.07 (m, 2H), 8.05–8.02 (m, 2H), 7.86 (d, J = 2.4 Hz, 1H), 7.76 (dd, J = 7.7, 1.7 Hz, 1H), 7.62–7.57 (m, 2H), 7.40–7.28 (m, 3H), 3.50 (s, 3H). 13C NMR (125 MHz, d6-DMSO) δ 165.3, 164.2, 159.9, 149.0, 138.5, 136.6, 135.3, 134.0, 132.5, 131.9, 128.5, 125.8, 125.5, 121.2, 119.9, 118.3, 118.2, 115.0, 114.0, 36.2.
N-(10-Methyl-11-oxo-10,11-dihydrodibenzo[b,f][1,4]oxazepin-8-yl)-4-(trifluoromethyl)benzamide 12. Prepared as for 11 from aryl amine 9 (50 mg, 0.208 mmol) and 4-trifluoromethylbenzoyl chloride (52 mg, 0.249 mmol). The mixture was poured into 1 M HCl and extracted with EtOAc (×3). The combined extracts were washed with H2O then brine, dried (MgSO4) and evaporated. The crude product was purified by column chromatography (EtOAc/CHCl3, 10:90 → 20:80) to give amide 12 (75 mg, 87%) as an off-white solid. Mp 224–231°C. LCMS: tR 3.23 min. (98.5% purity). HRMS (ESI, +ve ion) m/z calcd for C₂₂H₁O₃N₂F₃+ [M + H+] 413.1108, found 413.1109. 1H NMR (500 MHz, d6-DMSO) δ 10.59 (broad s, 1H), 8.16–8.12 (m, 2H), 7.94–7.90 (m, 2H), 7.87 (d, J = 2.4 Hz, 1H), 7.77 (dd, J = 7.8, 1.7 Hz, 1H), 7.63–7.57 (m, 2H), 7.38 (d, J = 8.8 Hz, 1H), 7.35 (dd, J = 8.1, 0.9 Hz, 1H), 7.33–7.29 (m, 1H), 3.51 (s, 3H). 13C NMR (125 MHz, d6-DMSO) δ 165.8, 164.9, 160.4, 149.4, 138.8, 137.1, 135.8, 134.5, 132.4, 132.0 (q, 2JCF = 32 Hz), 129.0, 126.3, 126.0–125.9 (m), 124.4 (q, 1JCF = 272 Hz), 121.7, 120.3, 118.7, 115.4, 36.7.
4-Methoxy-N-(10-methyl-11-oxo-10,11-dihydrodibenzo[b,f][1,4]oxazepin-8-yl)benzamide 13. Prepared as for 1 from aryl amine 9 (28 mg, 0.117 mmol) and p-anisic acid (20 mg, 0.128 mmol) using T3P. The crude product was purified by column chromatography (CHCl3) to give product 13 (38 mg, 87%) as a colourless solid. Mp 253–255°C. LCMS: tR 2.79 min. (96.9% purity). HRMS (ESI, +ve ion) m/z calcd for C₂₂H₁O₄N₂+ [M + H+] 375.1339, found 375.1340. 1H NMR (400 MHz, CDCl3) δ 7.88–7.78 (m, 5H), 7.45–7.40 (m, 1H), 7.22–7.15 (m, 4H), 6.98–6.92 (m, 2H), 3.85 (s, 3H), 3.59 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 166.7, 165.5, 162.9, 160.7, 150.1, 136.5, 136.0, 133.8, 132.6, 129.1, 126.7, 126.4, 125.5, 121.8, 119.9, 117.9, 115.0, 114.3, 55.7, 37.0.
