

Strategies for Red-Shifting Type I Photoinitiators: Internal Electric Fields versus Lewis Acids versus Increasing Conjugation*
Nicholas S. Hill A and Michelle L. Coote A BA ARC Centre of Excellence for Electromaterials Science, Research School of Chemistry, Australian National University, Canberra, ACT 2601, Australia.
B Corresponding author. Email: michelle.coote@anu.edu.au
Australian Journal of Chemistry 72(8) 627-632 https://doi.org/10.1071/CH19262
Submitted: 9 June 2019 Accepted: 12 July 2019 Published: 31 July 2019
Abstract
Time-dependent density functional theory calculations were performed on derivatives of Irgacure 2959, a water-soluble, acetophenone-type photoinitiator, in order to assess the relative merits and drawbacks of three distinct ways of modifying its photochemistry: Lewis acid complexation, changing the amount of conjugation in the molecule, and application of an internal electric field through inclusion of a remote charged functional group. The effectiveness of each of the three methods was evaluated against the magnitude of the change in energy of the excited states. Internal electric fields were shown to provide the best method for targeting specific excited states in a controlled and rational manner. The other strategies also had significant effects but it was more difficult to independently target different transitions. Nonetheless, for the specific case of Irgacure 2959, we predict that its complexation with Mg2+ ions in a range of solvents will both red-shift the initiator’s absorbance while improving its efficiency and it is thus a promising candidate for testing as a visible light photoinitiator.
Introduction
Over the past few years, much attention has focussed on harnessing electric fields – either externally or via the internal fields generated by charged functional groups – to modify chemical reactivity.[1,2] During the course of such work, it has been noted that polarizable molecules, such as resonance-stabilized radicals, show particularly large sensitivities to electric field effects.[3,4] As many excited states are highly polarizable, this raises the prospect of selectively manipulating these states with electric fields.
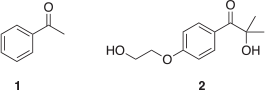
Recently, we used time-dependent density functional theory (TD-DFT) to investigate if electric fields offer a route for tuning the photochemical behaviour of acetophenone (1, Chart 1) and, by extension, other similar Type I photoinitiating compounds.[5] The study highlighted how the introduction of a non-conjugated charged functional group can either stabilize or destabilize nπ* and ππ* excited states. Moreover, the two types of state are affected in opposite directions depending on the sign of the charge, and there is a significant dependence of the magnitude of the electrostatic effect on ring position, thus allowing tuning of the effect. This is important if one wishes to, say, red-shift a photoinitiator without compromising its efficiency.
|
In the present work, we aim to assess the relative merits of using such electrostatic effects to modify photoinitiation behaviour when compared with more traditional approaches based on resonance. In particular, it is well known that extending the π-system of a photoactive compound will decrease the π–π* excitation energy and hence provide a means of red-shifting excitations into the π* system.[6] Moreover, aromatic systems with conjugated lone pair donors respond to pH changes, as these determine if the lone pair is available for resonance with the aromatic ring. One such example is the pH-dependent UV-vis spectrum of F420.[7] A related approach is to change the extent of resonance by coordinating Lewis acids with conjugated lone pair donor and/or π-acceptor groups. For example, we recently showed that Lewis acids could red-shift the absorption spectra and alter the photoinitiation behaviour of the popular Type I photoinitiators methyl-4′-(methylthio)-2-morpholinopropiophenone (MMMP) and 2,2-dimethoxy-2-phenylacetophenone (DMPA).[8]
To compare these different approaches, in the present work we attempt to improve the commercial Type I photoinitiator Irgacure 2959 (I2959, 2 in Chart 1). Recently, I2959 has found use in 3D bioprinting,[9–11] a process that involves the printing of biological material to form a specific tissue shape. Initially, the printable material is in the form of a ‘bio-ink’, a liquid-like mixture of cellular material, monomer, and photoinitiator. At the point of printing, the ink is irradiated with UV light in order to initiate a polymerization curing reaction that hardens the ink so that a three-dimensional tissue can be additively built. UV light does, however, have a detrimental effect on the cell viability within the 3D-printed tissue, as the high-energy photons can be absorbed by the cells with damaging consequences. Moreover, I2959 exhibits a reasonably low quantum yield of photolysis, at Φ = 0.29.[12] Although visible-light photoinitators exist,[13–15] to date, the initiators used are not as attractive as I2959 for bioprinting applications owing to their poor solubility in water.
