

The 9-Fluorenylmethoxycarbonyl (Fmoc) Group in Chemical Peptide Synthesis – Its Past, Present, and Future
Wenyi Li A , Neil M. O’Brien-Simpson A , Mohammed Akhter Hossain B C D and John D. Wade B C D
A , Neil M. O’Brien-Simpson A , Mohammed Akhter Hossain B C D and John D. Wade B C D
A Bio21 Institute and Melbourne Dental School, University of Melbourne, Melbourne, Vic. 3010, Australia.
B Florey Institute of Neuroscience and Mental Health, University of Melbourne, Melbourne, Vic. 3010, Australia.
C School of Chemistry University of Melbourne, Melbourne, Vic. 3010, Australia.
D Corresponding authors. Email: akhter.hossain@florey.edu.au; john.wade@florey.edu.au

Wenyi Li received his doctoral degree in 2016 from The University of Melbourne under the supervision of Professors John D. Wade and Frances Separovic. His doctoral thesis on antimicrobial peptide development won the Graham Johnston Best Thesis Award from the RACI, Australia. From 2016 to 2018, he was based at the Leibniz Institute of Molecular Pharmacology, Germany, where he was partially supported as a Leibniz-DAAD postdoctoral fellow working on protein semisynthesis and chemical ligation. In 2018, he returned to The University of Melbourne to work on antibacterial polymers and peptides in the groups of Professors Neil M. O’Brien-Simpson and Greg Qiao. |

Neil M. O’Brien-Simpson graduated from Edinburgh Napier University in 1992 with a B.Sc. degree with honours in science and management studies. He moved to Australia and completed a Ph.D. in peptide-polymer vaccines in 1998 at The University of Melbourne. He is currently a professor and his research interests include antimicrobial peptides/materials, nanoparticles for peptide and vaccine delivery, and bacterial outer membrane vesicles-host interactions. He has several editorial duties and is the current president of the Peptide Users Group of the Royal Australian Chemical Institute. |

Mohammed Akhter Hossain received his Ph.D. degree in 2001 from the Tokyo Institute of Technology, where he worked on chemosensors based on cyclodextrin-peptide conjugates with Professor Akihiko Ueno. After that, he was a postdoctoral fellow at the Josef Fourier University in France. In 2005, he joined the Florey Institute of Neuroscience and Mental Health in Melbourne, Australia, where his research interests involve the design and synthesis of novel insulin and relaxin peptidomimetics, and β-amyloid peptides. He is currently a Florey Principal Research Fellow and head of the Insulin Peptides Laboratory. |

John D. Wade obtained his Ph.D. degree in 1979 at Monash University, Australia, on the structural basis of the diabetogenic action of growth hormones. He received a Nuffield Foundation Fellowship to Cambridge, UK, to undertake post-doctoral studies in the laboratory of Dr R. C. Sheppard at the MRC Laboratory of Molecular Biology on the development of the Fmoc-solid phase peptide synthesis methodology. In 1983, he returned to Melbourne at the invitation of the now Florey Institute of Neuroscience and Mental Health, University of Melbourne, where he heads the Laboratory of Peptide and Protein Chemistry. His interests are in solid phase peptide synthesis of large, complex, functionalized, and often multi-chain peptides. Professor Wade is an NHMRC of Australia Principal Research Fellow and a Fellow of both the Royal Australian Chemical Institute and the Royal Society of Chemistry. |
Australian Journal of Chemistry 73(4) 271-276 https://doi.org/10.1071/CH19427
Submitted: 3 September 2019 Accepted: 9 October 2019 Published: 22 November 2019
Journal Compilation © CSIRO 2020 Open Access CC BY-NC-ND
Abstract
The chemical formation of the peptide bond has long fascinated and challenged organic chemists. It requires not only the activation of the carboxyl group of an amino acid but also the protection of the Nα-amino group. The more than a century of continuous development of ever-improved protecting group chemistry has been married to dramatic advances in the chemical synthesis of peptides that, itself, was substantially enhanced by the development of solid-phase peptide synthesis by R. B. Merrifield in the 1960s. While the latter technology has continued to undergo further refinement and improvement in both its chemistry and automation, the development of the base-labile 9-fluorenylmethoxycarbonyl (Fmoc) group and its integration into current synthesis methods is considered a major landmark in the history of the chemical synthesis of peptides. The many beneficial attributes of the Fmoc group, which have yet to be surpassed by any other Nα-protecting group, allow very rapid and highly efficient synthesis of peptides, including ones of significant size and complexity, making it an even more valuable resource for research in the post-genomic world. This review charts the development and use of this Nα-protecting group and its adaptation to address the need for more green chemical peptide synthesis processes.