3,4-Dimethyl-N-(10-methyl-11-oxo-10,11-dihydrodibenzo[b,f][1,4]oxazepin-8-yl)benzamide 14. Prepared as for 1 from aryl amine 9 (50 mg, 0.208 mmol) and 3,4-dimethylbenzoic acid (34 mg, 0.229 mmol) using T3P. The crude product was purified by column chromatography (MeOH/CHCl3, 0:100 → 2:98) to give amide 14 (47 mg, 61%) as an off-white solid. Mp 242–244°C. LCMS: tR 3.15 min (96.2% purity). HRMS (ESI, +ve ion) m/z calcd for C₂₃H₂₁O₃N₂+ [M + H+] 373.1547, found 373.1542. 1H NMR (400 MHz, d6-DMSO) δ 10.24 (broad s, 1H), 7.87 (d, J = 2.4 Hz, 1H), 7.76 (dd, J = 7.7, 1.7 Hz, 1H), 7.75–7.72 (m, 1H), 7.68 (dd, J = 7.8, 1.7 Hz, 1H), 7.64–7.56 (m, 2H), 7.37–7.27 (m, 4H), 3.50 (s, 3H), 2.30 (s, 3H), 2.29 (s, 3H). 13C NMR (d6-DMSO, 125 MHz) δ 165.5, 165.3, 160.0, 148.6, 140.6, 137.2, 136.4, 135.2, 134.0, 132.0, 131.9, 129.4, 128.6, 125.9, 125.4, 125.1, 121.0, 119.8, 118.0, 114.7, 36.2, 19.4.
10-Methyl-7-nitrodibenz[b,f][1,4]oxazepin-11(10H)-one 16: This method was adapted from the protocol previously described by Liu et al.[10] 2-Hydroxy-N-methylbenzamide 15 (240 mg, 1.59 mmol) was dissolved in DMF (10 mL), then 3,4-difluoronitrobenzene (176 µL, 1.59 mmol) and K2CO3 (549 mg, 3.98 mmol) were added. The hetereogeneous mixture was stirred vigorously for 1 h at 80°C. The yellow mixture was cooled and evaporated under reduced pressure and the residue was partitioned between EtOAc and water. The organic phase was washed with brine, dried (MgSO4), then concentrated to afford a light-yellow oil. Purification by flash column chromatography (CH2Cl2/heptane, 2:1) yielded 16 (313 mg, 73%) as a white solid. Mp 156–158°C (lit.[10] 158–163°C). 1H NMR (400 MHz, CDCl3) δ 8.12 (d, J = 2.6 Hz, 1H), 8.08 (dd, J = 8.9, 2.6 Hz, 1H), 7.90–7.86 (m, 1H), 7.53–7.49 (m, 1H), 7.34 (d, J = 8.9 Hz, 1H), 7.28–7.24 (m, 2H), 3.60 (s, 3H).
7-Amino-10-methyldibenzo[b,f][1,4]oxazepin-11(10H)-one 17: The nitro compound 16 (313 mg, 1.16 mmol) was hydrogenated (balloon) over 10% Pd/C (50% H2O) (63 mg, 0.030 mmol) in a mixture of ethanol (100 mL) and EtOAc (5 mL) at room temperature for 2 h. The mixture was filtered through Celite and evaporated under reduced pressure. The residue was dissolved in EtOAc and filtered through a plug of silica gel to give 17 (249 mg, 1.04 mmol, 90%) as a pale-yellow solid. Mp 188–192°C (lit.[11] 194–196°C). 1H NMR (400 MHz, CDCl3) δ 7.84 (dd, J = 7.8, 1.7 Hz, 1H), 7.43–7.37 (m, 1H), 7.21–7.15 (m, 1H), 7.12 (dd, J = 8.1, 1.0 Hz, 1H), 6.98 (d, J = 8.6 Hz, 1H), 6.55 (d, J = 2.6 Hz, 1H), 6.47 (dd, J = 8.6, 2.6 Hz, 1H), 3.67 (broad s, 2H), 3.51 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 166.4, 160.4, 154.7, 145.1, 133.1, 132.1, 126.8, 126.6, 125.1, 123.4, 119.8, 112.1, 107.3, 36.7.