Summing up, I2959 exhibits several properties that make it useful in 3D-printing applications; however, these properties could be improved. In particular, the necessity for damaging UV irradiation can be altered by lowering the energies of the reactive states; the efficiency of the electronic transitions could be improved, which would result in a higher value of Φ, and I2959’s solubility in water can be increased. Herein, we investigate if, and to what extent, this can be achieved through the use of remote charged functional groups, or through other strategies including Lewis acid complexation or more direct modifications to the chromophore such as extending the π system.
Theoretical Procedures
Although more accurate multireference methods are required to fully explore excited-state properties, here we are primarily interested in systematic effects on vertical excitation energies. Literature studies have shown that TD-DFT can reproduce these excitation energies for organic molecules in general[16] and acetophenone in particular.[17] Thus, as in our previous study,[5] the M06-2X density functional[18] was used for all ground- and excited-state calculations. Every species was extensively conformer-searched in order to locate the global minimum energy structure; structures were optimized with the 6-31+G(d,p) basis set, on which TD-DFT calculations were performed. Where relevant, solvent effects were considered with the SMD implicit continuum solvent model.[19] All calculations were performed with the Gaussian 16 Revision A electronic structure software package.[20]
Results and Discussion
Photochemistry of Unfunctionalized I2959
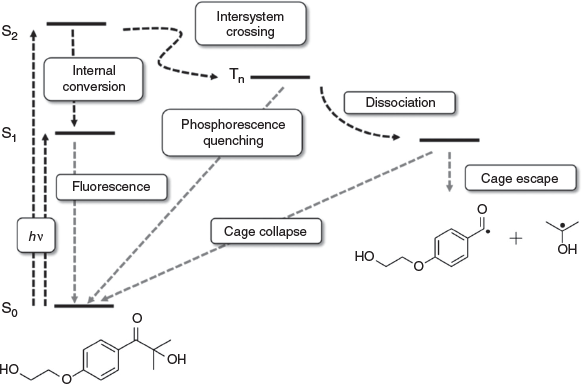
Like acetophenone, I2959 exhibits S1(nπ*) and S2(ππ*) excited states and several low-lying triplet excited states. Experimentally, I2959 exhibits λmax at 273 nm; at these wavelengths, the predominant excited state transition will be to the S2(ππ*) state, and the reactive triplet excited state has 3ππ* and 3nπ* character.[12] TD-DFT calculations suggest, however, that the S1(nπ*) state at ~305 nm has a nearby T2(nπ*) at ~337 nm and T1(ππ*) at 348 nm. According to El Sayed selection rules,[21] the most efficient intersystem crossing (ISC) will be take place from the S1(nπ*) to the T1(ππ*) surface. Fig. 1 is a simple diagram describing Type 1 photoinitiation, and the different internal processes that can take place on photoexcitation. A simple approach to improving the initiation efficiency of I2959 would therefore be to reduce the S1/T1 energy gap, resulting in a red-shift in the required wavelength of light for initiation (S1 rather than S2 is being targeted), and the different symmetries of the states would allow the efficient population of the T1 state.
