

Influence of Crystal Disorder in MoS2 Cathodes for Secondary Hybrid Mg-Li Batteries*
Jyah Strachan A , Lu Chen A , Thomas Ellis A , Anthony Masters A and Thomas Maschmeyer A B
A , Lu Chen A , Thomas Ellis A , Anthony Masters A and Thomas Maschmeyer A B
A Laboratory of Advanced Catalysis for Sustainability, School of Chemistry, The University of Sydney, Camperdown, NSW 2006, Australia.
B Corresponding author. Email: thomas.maschmeyer@sydney.edu.au
Australian Journal of Chemistry 74(11) 819-825 https://doi.org/10.1071/CH21187
Submitted: 4 August 2021 Accepted: 10 November 2021 Published: 30 November 2021
Abstract
The full extent to which the electrochemical properties of MoS2 electrodes are influenced by their morphological characteristics, such as crystalline disorder, remains unclear. Here, we report that disorder introduced by ball-milling decreases the Faradaic component of cell capacity and leads to increasingly pseudo-capacitive behaviour. After high temperature annealing, a more battery-like character of the cell is restored, consistent with a decrease in disorder. These findings aid the optimisation of MoS2 electrodes, which show promise in several battery technologies.
Keywords: MoS2, molybdenum sulfide, electrochemistry, battery, capacitor, pseudo-capacitor, materials chemistry, lithium.
Introduction
Lithium ion batteries are unmatched in portable energy storage applications for their high power and energy density.[1–3] A range of Li-intercalation cathodes has been studied, among which MoS2 excels in cost, capacity, and power.[4–7] Indeed, the first commercial rechargeable Li ion battery (MOLICEL) used a Li metal anode (Eqn 1) and a MoS2 cathode (Eqn 2). The battery did not achieve commercial success mainly due to uncontrollable Li dendrite formation during charging with a Li plating stripping mechanism. A MoS2 electrode stores charges via intercalation/deintercalation of Li+ ions. MoS2 was used mainly due to its low cost (e.g. sub-micrometre MoS2 powder retails for dollars per kg) and high capacity retention.[8] Like graphite, MoS2 consists of Bernal-stacked monolayers of covalently bound Mon(S1/3)6n, the layers being held together by weak Van der Waals forces. This layer structure allows for the intercalation of guest ions, such as Li+.[9–11] Historically, intercalation has been understood as a process whereby Li+ ions diffuse between the MoS2 layers and are stabilised in octahedral interstices between sulfur atoms.[12,13] The rate of diffusion along the gallery between the basal layers of MoS2 is rapid as the activation energy of Li+ ion migration between these layers is low.[12] The presence of intercalated Li+ ions do, however, cause expansion of the c-axis by ~5 %.[12,14] This expansion induces strain in the 2H-MoS2 layers, which leads to the formation of dislocations.[15] As the lithium concentration nears saturation, the resultant strain can cause particles to repeatedly break into ever smaller polycrystalline fragments, potentially creating alternative pathways for Li+ ion diffusion into the MoS2 particles.[16] Recently, Cui and co-workers have shown that intercalation can also occur through the basal planes of exfoliated MoS2.[17] The authors propose that Li+ ions may additionally diffuse through ‘natural defects’ in the MoS2 lattice, although this process occurs at a relatively lower rate than diffusion between basal planes. As a result, it might be expected that the kinetics of the cathodic reaction (Eqn 2) are enhanced in MoS2 particles with a high defect concentration, i.e. substantial disorder.