4-Methyl-N-(10-methyl-11-oxo-10,11-dihydrodibenzo[b,f][1,4]oxazepin-7-yl)benzamide 2: Aryl amine 17 (50 mg, 0.208 mmol) and p-toluic acid (31.2 mg, 0.229 mmol) were dissolved in pyridine (1 mL). After 10 min, T3P (50% w/w in EtOAc, 0.186 mL, 0.312 mmol) was added dropwise. The mixture was stirred at room temperature for 3 h, then poured into 1 M HCl (15 mL) and extracted with EtOAc (3 × 15 mL). The combined extracts were washed with brine, dried (MgSO4) and evaporated under reduced pressure. Silica gel chromatography (EtOAc/CHCl3, 10:90) afforded 2 (55 mg, 74%) as a colourless solid. Mp 211–213°C. LCMS: tR 2.96 min (99.6% purity). m/z 359.31 [M + H]+. HRMS (APCI, +ve ion) m/z calcd for C22H19N2O3+ [M + H]+ 359.1390, found 359.1388. 1H NMR (400 MHz, CDCl3) δ 7.88–7.84 (m, 1H); 7.77 (broad s, 1H), 7.76–7.71 (m, 3H), 7.46–7.40 (m, 1H), 7.37 (dd, J = 8.8, 2.5 Hz, 1H), 7.30–7.26 (m, 2H), 7.22–7.17 (m, 3H), 3.56 (s, 3H), 2.41 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 166.4, 165.6, 160.4, 153.9, 142.8, 136.2, 133.5, 132.2, 132.1, 131.6, 129.5, 127.0, 126.2, 125.3, 122.7, 120.0, 117.2, 113.2, 36.8, 21.5. A purchased (ChemDiv) sample of SN00797640 gave mp 211–212°C and spectral data in agreement with compound 2.
Cell lines and cultivation
Metronidazole-sensitive Giardia duodenalis assemblage B (BRIS/91/HEPU/1279)[12] parasites were maintained essentially as previously described in 8 mL borosilicate culture tubes, using modified Keister’s TYI-S-33 medium supplemented with 10% heat-inactivated fetal bovine serum, 100 units/mL penicillin, and 100 µg/mL streptomycin.[13]
Giardia dose-response assays
The activity of compounds was assessed by dose response to determine IC50 values as previously described.[14] In brief, compounds were serially diluted in triplicate, in DMSO-controlled media, and all test wells were inoculated with parasites (1.5 × 104 parasites/well; 100 µL/test). Albendazole (positive control) plates were prepared identically and run alongside each experiment. All plates were sealed in culture chambers, gassed with 3% O2, 5% CO2 in N2 and incubated at 37°C, with parasites imaged and enumerated at 48 h.[14] Growth inhibition was calculated as a percentage relative to vehicle controls minus any background, and IC50 values were determined using log-linear interpolation as previously described.[15] All assays were performed at least twice.
Supplementary material
Copies of 1H NMR, 13C NMR spectra and LCMS traces are available online. Supplementary material is available online.
Data availability
The data that support this study are available in the article and accompanying online supplementary material.
Conflicts of interest
The authors declare no conflicts of interest.
Declaration of funding
This work was funded by NHMRC Grant APP1141069w.
References
[1] JL Dahlin, MA Walters, The essential roles of chemistry in high-throughput screening triage. Future Med Chem 2014, 6, 1265.| The essential roles of chemistry in high-throughput screening triage.Crossref | GoogleScholarGoogle Scholar |
[2] JB Baell, Observations on screening-based research and some concerning trends in the literature. Future Med Chem 2010, 2, 1529.
| Observations on screening-based research and some concerning trends in the literature.Crossref | GoogleScholarGoogle Scholar |
[3] X Yi, L Xue, T Thomas, JB Baell, Action plan for hit identification (APHID): KAT6A as a case study. Future Med Chem 2020, 12, 423.
| Action plan for hit identification (APHID): KAT6A as a case study.Crossref | GoogleScholarGoogle Scholar |
[4] JH Ryan, KE Jarvis, RJ Mulder, CL Francis, GP Savage, O Dolezal, TS Peat, JJ Deadman, Unexpected isomerisation of a fragment analogue during fragment-based screening of HIV integrase catalytic core domain. Aust J Chem 2015, 68, 1871.