Introduction
A mandatory requirement in chemical peptide synthesis is the selection and use of a temporary protecting group for the Nα-group of an activated incoming amino acid. The C-terminus of the receiving amino acid to be acylated needs also be protected for liquid-phase peptide synthesis (LPPS) or, overwhelmingly and more commonly, anchored to a solid support as in solid-phase peptide synthesis (SPPS). It was not until 1932 that the first readily cleavable protecting group, the carbobenzoxy (Z) group of Bergman and Zervas, was developed that peptides of more than a few residues could be assembled.[1] However, the need for prolonged hydrogenolysis of the Z group for its removal significantly limited its wider utility. The development of the acid-labile urethane, tert-butyloxycarbonyl (Boc) group (Fig. 1), by Carpino in the late 1950s enabled a significant facilitation and advance in the conduct of chemical peptide synthesis.[2] Its adoption into the original scheme of SPPS by R. B. Merrifield enabled the first feasible general procedure for the chemical synthesis of peptides.[3] However, it was soon apparent that the need for differential acid lability for the Boc group and amino acid side chain protection and for the linkage to the resin presented significant problems of selectivity in cleavage, particularly with the final step requiring the use of toxic liquid hydrogen fluoride.[4] Additionally, the continuous acidic treatment for the amino deprotection throughout the synthesis can lead to premature side chain deprotection and unwanted side products. However in 1970, almost 50 years ago, Carpino described the preparation and chemical properties of a new urethane amino protecting group, the 9-fluorenylmethoxycarbonyl (Fmoc) group (Fig. 1) that possessed lability to bases.[5] Its unique and advantageous properties were not initially appreciated nor was the group adopted in synthetic strategies, probably owing to the original report describing its removal by refluxing with ammonia.[5] On subsequent recognition that the Fmoc group could also be cleaved by primary and secondary amines, the research groups of Sheppard and Meienhofer independently reported its use in SPPS in concert with mildly acid-labile tert-butyl side chain protecting groups and p-alkoxybenzyl ester resin linkage (Fig. 1).[6,7] The milder conditions endowed on this protocol decrease the risk of side reactions associated with the prolonged and strong acidic treatment in Boc-based synthesis and introduce orthogonality in the Nα-amino and side chain protecting groups with the former being base-labile and the latter acid-labile. This true orthogonality and ready adaptation to automation led to a ballooning increase in popularity and widespread use in both academia and industry that remains unabated today.

|
The recent passing of both Louis A. Carpino (University of Massachusetts Amherst, USA) and Robert (Bob) C. Sheppard (MRC Laboratory of Molecular Biology, Cambridge, UK),[8,9] unquestionably two of the most influential contributors to the chemical synthesis of peptides through their separate development and use of the Fmoc group, provides a timely opportunity to revisit the significant past and current studies with this still widely employed amine protecting moiety.