|
Effect of Increasing π-Conjugation
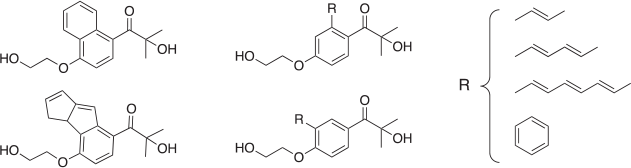
The effect of increasing conjugation of the photoactive moiety of I2959 was investigated with the structures shown in Fig. 2. Increasing the amount of π-conjugation present in a molecule is the most commonly adopted method to altering photochemistry. As this is a through-bond orbital effect, rather than a through-space electric field effect, this approach ought to be the most robust to solvent effects and so gas-phase energies were studied for simplicity. To explore these effects, three ways of increasing conjugation were evaluated; by fusing either an extra phenyl ring or an antiaromatic ring system to the original I2959 phenyl moiety, or by replacing the non-equivalent H-atoms with increasingly large unsaturated alkyl chains or a benzene ring. It is noted that these structural changes would likely decrease rather than increase water solubility but they are considered here to provide a baseline as to what effects are possible with changes to π-conjugation.
|
The changes in the gas-phase energies of the S1, S2, etc., states on the introduction of these functional groups relative to that of I2959 are shown in Fig. 3. Owing to the presence of the phenyl ring in I2959, introducing further conjugation in the form of an extra phenyl moiety, or a long alkyl chain, is found to have only a limited effect on the energies of the S1 states; in fact, at the meta positions, the effects are negligible (Fig. 3). In contrast, extending the π system greatly lowers the energy of the ππ* transitions. This is particularly pronounced with the fused anti-aromatic rings, which become aromatic in the excited state.

|
The net result of introducing more conjugation to I2959 is primarily to lower (red-shift) the energies for ππ* transitions, with minimal effect on the nπ* transitions, thus closing the gap with the ππ* states. Unfortunately, although this does red-shift the absorption of the initiator, it is unlikely to improve initiator efficiency. For instance, with the addition of butene at the meta position, for example, the S1(nπ*) to S2(ππ*) is reduced from 0.61 eV in I2959 to 0.15eV. This small energy gap may allow the efficient population of the nπ* state via the bright ππ* state; however, the corresponding triplet excited states have also been stabilized, and are now further away in energy from the singlet states. The red-shifting of the 1ππ* state is particularly profound on the addition of a fused antiaromatic unit, resulting in a 2.85 eV reduction in energy in the gas phase, and a 2.58 eV lowering in water. The nπ* states are stabilized to a much lower extent, and therefore whereas the absorption of I2959 in this case would be significant in the visible light region, TD-DFT calculations do not suggest this would lead to efficient photoinitiation.
Effect of Lewis Acid Complexation
In order to assess the potential application of Lewis acid (LA) complexation to alter the photochemistry of I2959, three metal chloride species were investigated: ZnCl2, MgCl2, and CaCl2. The counterions were chosen for computational efficiency and because they had been used in our previous experimental study of the effect of LA complexation on MMMP and DMPA.[8] Depending on the conditions, other counterions could be used to enhance solubility and, particularly if non-coordinating, will have little impact on the results. It should also be added some of the reaction conditions studied (e.g. gas phase) are not practical but are included to better understand trends in the data.
There are two possible binding modes between I2959 and LA species, and each LA was modelled in both LA2+ and LACl2 forms in order to determine the I2959-LA complex most prevalent in different solvent environments. The thermodynamic calculations are summarized in Tables S1–S4 in the Supplementary Material, and show that for each of the LA species, the most favourable complexation position is to the acyl oxygen site (Fig. 4).