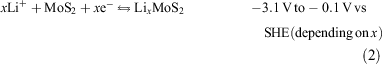

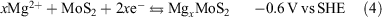
Defect introduction in MoS2 can be achieved both during and post-synthesis.[18] Although sulfur vacancies, grain boundaries, and strain can be controlled by varying synthesis parameters in techniques such as chemical vapour deposition,[18–20] in-situ approaches have limited scalability. Post-synthetic treatments are better suited to upscaling with lower cost bulk materials, as they allow MoS2 to be sourced rather than synthesised. In MoS2 crystals, sulfur vacancies can be induced with electron beam or argon plasma treatment,[21,22] Mo–O moieties can be incorporated by oxygen plasma treatment and hydrothermal treatment,[23,24] and lattice ripples induced by SF6, CF4, and CHF3 plasmas.[25] Ozone treatment and ion, proton, and α-particle bombardment also introduce defects, although the effects are less well understood.[18] An alternative to these high energy processes is mechanical milling, which is a scalable method to manipulate crystal morphology by applying impact and shear stress.[26] When applied to layered materials, ball-milling has been shown to cleave, exfoliate, and twist sheets,[26] introduce vacancies[27] and expose edge sites.[28] The coordinatively unsaturated atoms along exposed edge sites have been shown to preferentially bind Li+.[13,29] Thus, mechanical milling is expected to improve the capacity of MoS2 cathodes in Li+ ion batteries by introducing defects, and increasing the number of edge sites. In addition, ball-milling is expected to decrease the particle size, increasing surface area and reducing diffusion path length, all of which are known to further improve the capacity of electrode materials in general.[12]
Lithium anodes are prone to forming dendrites, which can short-circuit the cell, initiating an exothermic reaction between the electrode and electrolyte that can cause a battery to ignite.[30–32] Efforts to inhibit dendrite formation have focussed on modifying the electrode,[33,34] electrolyte,[35–38] separator,[39,40] and battery management system,[31] although at high current, low temperature, or low overcharge, dendrites still present a significant problem.[32] A possible solution might be changing the anode material while retaining Li+ as the charge carrier. Magnesium is a viable alternative anode (Eqn 3) as it is not prone to dendrite formation and has suitable electrochemical characteristics (Table 1). However, intercalation/deintercalation of Mg2+ into the MoS2 cathode (Eqn 4) is slow.[41,42] Although research into Mg batteries is active, they are not commercially available yet, in part due to a lack of suitable cathode materials;[43–48] the known cathode materials typically possess low capacity and/or poor intercalation kinetics.[48,49] An alternative approach is to use Mg/Li dual-ion hybrid batteries to exploit the benefits of both Mg and Li batteries by combining the dendrite-free Mg anode with the fast kinetics of a Li+ intercalation cathode.[50–52] An attractive application of this concept is using earth abundant molybdenum sulfide phases as cathodes.[42] In addition, the low redox potential of MoS2/Lix(MoS2)y also matches the anodic potential window of common magnesium electrolytes (e.g. +2.75 V versus Mg for electrolyte used in this work). Cathode materials such as Ni–Mn–Co oxide (NMC) and lithium iron phosphate (LFP) are not suitable in this case due to their higher redox potentials.[2]

|
As the rate of reversible Li+ ion intercalation is the kinetic bottleneck of the cathodic reaction, the modification of bulk MoS2 morphology by ball-milling and associated changes at the atomic level, as outlined above, can be expected to influence the electrochemical performance of batteries. This study, therefore, aims to investigate the effect of changes in the cathode disorder/crystallinity on the electrochemical performance of Mg/Li hybrid cells.
Results and Discussion
Structural Characterisation
Two commercial bulk MoS2 samples, herein designated 2 µm and 90 nm (from the average height in the c-axis direction – the direction perpendicular to the stacked S-Mo-S layers), were chosen for baseline measurements. Adjusting the treatment duration of mechanical milling led to samples with varying particle sizes and degrees of crystal defects. The 90 nm MoS2 was ball-milled for 4, 24, and 60 h to produce BM4-, BM24-, BM60-MoS2, respectively. Characterisations by transmission electron microscopy (TEM), powder X-ray diffraction (PXRD), and Raman spectroscopy are shown in Fig. 1 and Fig. 2 and summarised alongside N2-sorption analysis (fitted with the Brunauer–Emmett–Teller analysis; BET) in Table 2. Scanning electron microscopy (SEM) images are shown in Fig. S2 (Supplementary Material). As the ball-milling duration was lengthened (4, 24, and 60 h), the MoS2 flakes became increasingly more fissured and smaller in size (Fig. S2), the surface area rose from 8 to 90 m2 g−1 (Fig. S2), the XRD reflections broadened (Fig. 2), the layers in the crystallites became distorted with the introduction of defects (Fig. 1), and the defect-induced LA(M) Raman mode[53,54] increased in relative intensity (Fig. 2). These results are all consistent with the crystals of the samples breaking into smaller, increasingly disordered crystallites (by the introduction of defects and strain) as a result of the ball milling.