Nα-Fmoc Group
In his efforts to develop a urethane-based Nα-protecting group that could be efficiently and rapidly cleaved under mild, alkaline, non-hydrolytic conditions, Carpino initially examined the potential of the β-nitroethyloxycarbonyl group.[10] However, its lability towards base was deemed too high. After much further effort, the Fmoc group was then developed and shown to possess eminently suitable chemical properties.[11] It was initially introduced to an amino acid via Fmoc-chloroformate but, nowadays more commonly via Fmoc-succinimidyl carbonate because the former can cause Fmoc-dipeptide generation.[12] The remarkable lability of the Fmoc group to bases, particularly to secondary amines, results from activation of the ring proton β to the urethane oxygen by participation in a potential cyclopentadienide system. Cleavage likely follows an E1cb elimination mechanism (Scheme 1).[13,14]
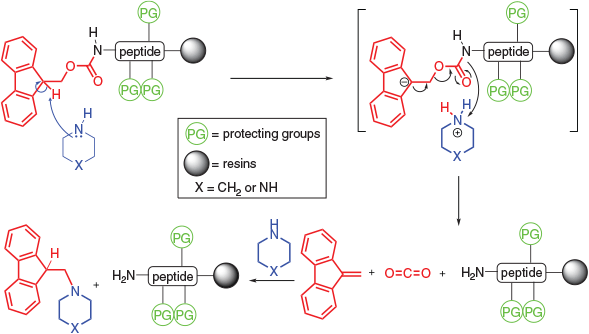
|
The attractive chemical features of the Nα-Fmoc group are many,[14,15] including the ease of preparation of Fmoc-amino acids in high yield, which are crystalline and are stable as the free acid when stored in the cold in a dry form. They also are generally freely soluble in solvents typically employed for chemical peptide synthesis such as N,N-dimethylformamide (DMF). Further, the Fmoc group is completely stable to treatment with trifluoroacetic acid (TFA) and hydrogen bromide/acetic acid. The fluorenyl group has a strong absorbance in the ultraviolet region (λmax 266 nm in DMF) that has proved very useful for spectrophotometrically monitoring coupling and deprotection reactions. However, the Fmoc group does possess disadvantageous properties. Its significant hydrophobicity means that amino acid derivatives are less soluble than the corresponding Boc-derivatives, which can place limitations on preparing highly concentrated solutions for use in automated synthesizers. Together with its steric bulk, it is also likely a contributory factor in the onset of peptide aggregation during SPPS of so-called ‘difficult peptides’.[16]
Nα-Fmoc Group Derivatives and Other Base-Labile Nα-Protecting Groups
Notwithstanding the positive attributes of the Fmoc group, research has been undertaken over the past decades into developing alternative base-labile Nα-protection groups. It is beyond the scope of this review to fully address this aspect but other interesting protecting groups to have been developed include 1-benzo[f]indenylmethoxycarbonyl (Bimoc)[15] and 2-chloro-1-indenylmethoxycarbonyl (Climoc),[15] which are both more base-labile than the Fmoc group. In contrast, the simple methylsulfonylethoxycarbonyl (Msc) group was considered to be insufficiently base labile.[17] The 9-(2-sulfo)fluorenylmethoxycarbonyl (Sulfmoc) group stems from chlorosulfonation of the Fmoc derivative.[17] It is stable to hydrogen fluoride and pyridine but readily removed by anhydrous morpholine or piperidine.[18] The 2-(4-nitrophenylsulfonyl)ethoxycarbonyl (Nsc)-protected amino acids are crystalline compounds, a physical property strongly preferred for both shelf storage and automated SPPS. Furthermore, the mechanism of removal of the Nsc group is believed to be similar to Fmoc deprotection, a base-catalyzed β-elimination reaction.[19] Unfortunately, it was subsequently shown to produce less pure peptides to those prepared using Fmoc protection.[20] The 2,2-[bis(4-nitrophenyl)]ethoxycarbonyl (Bnpeoc) group is also more base-labile than the Fmoc group.[21] However, none of the many reported base-labile protecting groups have supplanted Fmoc in terms of ease of preparation, cost-effectiveness, and subsequent use in chemical synthesis strategies, including SPPS.
