

Contemplating 1,2,4-Thiadiazole-Inspired Cyclic Peptide Mimics: A Computational Investigation
Sida Xie A B , Paul V. Bernhardt A , Lawrence R. Gahan A C and Craig M. Williams A CA School of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane, Qld 4072, Australia.
B College of Chemical Engineering, Southwest Forestry University, Kunming 650224, China.
C Corresponding authors. Email: gahan@uq.edu.au; c.williams3@uq.edu.au
Australian Journal of Chemistry 72(11) 894-899 https://doi.org/10.1071/CH19248
Submitted: 1 June 2019 Accepted: 1 July 2019 Published: 30 July 2019
Abstract
Marine derived cyclic peptides have inspired chemists for decades as the cavitand architecture can be compared with macrocyclic ligands, and hence easily conceived as mediators of metal-ion transport. Lissoclinamide 5 and ascidiacyclamide are two such cyclic peptides that have received much attention both for their metal ion complexation properties and biological activity; the metal ion binding properties of mimics of these two systems have been reported. Reported herein is a computational study aimed at evaluating the stability, and potential for copper(ii) ion binding by lissoclinamide 5 mimics that substitute the naturally occurring 4-carboxy-1,3-thiazole units for novel valine- and phenylalanine-derived 1,2,4-thiadiazole units. Our results suggest that one lissoclinamide 5 mimic, 1,2,4-thiadiazole (TDA)-lissoclinamide 9, may be capable of forming a complex with one CuII ion, [Cu(9-H)(H2O)]+. A complex with two CuII ions, [Cu2(9-H)(μ-OH)]2+, was also considered. These results set the stage for synthetic and experimental metal binding studies.
Cyclic peptides arising from the marine environment[1–4] continue to attract considerable fascination prompted in part by marine-based pharmaceuticals currently being utilised in the clinic,[5–8] which in turn is driving drug-discovery efforts,[9–14] and substantial pursuits in understanding their chemical ecology.[15,16] Prominent amongst these have been the azole-based cyclic peptides,[17] with an archetypal cavitand appearance, that have led chemists to consider metal ion coordination and transport as a potential mode of biological activity and/or a role in electron-transfer processes.[18–20] Potassium[21] and other metal ions have been observed to form complexes with azole cyclic peptides, but the most amenable to coordination has been copper.[22–24] To explore the metal binding features of these systems further we recently introduced the concept of heteroatom-interchanged (HI) cyclic peptides.[25,26] Lissoclinamide 5 (1)[27–31] was investigated first as a working template, followed subsequently by ascidiacyclamide (2),[32–34] which led to the synthesis of four HI-isomers (e.g. 3) of lissoclinamide 5 and one HI-isomer 4 of ascidiacyclamide (Fig. 1). Metal ion binding studies using mass spectrometry (MS), electronparamagnetic resonance (EPR), and density functional theory (DFT) revealed that the HI-lissoclinamides (e.g. 3) form complexes with one CuII ion, whereas the HI-ascidiacyclamide isomer 4 was a weaker ligand for CuII,[25,26] suggesting small changes can have a dramatic effect on co-ordination, conformation,[35] and biological activity.[25,35]

|
To enable the heteroatom-interchange concept, naturally occurring 4-carboxy-1,3-thiazole units (i.e. 5) were replaced by non-natural 5-carboxy-1,3-thiazole derivatives 6, which required new methodologies to be evaluated in pursuit of obtaining suitable 5-carboxy-1,3-thiazole building blocks.[25,26] Numerous synthetic methods to access 5-carboxy-1,3-thiazoles were evaluated, and in one case valine- and phenylalanine-derived 1,2,4-thiadiazoles (i.e. 7 and 8) were produced unexpectedly. Given our interest and on-going efforts in the azole cyclic peptide arena we considered whether the 1,2,4-thiadiazole system would underpin next generation cyclic peptide mimic design. However, in light of the HI-isomers creating a considerable synthetic challenge, an in silico investigation to assist in determining a likely synthetic candidate was justified.
Reported herein is the synthesis and characterisation of novel 1,2,4-thiadiazoles 7 and 8, along with an in silico assessment of lissoclinamide 5 mimics 9–12 (Fig. 1) in terms of ground state stability and the propensity to coordinate CuII ions.