|

|

|
Cell Preparation
The MoS2 samples, 2 µm-, 90 nm-, BM4-, BM24-, and BM60-MoS2, were suspended in N-methylpyrrolidone with polyvinylidene difluoride and carbon black (8:1:1 active/binder/carbon mass ratio), coated onto stainless steel, and dried at 120°C. The coated electrodes were transferred to an argon-filled glove box and assembled as part of conventional coin cells using a 0.8 M Mg + 1.0 M LiCl in tetrahydrofuran (THF) electrolyte (prepared from a 1.05:1 molar ratio, EtMgCl/t-AmOH mixture in THF) and a Mg metal anode. The excess anode material and the choice of electrolyte concentration, in addition to the kinetically favoured anodic reaction, ensured that the cathode was the kinetic and capacity limiting component of the cell (see Supplementary Material for details).
Electrochemical Performance
The gravimetric capacity density, reversibility, and stability of the coin cells were assessed. At a charging rate of 250 mA g−1, the capacity density of the samples increased from 170 to 266 mA h g−1 as the ball-milling duration of the electrode precursors increased (Table 3 with b indicating the charge storage mechanism, see also Eqn 5).

|
Analysis of the voltage plateau (the region in the voltage/capacity density plot where discharging voltage remains nearly constant across a given capacity range) was used to reveal the charge–storage mechanism of the system. For our samples, electrodes prepared with MoS2 samples that were not ball-milled exhibit voltage plateaus for approximately a third of the entire discharge capacity. As the ball-milling duration of the electrode precursors increases, the plateau begins to increase in slope, and then disappears entirely for the BM24- and BM60-MoS2 samples (Fig. 3). This behaviour is consistent with a change to the charge-storage mechanism from battery-like to pseudo-capacitor-like.[55]

|
Given the voltage profile of the ball-milled samples indicates a change in the relative extent with which the various charge storage mechanisms operate in parallel, the samples were further analysed by cyclic voltammetry to quantify the ratio of battery- to pseudo-capacitor-like behaviour. The cyclic voltammograms in Fig. 4 show two redox peaks A and B, which correspond to the two plateaus in the cycling plots (Fig. 3). These current peaks progressively broaden as the ball-milling duration of the electrode precursors is increased. The current was analysed as a function of scan rate to further investigate the charge storage mechanism according to Eqn 5:[56–58]

|

where i = current, v = scan rate, and a and b = adjustable parameters (with the magnitude of b being indicative of the storage mechanism).
Values of b of either 0.5 or 1 reflect typical battery (Faradaic) or pseudo-capacitor (non-Faradaic) behaviour, respectively, with possible solutions to Eqn 5 existing on a continuum between these two values.[59] The values for b, derived from fitting both current lines, are given in Fig. 4 and Table 3. Values for b for the BM24- and BM60-MoS2 could not be obtained as the maxima could not be resolved. The average values of b derived from fitting peak A and peak B in the plots of Fig. 4 are 0.82, 0.86, and 0.85 for 2 µm, 90 nm, and 90 nm BM4 MoS2 respectively. This progression generally shows the expected trend of b value with particle size. For 90 nm BM24- and 90 nm BM60-MoS2, the impact of the milling treatment on the charge storage mechanism is more pronounced, the voltage plateaus are barely noticeable, indicating the dominance of capacitance as a contributor to the total charge storage process.
The stability of an electrode is assessed by examining the capacity retention over a given number of charge/discharge cycles. For electrodes prepared from the crystalline samples, 2 µm- and 90 nm-MoS2, a capacity decay of ~20 % is observed after 500 cycles and this value increases to 50 % for electrodes prepared from ball-milled samples.
The Coulombic efficiency describes the reversibility of the cathodic reaction (Eqn 6), which, together with the capacity retention, reflects the lifespan of a battery.[60] Values of the Coulombic efficiency less than unity indicate some combination of irreversible Li+ intercalation, changes to the cathode, or the presence of side reactions.[60]
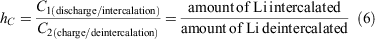
For all samples, after conditioning (~50 cycles), the coulombic efficiency per cycle is always very close to 100 %, however, small losses build up over time and cause the net capacity loss.