Nα-Fmoc Deprotection – The Early Studies
Use of thin layer chromatographic analysis or amino acid analysis showed that the Fmoc group is, in general, rapidly removed by some primary (e.g. ethanolamine) and some secondary (notably piperidine and piperazine) amines (Fig. 2), but significantly more slowly with tertiary amines (e.g. triethylamine, N,N-diisoethylpropylethylamine).[22] The deprotection reaction is much faster in polar solvents such as DMF and N-methylpyrrolidone (NMP) than in apolar solvents such as dichloromethane.[23] Table 1 lists examples of cleavage of Fmoc-amino acids by various bases. Importantly, it was demonstrated that the free amino group of the resin-bound amino acid does not have any significant effect on the Fmoc group of the incoming amino acid.[22] A greater concern is the possible decomposition in DMF or NMP. However, Fmoc-Gly was found to be deprotected after 7 days to the extent of only 5 and 14 % respectively in these two solvents as monitored by HPLC.[24]

|

|
Importantly, during Fmoc deprotection, following abstraction of the acidic proton at the 9-position of the fluorene ring, β-elimination occurs to give a reactive dibenzofulvene (DBF) intermediate.[15] DBF can be sequestered by excess amine cleavage reagent to form stable adducts (Scheme 1).[15] The factors governing the rates of adduct formation remain unclear but both basicity and nucleophilicity as well as steric factors are critical. Piperazine, piperidine, and morpholine undergo rapid adduct formation whereas the sterically hindered 2,6-lutidine and the primary amine propylamine do not.[15]
In a reexamination of selected secondary amines for preferential use in Fmoc-based SPPS, solution experiments using Fmoc-Val showed a cleavage rate half-life t1/2 of ~1 min in 50 % morpholine, ~30 s in 5 % piperazine, and ~6 s in 20 % piperidine, all in DMF solution. Piperidine has been used in a range of concentrations from 5 to 50 % (v/v) in DMF but 20 % (v/v) piperidine in DMF has been adopted as the general, default reagent for Fmoc deprotection in SPPS.[24]
Alternative Nα-Fmoc Deprotection Methods
Other interesting alternative deprotection methods that have been reported include the use of tetrabutylammonium fluoride (TBAF) in DMF. It was shown to rapidly remove the Fmoc group in SPPS (Fig. 2).[25] However, as TBAF cannot sequester the liberated DBF, it is best suited to continuous-flow synthesis where the DBF is continually forced from the resin to prevent its back-attachment. Its more general use is further limited owing to significant aspartimide formation during the assembly of sensitive peptide sequences.[26] Wade and colleagues showed that the non-nucleophilic base 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) at a concentration of 2 % in DMF efficiently and rapidly removes the Fmoc group (Fig. 2).[27] Using resin-bound S-trityl cysteine as a model, base-induced racemization is substantially reduced with DBU compared with 20 % (v/v) piperidine in DMF. However, like TBAF, this tertiary amidine is unable to scavenge the DBF that is formed on Fmoc removal, thus preventing alkylation of the newly liberated amino group. For this reason, use of this strong base is recommended for continuous flow synthesis systems.[27] For conventional batchwise synthesis, DBU can be used in combination with, typically, 5 % piperidine to quench the liberated DBF.[28] It was further shown to be particularly beneficial for the synthesis of so-called ‘difficult peptide’ sequences, which suggested that insufficient Fmoc deprotection is a significant contributory factor to the past failure of such syntheses in addition to incomplete amino acid acylation.[29] Use of DBU was shown to be particularly beneficial for the assembly of thioamide peptides, with lower levels of epimerization being observed compared with the use of piperidine.[30] Caution is especially advised when using DBU for the synthesis of sensitive Asp-containing sequences, with reports of aspartimide formation being a major issue.[26,31,32] This problem has been the subject of much investigation given it is an inherent problem with base-mediated Fmoc deprotection. The problem can be circumvented by use of N-(2-hydroxy-4-methoxybenzyl) (Hmb) backbone-protected dipeptides.[33] However, this approach is not always practical owing to the high cost of the dipeptides or lack of commercial availability. More recently, it was shown that aspartimide formation can be ameliorated by addition of small amounts (5 % v/v) of organic acids such as formic acid to the deprotection solution.[34] A recent study showed that combination of the weaker base, piperazine (5 % v/v), with 2 % (v/v) DBU in DMF provided superior Fmoc deprotection. Addition of 1 % formic acid to this combination prevented aspartimide formation.[35]