3,5-Disubstitiuted-1,2,4-thiadiazoles
As mentioned above, in the course of developing the synthesis of the HI-cyclic peptides (e.g. 3 and 4),[25,26] 5-carboxy-1,3-thiazole building blocks were required (e.g. 6), which at the time demanded de novo synthetic routes to be developed. To satisfy the required functionality, and stereochemistry, any newly developed method needed to accommodate amino acid derived substrates for practicality reasons. The method reported by Zhao[36] was initially chosen for potential modification because it described that exposure of mono-substituted thioureas (e.g. 13) to the in situ generated bromohydrin 14 (bromination of enol ether 15 with N-bromosuccinimide, NBS) afforded aminothiazoles (e.g. 16) in good yields.[37] Therefore, this method could likely be modified as valine- and phenylalanine-derived thioamides 17 and 18 were readily accessible,[38,39] and this proved to be successful giving thiazoles 20 and 19, respectively (Scheme 1). However, a by-product was observed in both cases, but in varying amounts. When using the phenylalanine thioamide 18, thiazole 19 was obtained in 46 % yield, in addition to 9 % of 1,2,4-thiadiazole 8, whereas the valine thioamide 17 afforded thiazole 20 and thiadiazole 7 in a higher yield of 34 % (Scheme 1). The latter 1,2,4-thiadiazole structure was confirmed by X-ray crystallographic analysis (Fig. 2).

|

|
A literature search revealed that 3,5-disubstitiuted-1,2,4-thiadiazoles have been previously synthesised from thioamides, but importantly not those derived from amino acids. Takikawa et al. reported that treatment of a range of thioamides directly with NBS afforded 1,2,4-thiadiazoles in yields ranging from 72–93 %,[40] which provided justification as to why 1,2,4-thiadiazoles were appearing as by-products in the modified Zhao synthesis of thiazoles.
Thiadiazole Cyclic Peptide Stability and CuII Binding
Given that methodology now existed to access 1,2,4-thiadiazoles derived from amino acids, and our interest in the lissoclinamide 5 system, mimics 9–12 were conceived (Fig. 1). In these cases, however, the 1,2,4-thiadiazoles do not have a N and C terminus to enable all peptide bonds seen in in lissoclinamide 5 to be formed. Therefore, an aspect of the design (i.e. 9–12) features two secondary amines that link both the two 1,2,4-thiadiazole units and the N-terminus of the polypeptide (i.e. left-hand fragment), of which the latter polypeptide has been conserved throughout our entire HI studies (i.e. right hand fragment).[25,26] These structures, which break traditional cyclic peptide mimicking seen previously (i.e. all peptide bonds conserved), are of interest considering the wealth of literature available with respect to the conformations and biological activity of similar cyclic peptides.[22–27] In addition, the metal complexes of these ligands, particularly the CuII complexes, have been studied as phosphoesterase and carbonic anhydrase mimics.[22,24,41–43] We therefore chose to explore, computationally, peptides 9–12 in terms of their structure and the potential for CuII binding.
Peptides 9–12 each exhibit eight chiral centres. To gain insight into structural effects of these chiral centres computational modelling was undertaken using a force field-based approach involving the Macrocycle Conformational Sampling algorithm of MacroModel 10.6 in conjunction with the MMFFs force field.[44,45] The minimum energy conformers for each peptide were identified and are shown in the Supplementary Material, although it should be noted that in each case some hundreds of possible conformations were identified. The most stable conformer of each peptide displayed the peptide NH groups and the nitrogen atoms of the oxazoline and 1,2,4-thiadiazoles rings pointing towards the interior of the macrocycle. This orientation of the 1,2,4-thiadiazole rings was in contrast to that seen for the ‘inverted’ analogues 3 and 4 where the sulfur atoms of the thiazoles were pointed towards the centre of the cyclic peptide.[25,26] A study of the relative energies of the most stable conformers of 9–12 was performed using the B3LYP/6-31G(d) DFT method.[46,47] The spread of energies calculated for the lowest energy conformers of 9–12 was small, of the order of 6.3 kJ mol−1, with 9 representing the lowest energy structure. The conformer studies suggested that these cyclic peptides may have the capacity to bind metal ions particularly, and in contrast to 3 and 4, as the potential donor N atom atoms are pointed towards the centre of the cavity.