|
With the complexation mode of the LA with I2959 established, TD-DFT calculations were performed in order to assess the effect of their introduction on I2959 excited states. Fig. 5 shows the change in vertical excitation energy of the three excited states of interest on complexation relative to uncomplexed I2959 (Tables S10–S12, Supplementary Material). The calculations show that the nπ* states are destabilized and the ππ* states are stabilized by the presence of LAs. This is consistent with our previous experimental and theoretical study of the effects of zinc chloride (ZnCl2) and aluminium chloride (AlCl3) on MMMP and DMPA.[8] In the present work, we find that MgCl2 complexation has the most significant effect on the stability of the excited states of I2959. The observed splitting in stability between the nπ* and ππ* states arises from the difference in their respective Lewis basicities;[8] nπ* transitions involve moving an electron from the ketone oxygen n-orbitals into the π*-system, resulting in a net less Lewis-basic ketone oxygen and conversely, ππ* transitions involve moving an electron from the π-system into π*-orbitals that have some ketone delocalization, resulting in an increase in Lewis basicity at the ketone oxygen.

|
Compared with the remote charged functional groups (below), the complexation of LAs does not allow specific states to be selectively altered in energy based on their position or direction of charge, with complexation limited to only destabilizing the nπ* and vice versa for the ππ* states. As a result, their effects can be a bit hit-and-miss. For instance, in our previous study,[8] LAs red-shifted the excitation energies but caused a deterioration in initiator efficiency by prolonging triplet lifetimes. Nonetheless, the calculations presented here suggest that introduction of MgCl2 offers a promising route to increasing photoinitiation efficiency of Irgacure. On closer inspection of the excited states of the MgCl2–I2959 complex in water, it is found the (de)stabilization of its excited states results in the ordering of the 1nπ* and 1ππ* states to swap, while the energy gap between the 1ππ* and 3nπ* states shrinks from 0.97 to 0.18 eV. If found experimentally to be accurate, this change in order of the excited states would allow the efficient population of the bright 1ππ* state, followed by efficient ISC to the 3nπ* to form radical species.
Effect of Remote Charged Functional Groups
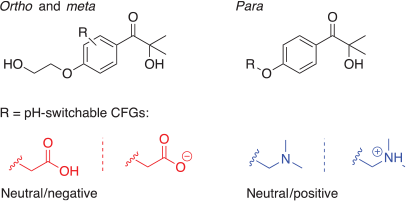
Previous work demonstrated that the attachment of non-conjugated charged functional groups (CFGs), i.e. acid and base groups that can be (de)protonated to form a charge, results in a ring-position-dependent electric field effect that can (de)stabilize nπ* and ππ* excited states, depending on the charge formed.[5] In I2959, the effect of attaching the carboxylic acid (CA) and tertiary amine (TA) groups at the ortho and meta positions of the phenyl ring, and replacing the alcohol group at the end of the alkyl chain as a pseudo-para position (Fig. 6) was investigated in solvents with increasingly large dielectric constants (Fig. 7). The para position was altered in a slightly different manner than the ortho and meta positions; in the case of the former, CFGs were introduced either by adding an acyl oxygen group to the –CH2OH moiety or swapping the alcohol group for a tertiary amine group. For the ortho and meta positions, –CH2COOH and –CH2NMe2 groups were used. The difference in pKa values of the CA and alkyl-OH groups of I2959 would be sufficient to allow selective deprotonation of the CA.
|

|
Fig. 7 includes the change in 1nπ*, 1ππ*, and 3ππ* vertical excitation energies on the formation of the charged species in the following solvent phases: gas, toluene, dichloromethane, acetonitrile, and water. Interestingly, there is a distinct ortho/meta/para (de)stabilization pattern different to that which has been reported previously.[5] Here, moving from the ortho to the meta positions results in a complete inversion in the effect of introducing either a positive or negative charged group. As well as this, the effect of applying a charged functional group is generally maximized at the para position. The reason for the observed anomalous results for the 1nπ* state on introduction of an NMe2H+ group is the presence of an internal hydrogen bond between it and the n-orbitals of the acyl moiety, which is countering the expected electrostatic destabilization of the nπ* excited state.