Interestingly, sodium azide in DMF has been shown to cleanly remove the Fmoc group without causing aspartimide formation or racemization. However, the need for conditions involving elevated temperature and long reaction times together with the known explosive potential of the sodium azide salt obviously limits its usefulness.[36] More recently, Di Giola and colleagues showed that the Fmoc group could be removed by a cheap, less toxic and readily available base such as triethylamine and an imidazolium-based ionic liquid such as 1-butyl-3-methylimidazolium tetrafluoroborate ([Bmim][BF4]) (Fig. 2). However, while effective for solution methods, it is an involved process requiring ether extraction and subsequent protonation of the amine to remove the DBF. It has not yet been adapted to SPPS.[37]
The enduring popularity of piperidine for Nα-Fmoc deprotection has been tempered in recent years by the fact that it is a controlled substance that is regulated by the US Drug Enforcement Agency owing to it being the basis for the synthesis of narcotic drugs and psychotropic compounds. Renewed efforts have been expended to reexamine previously assessed deprotection reagents as well as to identify and adopt alternative ones in addition to those mentioned above. In a detailed study, Albericio and colleagues compared the performance of three different bases, 4-methylpiperidine, piperidine, and piperazine, using different test peptides that were assembled under microwave-mediated SPPS conditions. They showed that all three bases generally behaved similarly, with Fmoc-group removal being rapid. However, when cost-effectiveness was considered, piperazine was superior, more so given it is a solid reagent, which is advantageous for ease of transportation.[38] In another study, piperidine was compared with 2-, 3-, and 4-methylpiperidine together with 4-methylpiperazine and piperidine under standard SPPS conditions. 4-Methylpiperidine performed as well as piperidine and is now gaining widespread popularity.[39] More recently, Rodriguez and colleagues also confirmed the effectiveness of 4-methylpiperidine but recommended the use of lower concentrations (2.5 % v/v in DMF), which is as effective but has reduced toxicity.[40]
The Nα-Fmoc Group in ‘Green’ Chemical Peptide Synthesis Strategies
Much recent effort has been devoted by both academia and industry to significantly improve the environmental impact of chemical peptide synthesis. The latter includes substantial amounts of toxic process solvents such as DMF and NMP due to the large number of repetitive chemical steps required to produce a peptide together with intermediate washing. As an example, the industrial production of 500 kg of a nonapeptide requires an extraordinary 2500 t of NMP, creating additional issues such as the need for both waste tank farms and rail car transport.[41] Critically, owing to their reproductive toxicity, DMF and NMP have recently been labelled by Registration, Evaluation, Authorisation and Restriction of Chemicals – European Chemicals Agency (REACH-ECHA) as substances of very high concern (SVHC), leading to a concerted effort to replace these with environmentally acceptable alternatives.[42] Albericio and colleagues have undertaken substantial research into alternatives to these organic solvents and reported that greener solvents such as 2-methyltetrahydrofuran (2-MeTHF) and cyclopentyl methyl ether (CPME) were more attractive alternatives.[43] However, the Fmoc-removal steps with these solvents were not optimal following SPS of a ‘difficult peptide’ in which both deletion peptides and Nα-Fmoc-peptides were obtained after liberation from the solid support. A systematic evaluation of a range of green solvents showed that Fmoc deprotection worked best in γ-valerolactone when 20 % piperidine was employed. N-Formylmorpholine also showed good results.[44]