Two models for putative CuII complexes were investigated, [Cu(9-H)(H2O)]+ and [Cu2(μ-OH)(9-H)]2+; here 9-H represents the cyclic peptide deprotonated at the amide. Geometries of the two complexes were optimised with M06/6-31G(d)-LANL2DZ in an implicit (SMD) methanol solvent model.[46,48,49] For the [Cu2(μ-OH)(9-H)]2+ complex, the two CuII ions exhibited different coordination geometries. In one site a five-coordinate, square based pyramid CuII ion was coordinated through the nitrogen donors of a 1,2,4-thiadiazole (2.21 Å), the (R)-5-methyl-4,5-dihydrooxazole (2.16 Å), and the deprotonated amide (1.92 Å), as well as the oxygen of the carbonyl group adjacent to the pyrrolidine (2.42 Å) and the μ-OH (1.93 Å). The coordination sphere of the second, four-coordinate, square planar CuII site, was composed of the nitrogen donors of two secondary amines (2.24 and 2.54 Å), the nitrogen of a second 1,2,4-thiadiazole (1.92 Å), and the coordination sphere completed by the μ-OH (1.87 Å). The Cu–Cu distance was 3.05 Å with the Cu–OH–Cu angle 106.7°. For the [Cu(9-H)(H2O)]+ complex the coordination sphere was modelled as comprising the deprotonated amide (1.92 Å), the nitrogen donors of 1,2,4-thiadiazole (2.17 Å) and (R)-5-methyl-4,5-dihydrooxazole (2.11 Å), and a long interaction with the carbonyl oxygen adjacent to the pyrrolidine (2.57 Å) and the H2O (2.01 Å). The proposed structures are shown in Fig. 3 (and Fig. S2, Supplementary Material).

|
Interesting comparisons can be drawn between the above systems and the CuII complexes of the pseudo-octapeptide H4pat1, an analogue of the patellamides (Fig. 4).[50] H4pat1 contains four chiral centres and both [Cu(H3pat1)(H2O)2]+ and [Cu2(H2pat1)(μ-OH)(H2O)2]+) complexes have been characterised through MS, DFT calculations and, for the latter, X-ray crystallography.[50] For [Cu2(H2pat1)(μ-OH)(H2O)2]+ the Cu Cu separation was 3.76 Å with a Cu–OH–Cu angle of 136.8°; the other metal–donor distances were comparable in [Cu2(μ-OH)(9-H)]2+ and [Cu2(H2pat1)(μ-OH)(H2O)2]+. The structure of [Cu(H3pat1)(H2O)2]+, calculated with DFT, proposed a five-coordinate CuII site, the CuII coordinated through the nitrogen atoms of two 1,5-dimethylimidazoles and a deprotonated amide, with two water molecules completing the coordination sphere. The DFT analysis for [Cu2(H2pat1)(μ-OH)(H2O)2]+ and [Cu(H3pat1)(H2O)2]+ suggested that the former complex was 25 kJ mol−1 more stable than the latter.[50]
Cu separation was 3.76 Å with a Cu–OH–Cu angle of 136.8°; the other metal–donor distances were comparable in [Cu2(μ-OH)(9-H)]2+ and [Cu2(H2pat1)(μ-OH)(H2O)2]+. The structure of [Cu(H3pat1)(H2O)2]+, calculated with DFT, proposed a five-coordinate CuII site, the CuII coordinated through the nitrogen atoms of two 1,5-dimethylimidazoles and a deprotonated amide, with two water molecules completing the coordination sphere. The DFT analysis for [Cu2(H2pat1)(μ-OH)(H2O)2]+ and [Cu(H3pat1)(H2O)2]+ suggested that the former complex was 25 kJ mol−1 more stable than the latter.[50]
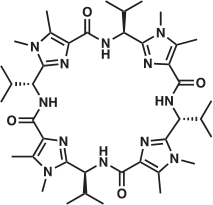
The question then arises – do the computations reported herein provide an indication as to whether it is possible to prepare the [Cu(9-H)(H2O)]+ and [Cu2(μ-OH)(9-H)]2+ complexes? In order to provide a clue as to this question, the energy required to reorganise the macrocyclic peptide from its ground-state geometry into its coordinated geometry was calculated for the two complexes. The calculations suggested that 12.5 kJ mol−1 was required for 9-H to reorganise to accommodate the formation of [Cu(9-H)(H2O)]+ whereas for the formation of the [Cu2(μ-OH)(9-H)]2+ complex the energy requirement for the reorganisation of the peptide was larger, of the order of 71 kJ mol−1. In addition, the shorter CuCu distance and more acute Cu–OH–Cu angle calculated for [Cu2(μ-OH)(9-H)]2+ (3.05 Å, 106.7°, respectively) compared with [Cu2(H2pat1)(μ-OH)(H2O)2]+ (3.76 Å, 136.8°, respectively)[50] suggest that the mono-CuII complex of cyclic peptide 9 may form preferentially.