Given we wish to increase photoinitiation efficiency by lowering the S1(nπ*) and raising the T1(ππ*) states in energy, this is best achieved with a negative charge at the ortho position. Indeed, this results in a pH switch significant enough to make the 1nπ* and 3ππ* states degenerate in all solvents except water; even then, the resulting singlet–triplet energy gap ΔE is reduced from ~0.5 to ~0.1 eV (see Supplementary Material for relevant vertical excitation energies). The convergence of these chosen excited states suggests that introducing remote charge to I2959 or, in fact, any molecule with photochemistry arising from excited states of differing symmetries is a practical and powerful method for manipulating photochemistry.
Finally, although the introduction of a positively charged group by the protonation of an amine substituent appears to be an ineffective approach to internal electric field introduction owing to spurious hydrogen-bond formation, it is important to remember that the amine group could instead be replaced with a quaternary ammonium salt. Such structures would not exhibit the unwanted internal hydrogen bond, and could be employed when a low pH environment (required to form COO−) is inappropriate.
Conclusions
TD-DFT calculations were performed on functionalized Irgacure I2959, a water-soluble Type I photoinitiator, in order to identify practical strategies for red-shifting its excitation energies while maintaining or improving its photoinitiation efficiency. Fig. 8 summarizes the main results and recommendations, which are also outlined below:
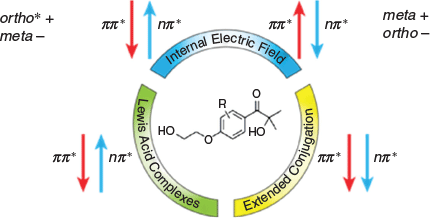
|
-
Extending the π-system will red-shift the UV-vis spectrum, but owing to the ππ* states being stabilized and the nπ* being relatively unaffected, it would lead to a decline in photoinitiation efficiency.
-
Including LAs to complex with the photoinitiator is a simple strategy that red-shifts ππ* transitions and blue-shifts nπ* transitions. The net effect on initiator efficiency varies considerably with the system but we predict that the Mg2+ complex of I2959 should exhibit the most significantly affected excited state energies, and therefore be a promising candidate as an efficient visible light photoinitiator.
-
Including remote (non-conjugated) charged functional groups to electrostatically alter the transition energies is a very powerful strategy that can give more precise control through choice of charge and location. For I2959, best results would be achieved by inclusion of a CH2C(O)O− group in the ortho position.
Work is under way to investigate these designs experimentally for the specific case of Irgacure I2959, but we also stress that the general findings can be applied to manipulation of other photochemical systems.
Supplementary Material
Further computational results, including total energies and optimized geometries of all species, are available on the Journal’s website.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
MLC gratefully acknowledges a Georgina Sweet ARC Laureate Fellowship (FL170100041), financial support from the ARC Centre of Excellence for Electromaterials Science (CE140100012), and generous allocations of supercomputing time on the National Facility of the Australian National Computational Infrastructure.
References
[1] S. Shaik, R. Ramanan, D. Danovich, D. Mandal, Chem. Soc. Rev. 2018, 47, 5125.| Crossref | GoogleScholarGoogle Scholar | 29979456PubMed |
[2] S. Ciampi, N. Darwish, H. M. Aitken, I. Díez-Perez, M. L. Coote, Chem. Soc. Rev. 2018, 47, 5146.
| Crossref | GoogleScholarGoogle Scholar | 29947390PubMed |
[3] G. Gryn’ova, M. L. Coote, J. Am. Chem. Soc. 2013, 135, 15392.
| Crossref | GoogleScholarGoogle Scholar | 24090128PubMed |
[4] G. Gryn’ova, M. L. Coote, Aust. J. Chem. 2017, 70, 367.
| Crossref | GoogleScholarGoogle Scholar |
[5] N. S. Hill, M. L. Coote, J. Am. Chem. Soc. 2018, 140, 17800.