A recent round-table assessment of best practices in peptide synthesis, particularly in relation to pharmaceutical scale, identified the need for greater sustainability across all spheres of current methodology, including solid supports, protecting groups, and solvents including cleavage reagents.[44] The need for greener solvents was highlighted and it was noted that propylene carbonate has been successfully employed as an alternative to DMF in SPPS. N-Butylpyrrolidinone (NBP) was also shown to be a suitable alternative for certain applications despite its higher viscosity and greater cost. The consensus appears to be that use of green solvents will eventually be more widely adopted but that further optimization and increased compatibility of each of the individual components associated with chemical peptide synthesis, including Fmoc deprotection, will be required.[45]
Concluding Remarks
With the explosive increase in genomic information and the concurrent identification of numerous novel peptide sequences across all species, there has come a demand for faster and more efficient means of chemically synthesizing these biomolecules or their fragments that allow ligation of these to be assembled into large peptides and small proteins.[46] The need for the chemical preparation of peptidomimetics[47] and of related biomolecules such as peptide nucleic acids[48] has similarly increased given that these are often not amenable to recombinant DNA expression methods. The development of the Fmoc group and its successful use together with modern SPPS strategies have unquestionably contributed enormously to meeting this demand, particularly in now allowing the assembly of peptide-ester segments with the establishment of methods for native chemical or enzymatic ligation of peptide segments. The enduring popularity and overwhelming use of the Fmoc group in chemical peptide synthesis show no sign of abating in the foreseeable future and are an enduring legacy of the remarkable contributions of both Louis Carpino and Bob Sheppard. They will also likely ensure that it will remain the preeminent Nα-protecting group for decades to come.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
The National Health and Medical Research Council (NHMRC) of Australia and Australian Research Council (ARC) are thanked for financial support over many years for the peptide chemistry and chemical biology studies reported in the authors’ laboratories. WL is the recipient of the 2019 Weary Dunlop Foundation grant of the University of Melbourne. JDW is an NHMRC Principal Research Fellow (project numbers APP628404 and 1117483). NMOBS is the recipient of NHMRC funding (project numbers APP1142472, APP1158841), Cancer Council of Victoria funding (project number APP1163285), and Australian Dental Research Foundation Funding in antimicrobial materials. Research at The Florey Institute of Neuroscience and Mental Health is supported by the Victorian Government Operational Infrastructure Support Program. The authors salute Professor Paul Alewood’s many years of outstanding collaboration and friendship and wish him well in his retirement.
References
[1] M. Bergmann, L. Zervas, Ber. Dtsch. Chem. Ges. 1932, 65, 1192.| Crossref | GoogleScholarGoogle Scholar |
[2] L. A. Carpino, J. Am. Chem. Soc. 1957, 79, 98.
| Crossref | GoogleScholarGoogle Scholar |
[3] R. B. Merrifield, J. Am. Chem. Soc. 1963, 85, 2149.
| Crossref | GoogleScholarGoogle Scholar |
[4] J. Meienhofer, Biopolymers 1981, 20, 1761.
| Crossref | GoogleScholarGoogle Scholar |
[5] L. A. Carpino, G. Y. Han, J. Am. Chem. Soc. 1970, 92, 5748.
| Crossref | GoogleScholarGoogle Scholar |
[6] E. Atherton, H. Fox, D. Harkiss, C. J. Logan, R. C. Sheppard, B. J. Williams, J. Chem. Soc. Chem. Commun. 1978, 537.
| Crossref | GoogleScholarGoogle Scholar |
[7] C.-D. Chang, J. Meienhofer, Int. J. Pept. Protein Res. 1978, 11, 246.
| Crossref | GoogleScholarGoogle Scholar | 649259PubMed |
[8] M. Beyermann, J. Pept. Sci. 2019, 25, e3168.
| Crossref | GoogleScholarGoogle Scholar | 30989739PubMed |
[9] E. Atherton, J. D. Wade, J. Pept. Sci. 2019, 25, e3171.
| Crossref | GoogleScholarGoogle Scholar | 30989738PubMed |
[10] L. A. Carpino, G. Y. Han, J. Am. Chem. Soc. 1970, 92, 5748.