In conclusion, this study has conceived novel cyclic peptides designed on the naturally occurring lissoclinamide 5 template, taking inspiration from preliminary synthetic studies that produced 1,2,4-thiadiazoles. Subsequently, these macrocyclic ring systems were explored in terms of determining both their ground state energies and the potential of the lowest energy conformer to bind copper(ii). 1,2,4-Thiadiazole (TDA)-lissoclinamide 9 was found to have the highest likelihood of forming a mono-copper complex i.e. [Cu(9-H)(H2O)]+, but not with two CuII ions. These combined results provide encouragement to initiate a synthetic program in this area and determine whether metal binding is observable.
Experimental
Synthesis
General experimental procedures along with the synthetic procedures for 17–20 have been previously reported.[25,26]
((1S,1'S)-(1,2,4-Thiadiazole-3,5-diyl)bis(2-methylpropane-1,1-diyl))dicarbamate (7) and Methyl 2-(1-[(tert-butoxycarbonyl)amino]-2-methylpropyl)thiazole-5-carboxylate (20)
To a mixture of methyl trans-3-methoxyacrylate (15) (3.8 mL, 35.3 mmol) in water (18 mL) and dioxane (18 mL) at −10°C was added recrystallised NBS (6.9 g, 38.8 mmol). The reaction mixture was stirred at room temperature for 1 h. After cooling the reaction to 0°C, (S)-N-tert-(butoxycarbonyl)thiovalinamide (17) 8.2 g, 35.3 mmol) was added and the reaction allowed to warm to room temperature overnight. Concentrated NH4OH (7 mL) was then added dropwise and the mixture stirred for 10 min. The resulting slurry was diluted with Et2O (180 mL) and washed with water (3 × 360 mL). The Et2O layer was dried with Na2SO4 and concentrated under vacuum. Flash chromatography (silica gel, petroleum ether/ethyl acetate 4 : 1, Rf 0.45) provided thiazole 20 as a white foam (4.6 g, 8 %), and thiadiazole 7 (1.3 g, 34 %). δH (400 MHz, CDCl3) 5.41 (d, J 9.0, 1H), 5.17 (d, J 8.3, 1H), 5.02 (m, 2H), 2.09 (m, 1H), 1.97 (m, 1H), 1.43 (s, 18H), 0.97 (d, J 6.8, 3H), 0.87 (d, J 6.9, 6H), 0.83 (d, J 6.8, 3H). δC (100 MHz, CDCl3) 193.4, 176.2, 155.5, 155.3, 80.5, 79.5, 40.1, 39.5, 28.3, 28.3, 24.9, 24.6, 15.4, 15.4, 11.6. m/z (ESI) 451.2364; calcd for C20H36N4O4SNa+ 451.2355.
((1S,1'S)-(1,2,4-Thiadiazole-3,5-diyl)bis(2-benzyl-1,1-diyl))dicarbamate (8) and Methyl 2-{1-[(tert-butoxycarbonyl)amino]-2-phenylethyl}thiazole-5-carboxylate (19)
To a mixture of methyl trans-3-methoxyacrylate (15) (5.29 mL, 42.4 mmol) in water (21 mL) and dioxane (21 mL) at −10°C was added recrystallised NBS (8.33 g, 46.6 mmol). The reaction mixture was stirred at room temperature for 1 h. After cooling the reaction to 0°C, (S)-N-tert-(butoxycarbonyl)thiophenylalaninamide (18) (11.90 g, 42.42 mmol) was added and the reaction allowed to warm to room temperature overnight. Concentrated NH4OH (8 mL) was then added dropwise and the mixture stirred for 10 min. The resulting slurry was diluted with Et2O (200 mL) and washed with water (3 × 400 mL). The Et2O layer was dried with Na2SO4 and concentrated under vacuum. Flash chromatography (silica gel, petroleum ether/ethyl acetate 4 : 1, Rf 0.61) provided methyl 2-{1-[(tert-butoxycarbonyl)amino]-2-phenylethyl}thiazole-5-carboxylate (19) as a white foam (5.7 g, 37 %), and thiadiazole 8 (1.4 g, 9 %). δH (400 MHz, CDCl3) 7.29 −7.25 (m, 2H), 7.22 −7.13 (m, 5H), 7.11 −7.03 (m, 2H), 6.94 (m, 1H), 5.37 (m, 1H), 5.23 (s, 1H), 3.27 (m, 4H), 1.41 (s, 9H), 1.39 (s, 9H). δC (100 MHz, CDCl3) 193.8, 175.7, 155.0, 154.9, 136.5, 135.5, 129.5, 129.2, 128.7, 128.2, 127.2, 126.6, 80.6, 79.6, 53.7, 53.6, 52.8, 52.7, 41.1, 41.0, 29.6, 28.2. m/z (ESI) 547.2340; calcd for C28H36N4O4SNa+ 547.2457.