Influence of Defects
Mechanical milling had a significant effect on the morphology and electrochemical performance of the MoS2 electrodes in this Mg/Li hybrid battery. The morphology of the samples becomes increasingly disordered (Table 2 and Fig. 2) and the charge storage mechanism shifts from battery-like to pseudo capacitor-like as the ball-milling duration for the electrode precursors is increased (Table 3).
The effect of ball-milling on crystalline samples is established; phenomena such as sheet exfoliation, particle size decrease, and defect generation are well precedented.[61] In the current study, the creation of defects increases the variety of MoS2 sites that can accept a Li+ ion.[62] These sites may be any combination of edges, dislocations, and vacancies both on the surface and within the layers of MoS2. Each unique site possesses a different thermodynamic potential for the cathodic reaction (Eqn 2), the ordering of which results in a slope in the voltage profile of the ball-milled samples (Fig. 3).[62] These results are contrasted by the voltage profiles of the non-ball-milled samples, where most of the sites of the cathodic reaction (i.e. intercalation) are equivalent because of the crystalline nature of the sample. This results in the voltage plateaus observed for 90 nm- and 2 µm-MoS2 (see, for example, Fig. S4a, Supplementary Material). In addition, because of the creation of new Li+ accepting sites upon milling, the capacity density was increased (see, for example, Fig. S4b, Supplementary Material). Similar behaviour has been previously observed in graphite[62] and graphene.[63]
Since cells incorporating electrodes fabricated from 2 μm-, 90 nm-, and BM4-MoS2 show relatively good capacity retention upon cycling, they were assessed at different current densities to investigate the influence of the current density on the resulting capacity of the cathode material. The discharging capacity was recorded at increasing current densities up to a maximum current density of 5 A g−1. A final scan at the original current density of 0.25 A g−1 was then performed to check whether the capacity could recover. Fig. S4b (Supplementary Material) plots the discharge capacity of cells with 2 μm-, 90 nm-, and BM4-MoS2 electrodes as a function of current density. All these three batteries exhibit similar features: the discharging capacity gradually decreases as the current density is increased and returns to the original value when the current density is lowered. At the lowest current density (0.25 A g−1), the cell with the electrode prepared from 2 μm-MoS2 exhibits the lowest specific capacity (140 mA h g−1) of the three and that cell with the electrode prepared from BM4-MoS2 shows the highest specific capacity (170 mA h g−1). At the highest current density of 5 A g−1, the ranking of the discharging capacity reverses: the cell with the electrode prepared from 2 μm-MoS2 is least affected (with a capacity decay of 60 %) and that with an electrode prepared from BM4-MoS2 is most affected (with a capacity decay of 80 %). We conclude that this is due to the undamaged crystal structure, which enables intercalation to be maintained more efficiently even at the higher charging rates. The ball-milled samples are more disordered as well as defect-rich and simply have a lower inherent intercalation capacity, being dominated by more capacitive characteristics.[17,64]
Thus, we propose that to optimise Li+-ion MoS2 electrodes, the crystallinity should be maximised and the particle size minimised. The crystallinity determines the capacity density of the intercalation region and the particle size determines the rate of Li+ intercalation. In order to test this hypothesis, we annealed the BM60-MoS2 sample at 800°C in a reducing atmosphere for 24 h – the resultant sample was designated Ann’d-MoS2. The physical characterisation data for the Ann’d-MoS2 are shown in Fig. 5. In summary, the sample exhibited sharper XRD reflections (which corresponded to a crystallite size of 100 nm; up from 20 nm) and the LA(M) signal in the Raman spectrum decreased in relative intensity compared with the un-annealed sample (to an LA(M) to E2g ratio of 0.33; down from 0.51). These changes are consistent with improved particle crystallinity.[53,54,65]

|
By checking whether the cycling curve of the cell incorporating the Ann’d-MoS2 sample possesses a plateau, one can determine whether the battery-like behaviour of the cell has increased (or correspondingly, whether the capacitor-like behaviour has decreased). As seen in Fig. 5 (right) the departure of the curve from linearity indicates an increased contribution from battery-like intercalation processes and decreased contribution from the capacitor-like adsorption processes. Relative to the non-annealed, ball-milled samples, which did not exhibit a plateau, this is a significant improvement. The data are consistent with our hypothesis, i.e. that defects introduced by ball-milling decrease the Faradaic contribution of a cell’s capacity and lead to increasingly pseudo-capacitive behaviour. When the defects are removed by high temperature annealing, the battery-like character of the cell is restored, further supporting our hypothesis above.