| Crossref | GoogleScholarGoogle Scholar |
[11] M. Tessier, F. Albericio, E. Pedroso, A. Grandas, R. Eritja, E. Giralt, C. Granier, J. Van Rietschoten, Int. J. Pept. Protein Res. 1983, 22, 125.
| Crossref | GoogleScholarGoogle Scholar | 6885246PubMed |
[12] G. F. Sigler, W. D. Fuller, N. C. Chaturvedi, M. S. Verlander, Biopolymers 1983, 22, 2157.
| Crossref | GoogleScholarGoogle Scholar |
[13] L. A. Carpino, G. Y. Han, J. Org. Chem. 1972, 37, 3404.
| Crossref | GoogleScholarGoogle Scholar |
[14] G. B. Fields, R. L. Noble, Int. J. Pept. Protein Res. 1990, 35, 161.
| Crossref | GoogleScholarGoogle Scholar | 2191922PubMed |
[15] L. A. Carpino, Acc. Chem. Res. 1987, 20, 401.
| Crossref | GoogleScholarGoogle Scholar |
[16] R. C. Sheppard, J. Pept. Sci. 2003, 9, 545.
| Crossref | GoogleScholarGoogle Scholar |
[17] G. I. Tesser, I. C. Balvert-Geers, Int. J. Pept. Protein Res. 1975, 7, 295.
| Crossref | GoogleScholarGoogle Scholar | 241727PubMed |
[18] R. B. Merrifield, A. E. Bach, J. Org. Chem. 1978, 43, 4808.
| Crossref | GoogleScholarGoogle Scholar |
[19] C. G. J. Verhart, G. I. Tesser, Recl. Trav. Chim. Pays Bas 1988, 107, 621.
| Crossref | GoogleScholarGoogle Scholar |
[20] P. M. Balse, G. Han, V. J. Hruby, H. J. Kim, J. Pept. Res. 2000, 56, 70.
| Crossref | GoogleScholarGoogle Scholar | 10961541PubMed |
[21] R. Ramage, J. Green, M. R. Florence, in Peptides – Chemistry and Biology (Ed. G. R. Marshall) 1988, pp. 157–158 (ESCOM: Leiden).
[22] C.-D. Chang, M. Waki, M. Ahmad, J. Meienhofer, E. O. Lundell, J. D. Huag, Int. J. Pept. Protein Res. 1980, 15, 59.
| 7358458PubMed |
[23] E. Atherton, C. J. Logan, R. C. Sheppard, J. Chem. Soc. Perkin Trans. 1 1981, I, 538.
| Crossref | GoogleScholarGoogle Scholar |
[24] E. Atherton, C. Bury, R. C. Sheppard, B. J. Williams, Tetrahedron Lett. 1979, 20, 3041.
| Crossref | GoogleScholarGoogle Scholar |
[25] M. Ueki, M. Amemiya, Tetrahedron Lett. 1987, 28, 6617.
| Crossref | GoogleScholarGoogle Scholar |
[26] J. D. Wade, M. N. Mathieu, M. Macris, G. W. Tregear, Lett. Pept. Sci. 2000, 7, 107.
[27] J. D. Wade, J. Bedford, R. C. Sheppard, G. W. Tregear, Pept. Res. 1991, 4, 194.
| 1823190PubMed |
[28] S. A. Kates, N. A. Solé, M. Beyermann, G. Barany, F. Albericio, Pept. Res. 1996, 9, 106.
| 8875589PubMed |
[29] A. K. Tickler, C. J. Barrow, J. D. Wade, J. Pept. Sci. 2001, 7, 488.
| Crossref | GoogleScholarGoogle Scholar | 11587187PubMed |
[30] D. M. Szantai-Kis, C. R. Walters, T. M. Barrett, E. M. Hoang, E. J. Petersson, Synlett 2017, 1789.
| Crossref | GoogleScholarGoogle Scholar |
[31] E. Kitas, R. Knorr, A. Trzeciak, W. Bannwarth, Helv. Chim. Acta 1991, 74, 1314.