Calculations
The conformations of cyclic peptides were investigated using the Macrocycle Conformational Sampling algorithm of MacroModel 10.6.[44,45] The conformer sampling employed the MMFFs (MMFF94s) force field and GB/SA (water) solvent model. The process utilised 5000 cycles of large-scale low mode searches performed on a set of seed structures obtained from 10000 cycles of MD simulated annealing, with eigenvectors calculated for each new global minimum, and included the ‘enhanced’ option for torsional sampling of amide bonds (which samples the amide C–N and C–O single bonds). The lowest-energy conformers of 9–12 obtained from the MMFFs–GB/SA forcefield-based conformer sampling were reoptimised with DFT. The DFT calculations were performed with Gaussian 16.[46] For the study of the relative energies of 9–12 the B3LYP functional was used in conjunction with the 6-31G(d) basis set, the studies undertaken in the gas phase.[46–49] For the CuII complexes, the M06 functional in conjunction with a mixed basis set consisting of LANL2DZ on Cu and 6-31G(d) on other atoms was used.[49] Methanol solvent was modelled with the SMD implicit model.[46–49]
Crystal Data for 7
C20H36N4O4S: M 428.59, T 293(2) K, Monoclinic, space group P21, a 11.1529(3), b 10.1893(2), c 11.1550(3) Å, β 93.620(2)°, V 1265.13(5) Å3, Z 2, F(000) 464, Dc 1.125 g cm−3, μ (Cu-Kα) 1.375 mm−1, 3989 unique data, R1 0.0331 (for 3738 observed reflections I > 2σ(I)), wR2 0.0945 (all data).
Single crystal intensity data were collected on an Oxford Diffraction Gemini S Ultra CCD diffractometer using graphite monochromatic Cu-Kα radiation (λ 1.5418 Å) for 1,2,4-thiadiazole 7 operating in the ω-scan mode. Data reduction and empirical absorption corrections were performed with the CrysAlis program (Oxford Diffraction, version 171.35.11), while all other computations were performed within the WinGX suite of programs.[51] The structure was solved by direct methods with SHELXS and refined by full matrix least-squares analysis with SHELXL97.[52] All non-H atoms were refined with anisotropic thermal parameters, and H-atoms were constrained at estimated positions using a riding model. The absolute configuration was established by anomalous dispersion effects. The atomic nomenclature is defined in Fig. 2 and drawn with ORTEP3.[53] Crystallographic data in CIF format is available from the Cambridge Crystallographic Data Base with CCDC deposition number 1903408.
Supplementary Material
Full crystal data, atomic coordinates, bond angles, bond lengths, and copies of 1H and 13C NMR data are available on the Journal’s website.
Conflicts of Interest
The authors declare no conflict of interest.
Acknowledgements
The authors thank the University of Queensland (UQ) for financial support and the Australian Research Council is gratefully acknowledged for a Future Fellowship award to C. M. W. (FT110100851). Computer resources were provided by the National Facility of the Australian National Computational Infrastructure and by the University of Queensland Research Computing Centre. They also thank Professor P. Comba and Dr B. Martin, Universitat Heidelberg, Anorganisch-Chemisches Institut, for access to data regarding the H4pat1 complexes and Associate Professor E. Krenske, School of Chemistry and Molecular Biosciences, The University of Queensland, for assistance.