Conclusions
We have shown that the electrochemical performance of MoS2 cathodes in Mg/Li batteries is primarily determined by two factors: the particle size and crystallinity. Commercial MoS2 samples were ball-milled to manipulate these two factors. To separate the effects of sample crystallinity from particle size, the ball milled samples were annealed and the electrochemistry of the two sample treatments compared. The annealed samples exhibited a larger capacity density plateau, consistent with the homogenisation of the active sites, in turn, consistent with increased crystallinity. These samples also exhibited a greater capacity than the samples that had not undergone ball-milling, which was attributed to the increased surface area of the ball milled particles.
Supplementary Material
Supplementary information, including materials, methods, further morphological characterisation, and cycling data are available on the Journal’s website.
Data Availability Statement
The data that support this study will be shared upon reasonable request to the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
Declaration of Funding
This research was in part supported by ARC DP200103099.
References
[1] N. Nitta, F. Wu, J. T. Lee, G. Yushin, Mater. Today 2015, 18, 252.| Crossref | GoogleScholarGoogle Scholar |
[2] M. Li, J. Lu, Z. Chen, K. Amine, Adv. Mater. 2018, 30, 1800561.
| Crossref | GoogleScholarGoogle Scholar |
[3] B. Scrosati, J. Hassoun, Y.-K. Sun, Energy Environ. Sci. 2011, 4, 3287.
| Crossref | GoogleScholarGoogle Scholar |
[4] C.-J. Hsu, C.-Y. Chou, C.-H. Yang, T.-C. Lee, J.-K. Chang, Chem. Commun. 2016, 52, 1701.
| Crossref | GoogleScholarGoogle Scholar |
[5] Y. Teng, H. Zhao, Z. Zhang, Z. Li, Q. Xia, Y. Zhang, L. Zhao, X. Du, Z. Du, P. Lv, K. Świerczek, ACS Nano 2016, 10, 8526.
| Crossref | GoogleScholarGoogle Scholar | 27556425PubMed |
[6] M. Azhagurajan, T. Kajita, T. Itoh, Y.-G. Kim, K. Itaya, J. Am. Chem. Soc. 2016, 138, 3355.
| Crossref | GoogleScholarGoogle Scholar | 26883789PubMed |
[7] F. Xiong, H. Wang, X. Liu, J. Sun, M. Brongersma, E. Pop, Y. Cui, Nano Lett. 2015, 15, 6777.
| Crossref | GoogleScholarGoogle Scholar | 26352295PubMed |
[8] T. Stephenson, Z. Li, B. Olsen, D. Mitlin, Energy Environ. Sci. 2014, 7, 209.
| Crossref | GoogleScholarGoogle Scholar |
[9] J. Zhang, A. Yang, X. Wu, J. van de Groep, P. Tang, S. Li, B. Liu, F. Shi, J. Wan, Q. Li, Y. Sun, Z. Lu, X. Zheng, G. Zhou, C.-L. Wu, S.-C. Zhang, M. L. Brongersma, J. Li, Y. Cui, Nat. Commun. 2018, 9, 5289.
| Crossref | GoogleScholarGoogle Scholar | 30538249PubMed |
[10] D. Sun, D. Ye, P. Liu, Y. Tang, J. Guo, L. Wang, H. Wang, Adv. Energy Mater. 2018, 8, 1702383.
| Crossref | GoogleScholarGoogle Scholar |
[11] J. A. Spirko, M. L. Neiman, A. M. Oelker, K. Klier, Surf. Sci. 2003, 542, 192.
| Crossref | GoogleScholarGoogle Scholar |
[12] T. Stephenson, Z. Li, B. Olsen, D. Mitlin, Energy Environ. Sci. 2014, 7, 209.
| Crossref | GoogleScholarGoogle Scholar |
[13] Y. Li, D. Wu, Z. Zhou, C. R. Cabrera, Z. Chen, J. Phys. Chem. Lett. 2012, 3, 2221.
| Crossref | GoogleScholarGoogle Scholar | 26295774PubMed |
[14] M. S. Whittingham, F. R. Gamble, Mater. Res. Bull. 1975, 10, 363.
| Crossref | GoogleScholarGoogle Scholar |
[15] P. J. Mulhern, Can. J. Phys. 1989, 67, 1049.