| Crossref | GoogleScholarGoogle Scholar |
[32] J. L. Lauer, C. G. Fields, G. B. Fields, Lett. Pept. Sci. 1995, 1, 197.
| Crossref | GoogleScholarGoogle Scholar |
[33] J. Offer, M. Quibell, T. Johnson, J. Chem. Soc., Perkin Trans. 1 1996, 175.
| Crossref | GoogleScholarGoogle Scholar |
[34] T. Michels, R. Dölling, U. Haberkorn, W. Mier, Org. Lett. 2012, 14, 5218.
| Crossref | GoogleScholarGoogle Scholar | 23025410PubMed |
[35] K. Ralhan, V. G. KrishnaKumar, S. Gupta, RSC Adv. 2015, 5, 104417.
| Crossref | GoogleScholarGoogle Scholar |
[36] C.-C. Chen, B. Rajagopal, X. Y. Liu, K. L. Chen, Y.-C. Tyan, F. Lin, P.-C. Lin, Amino Acids 2014, 46, 367.
| Crossref | GoogleScholarGoogle Scholar | 24306456PubMed |
[37] M. L. Di Gioia, P. Costanzo, A. De Nino, L. Maiuolo, M. Nardi, F. Olivito, A. Procopio, RSC Adv. 2017, 7, 36482.
| Crossref | GoogleScholarGoogle Scholar |
[38] O. F. Luna, J. Gomez, C. Cárdenas, F. Albericio, S. H. Marshall, F. Guzmán, Molecules 2016, 21, 1542.
| Crossref | GoogleScholarGoogle Scholar |
[39] J. Hachmann, M. Lebl, J. Comb. Chem. 2006, 8, 149.
| Crossref | GoogleScholarGoogle Scholar | 16529506PubMed |
[40] V. Rodriguez, H. Pineda, N. Ardila, D. Insuasty, K. Cárdenas, J. Román, M. Urrea, D. Ramírez, R. Fierro, Z. Rivera, J. García, Int. J. Peptide Res. Ther. 2019.,
| Crossref | GoogleScholarGoogle Scholar |
[41] J. Lopez, S. Pletscher, A. Aemissegger, C. Bucher, F. Gallou, Org. Process Res. Dev. 2018, 22, 494.
| Crossref | GoogleScholarGoogle Scholar |
[42] L. Bergkamp, N. Herbatschek, Rev. Eur. Comp. Int. Environ. Law 2014, 23, 221.
| Crossref | GoogleScholarGoogle Scholar |
[43] Y. E. Jad, G. A. Acosta, T. Govender, H. G. Kruger, A. El-Faham, B. G. de la Torre, F. Albericio, ACS Sustain. Chem.& Eng. 2016, 4, 6809.
| Crossref | GoogleScholarGoogle Scholar |
[44] Y. E. Jad, T. Govender, H. G. Kruger, A. El-Faham, B. G. de la Torre, F. Albericio, Org. Process Res. Dev. 2017, 21, 365.
| Crossref | GoogleScholarGoogle Scholar |
[45] A. Isidro-Llobet, M. N. Kenworthy, S. Mukherjee, M. E. Kopach, K. Wegner, F. Gallou, A. G. Smith, F. Roschangar, J. Org. Chem. 2019, 84, 4615.
| Crossref | GoogleScholarGoogle Scholar | 30900880PubMed |
[46] V. Agouridas, O. El Mahdi, V. Diemer, M. Cargoët, J. M. Monbaliu, O. Melnyk, Chem. Rev. 2019, 119, 7328.
| Crossref | GoogleScholarGoogle Scholar | 31050890PubMed |
[47] X. Liu, R. P. B. Emes, K. A. Jollifee, Aust. J. Chem. 2017, 70, 201.
| Crossref | GoogleScholarGoogle Scholar |
[48] E. C. Browne, S. J. Langford, B. M. Abbott, Aust. J. Chem. 2012, 65, 539.
| Crossref | GoogleScholarGoogle Scholar |