| Unexpected isomerisation of a fragment analogue during fragment-based screening of HIV integrase catalytic core domain.Crossref | GoogleScholarGoogle Scholar |
[5] A Riches, CJS Hart, KR Trenholme, TS Skinner-Adams, Anti-Giardia drug discovery: current status and gut feelings. J Med Chem 2020, 63, 13330.
| Anti-Giardia drug discovery: current status and gut feelings.Crossref | GoogleScholarGoogle Scholar |
[6] BT Tozer, S Smiles, A rearrangement of o-carbamyl derivatives of diphenyl ether. J Chem Soc 1938, 2052.
| A rearrangement of o-carbamyl derivatives of diphenyl ether.Crossref | GoogleScholarGoogle Scholar |
[7] WGA Ibrom, AW Frahm, Synthesis and antimycobacterial activity of nitroxanthones. 1st communication: synthesis and differential scanning calorimetry analysis. Arzneimittelforschung 1997, 47, 662.
[8] Go YJ, Kim MH, Shin JS, Lee GH, Lim HJ, Lee SH, Lee WH. WO 2018/174442 A1. (Ko YY, Kim MH, Shin JS, Ku GB, Lee GH, Lim HJ, Lee SH. Korean Patent 0106599. 2018.)
[9] K Nagarajan, A Venkateswarlu, CL Kulkarni, A Nagana Goud, RK Shah, Condensed heterotricycles: aminolkyl and aminodibenz[b,f][1,4]oxazepinon-11-(10)H-ones. Indian J Chem 1974, 12, 236.
[10] Y Liu, C Chu, A Huang, C Zhan, Y Ma, C Ma, Regioselective synthesis of fused oxazepinone scaffolds through one-pot Smiles rearrangement tandem reaction. ACS Comb Sci 2011, 13, 547.
| Regioselective synthesis of fused oxazepinone scaffolds through one-pot Smiles rearrangement tandem reaction.Crossref | GoogleScholarGoogle Scholar |
[11] JM Klunder, KD Hargrave, MA West, E Cullen, K Pal, ML Behnke, SR Kapadia, DW McNeil, JC Wu, GC Chow, J Adams, Novel non-nucleoside inhibitors of HIV-1 reverse transcriptase. 2. Tricyclic pyridobenzoxazepinones and dibenzoxazepinones. J Med Chem 1992, 35, 1887.
| Novel non-nucleoside inhibitors of HIV-1 reverse transcriptase. 2. Tricyclic pyridobenzoxazepinones and dibenzoxazepinones.Crossref | GoogleScholarGoogle Scholar |
[12] JA Upcroft, PFL Boreham, RW Campbell, RW Shepherd, P Upcroft, Biological and genetic analysis of a longitudinal collection of Giardia samples derived from humans. Acta Trop 1995, 60, 35.
| Biological and genetic analysis of a longitudinal collection of Giardia samples derived from humans.Crossref | GoogleScholarGoogle Scholar |
[13] DB Keister, Axenic culture of Giardia lamblia in TYI-S-33 medium supplemented with bile. Trans R Soc Trop Med Hyg 1983, 77, 487.
| Axenic culture of Giardia lamblia in TYI-S-33 medium supplemented with bile.Crossref | GoogleScholarGoogle Scholar |
[14] CJS Hart, T Munro, KT Andrews, JH Ryan, AG Riches, TS Skinner-Adams, A novel in vitro image-based assay identifies new drug leads for giardiasis. Int J Parasitol Drugs Drug Resist 2017, 7, 83.
| A novel in vitro image-based assay identifies new drug leads for giardiasis.Crossref | GoogleScholarGoogle Scholar |
[15] W Huber, JC Koella, A comparison of three methods of estimating EC50 in studies of drug resistance of malaria parasites. Acta Trop 1993, 55, 257.
| A comparison of three methods of estimating EC50 in studies of drug resistance of malaria parasites.Crossref | GoogleScholarGoogle Scholar |