| Crossref | GoogleScholarGoogle Scholar |
[16] K. Chrissafis, M. Zamani, K. Kambas, J. Stoemenos, N. Economou, I. Samaras, C. Julien, Mater. Sci. Eng. B 1989, 3, 145.
| Crossref | GoogleScholarGoogle Scholar |
[17] J. Zhang, A. Yang, X. Wu, J. van de Groep, P. Tang, S. Li, B. Liu, F. Shi, J. Wan, Q. Li, Nat. Commun. 2018, 9, 5289.
| Crossref | GoogleScholarGoogle Scholar | 30538249PubMed |
[18] Z. Lin, B. R. Carvalho, E. Kahn, R. Lv, R. Rao, H. Terrones, M. A. Pimenta, M. Terrones, 2D Mater. 2016, 3, 022002.
| Crossref | GoogleScholarGoogle Scholar |
[19] C. Huang, S. Wu, A. M. Sanchez, J. J. Peters, R. Beanland, J. S. Ross, P. Rivera, W. Yao, D. H. Cobden, X. Xu, Nat. Mater. 2014, 13, 1096.
| Crossref | GoogleScholarGoogle Scholar | 25150560PubMed |
[20] A. Castellanos-Gomez, R. Roldán, E. Cappelluti, M. Buscema, F. Guinea, H. S. van der Zant, G. A. Steele, Nano Lett. 2013, 13, 5361.
| Crossref | GoogleScholarGoogle Scholar | 24083520PubMed |
[21] H.-P. Komsa, S. Kurasch, O. Lehtinen, U. Kaiser, A. V. Krasheninnikov, Phys. Rev. B 2013, 88, 035301.
[22] P. K. Chow, R. B. Jacobs-Gedrim, J. Gao, T.-M. Lu, B. Yu, H. Terrones, N. Koratkar, ACS Nano 2015, 9, 1520.
| Crossref | GoogleScholarGoogle Scholar | 25603228PubMed |
[23] J. Xie, J. Zhang, S. Li, F. Grote, X. Zhang, H. Zhang, R. Wang, Y. Lei, B. Pan, Y. Xie, J. Am. Chem. Soc. 2013, 135, 17881.
| Crossref | GoogleScholarGoogle Scholar | 24191645PubMed |
[24] N. Kang, H. P. Paudel, M. N. Leuenberger, L. Tetard, S. I. Khondaker, J. Phys. Chem. C 2014, 118, 21258.
| Crossref | GoogleScholarGoogle Scholar |
[25] M. Chen, H. Nam, S. Wi, G. Priessnitz, I. M. Gunawan, X. Liang, ACS Nano 2014, 8, 4023.
| Crossref | GoogleScholarGoogle Scholar | 24680193PubMed |
[26] S. Zhang, Y. Cui, B. Wu, R. Song, H. Song, J. Zhou, X. Chen, J. Liu, L. Cao, RSC Adv. 2014, 4, 505.
| Crossref | GoogleScholarGoogle Scholar |
[27] B. Zhang, L. Lu, M. Lai, Physica B 2003, 325, 120.
| Crossref | GoogleScholarGoogle Scholar |
[28] D. Wang, Z. Wang, C. Wang, P. Zhou, Z. Wu, Z. Liu, Electrochem. Commun. 2013, 34, 219.
| Crossref | GoogleScholarGoogle Scholar |
[29] H. Wang, Q. Zhang, H. Yao, Z. Liang, H.-W. Lee, P.-C. Hsu, G. Zheng, Y. Cui, Nano Lett. 2014, 14, 7138.
| Crossref | GoogleScholarGoogle Scholar | 25372985PubMed |
[30] B. Scrosati, J. Garche, J. Power Sources 2010, 195, 2419.
| Crossref | GoogleScholarGoogle Scholar |
[31] J. Wen, Y. Yu, C. Chen, Mater. Express 2012, 2, 197.
| Crossref | GoogleScholarGoogle Scholar |
[32] H. Wu, D. Zhuo, D. Kong, Y. Cui, Nat. Commun. 2014, 5, 5193.
| Crossref | GoogleScholarGoogle Scholar | 25308055PubMed |
[33] K.-S. Park, D. Im, A. Benayad, A. Dylla, K. J. Stevenson, J. B. Goodenough, Chem. Mater. 2012, 24, 2673.