| Crossref | GoogleScholarGoogle Scholar | 30468576PubMed |
[6] D. P. Hagberg, T. Marinado, K. M. Karlsson, K. Nonomura, P. Qin, G. Boschloo, T. Brinck, A. Hagfeldt, L. Sun, J. Org. Chem. 2007, 72, 9550.
| Crossref | GoogleScholarGoogle Scholar | 17979286PubMed |
[7] A. E. Mohamed, F. H. Ahmed, S. Arulmozhiraja, C. Y. Lin, M. C. Taylor, E. R. Krausz, C. J. Jackson, M. L. Coote, Mol. Biosyst. 2016, 12, 1110.
| Crossref | GoogleScholarGoogle Scholar | 26876228PubMed |
[8] B. B. Noble, A. C. Mater, L. M. Smith, M. L. Coote, Polym. Chem. 2016, 7, 6400.
| Crossref | GoogleScholarGoogle Scholar |
[9] W. Schuurman, P. A. Levett, M. W. Pot, P. R. van Weeren, W. J. A. Dhert, D. W. Hutmacher, F. P. W. Melchels, T. J. Klein, J. Malda, Macromol. Biosci. 2013, 13, 551.
| Crossref | GoogleScholarGoogle Scholar | 23420700PubMed |
[10] B. J. Klotz, D. Gawlitta, A. J. W. P. Rosenberg, J. Malda, F. P. W. Melchels, Trends Biotechnol. 2016, 34, 394.
| Crossref | GoogleScholarGoogle Scholar | 26867787PubMed |
[11] K. S. Lim, B. S. Schon, N. V. Mekhileri, G. C. J. Brown, C. M. Chia, S. Prabakar, G. J. Hooper, T. B. F. Woodfield, ACS Biomater. Sci. Eng. 2016, 2, 1752.
| Crossref | GoogleScholarGoogle Scholar |
[12] S. Jockusch, M. S. Landis, B. Freiermuth, N. J. Turro, Macromolecules 2001, 34, 1619.
| Crossref | GoogleScholarGoogle Scholar |
[13] J. Zhang, M. Frigoli, F. Dumur, P. Xiao, L. Ronchi, B. Graff, F. Morlet-Savary, J. P. Fouassier, D. Gigmes, J. Lalevée, Macromolecules 2014, 47, 2811.
| Crossref | GoogleScholarGoogle Scholar |
[14] S. Shi, C. Croutxé-Barghorn, X. Allonas, Prog. Polym. Sci. 2017, 65, 1.
| Crossref | GoogleScholarGoogle Scholar |
[15] J. Kabatc, M. Zasada, J. Paczkowski, J. Polym. Sci. A Polym. Chem. 2007, 45, 3626.
| Crossref | GoogleScholarGoogle Scholar |
[16] D. Jacquemin, I. Duchemin, X. Blase, J. Chem. Theory Comput. 2015, 11, 5340.
| Crossref | GoogleScholarGoogle Scholar | 26574326PubMed |
[17] M. Huix-Rotllant, N. Ferre, J. Chem. Phys. 2014, 140, 134305.
| Crossref | GoogleScholarGoogle Scholar | 24712791PubMed |
[18] Y. Zhao, D. G. Truhlar, Theor. Chem. Acc. 2008, 120, 215.
| Crossref | GoogleScholarGoogle Scholar |
[19] A. V. Marenich, C. J. Cramer, D. G. Truhlar, J. Phys. Chem. B 2009, 113, 6378.
| Crossref | GoogleScholarGoogle Scholar | 19366259PubMed |
[20] M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, D. J. Fox, Gaussian 16 Revision A.03 2016 (Gaussian Inc.: Wallingford, CT)
[21] A. D. McNaught, A. Wilkinson, IUPAC Compendium of Chemical Terminology, 2nd edn 1997 (Blackwell Scientific Publications: Oxford).
* Michelle L. Coote is the winner of the 2019 RACI Physical Division Medal.