| Crossref | GoogleScholarGoogle Scholar |
[34] F. Ding, W. Xu, G. L. Graff, J. Zhang, M. L. Sushko, X. Chen, Y. Shao, M. H. Engelhard, Z. Nie, J. Xiao, J. Am. Chem. Soc. 2013, 135, 4450.
| Crossref | GoogleScholarGoogle Scholar | 23448508PubMed |
[35] S. S. Zhang, J. Power Sources 2006, 162, 1379.
| Crossref | GoogleScholarGoogle Scholar |
[36] C. Yang, J. Chen, X. Ji, T. P. Pollard, X. Lü, C.-J. Sun, S. Hou, Q. Liu, C. Liu, T. Qing, Y. Wang, O. Borodin, Y. Ren, K. Xu, C. Wang, Nature 2019, 569, 245.
| Crossref | GoogleScholarGoogle Scholar | 31068723PubMed |
[37] K. Xu, Chem. Rev. 2014, 114, 11503.
| Crossref | GoogleScholarGoogle Scholar | 25351820PubMed |
[38] K. Xu, Chem. Rev. 2004, 104, 4303.
| Crossref | GoogleScholarGoogle Scholar | 15669157PubMed |
[39] P. Arora, Z. Zhang, Chem. Rev. 2004, 104, 4419.
| Crossref | GoogleScholarGoogle Scholar | 15669158PubMed |
[40] S. M. Kang, M.-H. Ryou, J. W. Choi, H. Lee, Chem. Mater. 2012, 24, 3481.
| Crossref | GoogleScholarGoogle Scholar |
[41] Y. Cheng, Y. Shao, J.-G. Zhang, V. L. Sprenkle, J. Liu, G. Li, Chem. Commun. 2014, 50, 9644.
| Crossref | GoogleScholarGoogle Scholar |
[42] Y. Ju, Y. Meng, Y. Wei, X. Bian, Q. Pang, Y. Gao, F. Du, B. Liu, G. Chen, Chem. – Eur. J. 2016, 22, 18073.
| Crossref | GoogleScholarGoogle Scholar | 27727501PubMed |
[43] R. Zhang, C. Ling, MRS Energy & Sustainability 2016, 3, E1.
[44] M. Mao, T. Gao, S. Hou, C. Wang, Chem. Soc. Rev. 2018, 47, 8804.
| Crossref | GoogleScholarGoogle Scholar | 30339171PubMed |
[45] J. Muldoon, C. B. Bucur, T. Gregory, Angew. Chem. 2017, 56, 12064.
[46] P. Canepa, G. Sai Gautam, D. C. Hannah, R. Malik, M. Liu, K. G. Gallagher, K. A. Persson, G. Ceder, Chem. Rev. 2017, 117, 4287.
| Crossref | GoogleScholarGoogle Scholar | 28269988PubMed |
[47] J. Song, E. Sahadeo, M. Noked, S. B. Lee, J. Phys. Chem. Lett. 2016, 7, 1736.
| Crossref | GoogleScholarGoogle Scholar | 27088555PubMed |
[48] M. M. Huie, D. C. Bock, E. S. Takeuchi, A. C. Marschilok, K. J. Takeuchi, Coord. Chem. Rev. 2015, 287, 15.
| Crossref | GoogleScholarGoogle Scholar |
[49] Y. Liang, H. D. Yoo, Y. Li, J. Shuai, H. A. Calderon, F. C. Robles Hernandez, L. C. Grabow, Y. Yao, Nano Lett. 2015, 15, 2194.
| Crossref | GoogleScholarGoogle Scholar | 25706101PubMed |
[50] H.-R. Yao, Y. You, Y.-X. Yin, L.-J. Wan, Y.-G. Guo, Phys. Chem. Chem. Phys. 2016, 18, 9326.
| Crossref | GoogleScholarGoogle Scholar | 26996438PubMed |
[51] M. Rashad, X. Li, H. Zhang, ACS Appl. Mater. Interfaces 2018, 10, 21313.
| Crossref | GoogleScholarGoogle Scholar | 29862802PubMed |
[52] H. Li, N. L. Okamoto, T. Hatakeyama, Y. Kumagai, F. Oba, T. Ichitsubo, Adv. Energy Mater. 2018, 8, 1801475.
| Crossref | GoogleScholarGoogle Scholar |
[53] H. Li, Q. Zhang, C. C. R. Yap, B. K. Tay, T. H. T. Edwin, A. Olivier, D. Baillargeat, Adv. Funct. Mater. 2012, 22, 1385.
| Crossref | GoogleScholarGoogle Scholar |
[54] N. McDevitt, J. Zabinski, M. Donley, J. Bultman, Appl. Spectrosc. 1994, 48, 733.
| Crossref | GoogleScholarGoogle Scholar |
[55] F. Yu, Z. Liu, R. Zhou, D. Tan, H. Wang, F. Wang, Mater. Horiz. 2018, 5, 529.
| Crossref | GoogleScholarGoogle Scholar |
[56] Y. Jiang, J. Liu, Energy Environ. Mater. 2019, 2, 30.
| Crossref | GoogleScholarGoogle Scholar |
[57] J. Wang, J. Polleux, J. Lim, B. Dunn, J. Phys. Chem. C 2007, 111, 14925.
| Crossref | GoogleScholarGoogle Scholar |
[58] H. Lindström, S. Södergren, A. Solbrand, H. Rensmo, J. Hjelm, A. Hagfeldt, S.-E. Lindquist, J. Phys. Chem. B 1997, 101, 7717.
| Crossref | GoogleScholarGoogle Scholar |
[59] P. Simon, Y. Gogotsi, B. Dunn, Science 2014, 343, 1210.
| Crossref | GoogleScholarGoogle Scholar | 24626920PubMed |
[60] J. Dahn, J. Burns, D. Stevens, Electrochem. Soc. Interface 2016, 25, 75.
[61] Y. Zhang, L. Tao, C. Xie, D. Wang, Y. Zou, R. Chen, Y. Wang, C. Jia, S. Wang, Adv. Mater. 2020, 32, 1905923.
| Crossref | GoogleScholarGoogle Scholar |
[62] S. R. Sivakkumar, A. S. Milev, A. G. Pandolfo, Electrochim. Acta 2011, 56, 9700.
| Crossref | GoogleScholarGoogle Scholar |
[63] Y. Dong, S. Zhang, X. Du, S. Hong, S. Zhao, Y. Chen, X. Chen, H. Song, Adv. Funct. Mater. 2019, 29, 1901127.
| Crossref | GoogleScholarGoogle Scholar |
[64] H. Hwang, H. Kim, J. Cho, Nano Lett. 2011, 11, 4826.
| Crossref | GoogleScholarGoogle Scholar | 21958327PubMed |
[65] E. J. Mittemeijer, U. Welzel, Z. Kristallogr. Cryst. Mater. 2008, 223, 552.
| Crossref | GoogleScholarGoogle Scholar |
[66] Y. Cheng, H. J. Chang, H. Dong, D. Choi, V. L. Sprenkle, J. Liu, Y. Yao, G. Li, J. Mater. Res. 2016, 31, 3125.
| Crossref | GoogleScholarGoogle Scholar |
[67] L. Chen, S. Zhao, Y. Liu, M. Horne, A. M. Bond, J. Zhang, J. Electrochem. Soc. 2016, 163, H3043.
| Crossref | GoogleScholarGoogle Scholar |
[68] M. Matsui, J. Power Sources 2011, 196, 7048.
| Crossref | GoogleScholarGoogle Scholar |
[69] C. Ling, D. Banerjee, M. Matsui, Electrochim. Acta 2012, 76, 270.
| Crossref | GoogleScholarGoogle Scholar |
[70] D. Larcher, J.-M. Tarascon, Nat. Chem. 2015, 7, 19.
| Crossref | GoogleScholarGoogle Scholar | 25515886PubMed |
[71] J. R. Rumble, D. R. Lide, T. J. Bruno, Abundance of Elements in the Earth’s Crust and in the Sea - CRC Handbook of Chemistry and Physics 2017 (Taylor & Francis Group: London)
[72] M. El Garah, S. Bertolazzi, S. Ippolito, M. Eredia, I. Janica, G. Melinte, O. Ersen, G. Marletta, A. Ciesielski, P. Samorì, FlatChem 2018, 9, 33.
| Crossref | GoogleScholarGoogle Scholar |
* Thomas Maschmeyer is the recipient of the 2021 David Craig Medal from the Australian Academy of Science.