References
[1] J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. G. Munro, M. R. Prinsep, Nat. Prod. Rep. 2015, 32, 116.| Crossref | GoogleScholarGoogle Scholar | 25620233PubMed |
[2] D. J. Faulkner, Nat. Prod. Rep. 2002, 19, 1.
| 11902436PubMed |
[3] D. Skropeta, L. Wei, Nat. Prod. Rep. 2014, 31, 999.
| Crossref | GoogleScholarGoogle Scholar | 24871201PubMed |
[4] J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. G. Munro, M. R. Prinsep, Nat. Prod. Rep. 2014, 31, 160.
| Crossref | GoogleScholarGoogle Scholar | 24389707PubMed |
[5] M. S. Butler, A. A. B. Robertson, M. A. Cooper, Nat. Prod. Rep. 2014, 31, 1612.
| Crossref | GoogleScholarGoogle Scholar | 25204227PubMed |
[6] T. L. Simmons, E. Andrianasolo, K. McPhail, P. Flatt, W. H. Gerwick, Mol. Cancer Ther. 2005, 4, 333.
| 15713904PubMed |
[7] S. A. Dyshlovoy, F. Honecker, Mar. Drugs 2015, 13, 5657.
| Crossref | GoogleScholarGoogle Scholar | 26540740PubMed |
[8] M. Jaspars, D. De Pascale, J. H. Andersen, F. Reyes, A. Crawford, A. Ianora, J. Mar. Biol. Assoc. U. K. 2016, 96, 151.
| Crossref | GoogleScholarGoogle Scholar |
[9] V. A. Stonik, S. N. Fedorov, Mar. Drugs 2014, 12, 636.
| Crossref | GoogleScholarGoogle Scholar | 24473167PubMed |
[10] Y. Zhou, Curr. Org. Chem. 2014, 18, 918.
| Crossref | GoogleScholarGoogle Scholar |
[11] P. Kiuru, M. V. D’Auria, C. D. Muller, P. Tammela, H. Vuorela, J. Yli-Kauhaluoma, Planta Med. 2014, 80, 1234.
| 25203732PubMed |
[12] L. A. Salvador-Reyes, H. Luesch, Nat. Prod. Rep. 2015, 32, 478.
| Crossref | GoogleScholarGoogle Scholar | 25571978PubMed |
[13] L. A. Salvador-Reyes, N. Engene, V. J. Paul, H. Luesch, J. Nat. Prod. 2015, 78, 486.
| Crossref | GoogleScholarGoogle Scholar | 25635943PubMed |
[14] L. T. Tan, Drug Discov. Today 2013, 18, 863.
| Crossref | GoogleScholarGoogle Scholar | 23711931PubMed |
[15] P. N. Leao, N. Engene, A. Antunes, W. H. Gerwick, V. Vasconcelos, Nat. Prod. Rep. 2012, 29, 372.
| Crossref | GoogleScholarGoogle Scholar | 22237837PubMed |
[16] J. Piel, Nat. Prod. Rep. 2009, 26, 338.
| Crossref | GoogleScholarGoogle Scholar | 19240945PubMed |
[17] Z. Jin, Nat. Prod. Rep. 2006, 23, 464.
| Crossref | GoogleScholarGoogle Scholar | 16741589PubMed |
[18] A. Bertram, G. Pattenden, Nat. Prod. Rep. 2007, 24, 18.
| Crossref | GoogleScholarGoogle Scholar | 17268606PubMed |
[19] J. P. Michael, G. Pattenden, Angew. Chem. Int. Ed. Engl. 1993, 32, 1.[Angew. Chem. 1993, 105, 1].
| Crossref | GoogleScholarGoogle Scholar |
[20] Y. Shi, W. Jiang, B. N. Auckloo, B. Wu, Curr. Org. Chem. 2015, 19, 1935.
| Crossref | GoogleScholarGoogle Scholar |
[21] A. L. van den Brenk, D. P. Fairlie, L. R. Gahan, G. R. Hanson, T. W. Hambley, Inorg. Chem. 1996, 35, 1095.
| Crossref | GoogleScholarGoogle Scholar | 11666295PubMed |
[22] P. Comba, N. Dovalil, L. R. Gahan, G. R. Hanson, M. Westphal, Dalton Trans. 2014, 43, 1935.and references therein.
| Crossref | GoogleScholarGoogle Scholar | 24202205PubMed |
[23] P. Comba, A. Eisenschmidt, L. R. Gahan, D.-P. Herten, G. Nette, G. Schenk, M. Seefeld, Chem. – Eur. J. 2017, 23, 12264.
| Crossref | GoogleScholarGoogle Scholar | 28339125PubMed |
[24] P. Comba, A. Eisenschmidt, L. R. Gahan, G. R. Hanson, N. Mehrkens, M. Westphal, Dalton Trans. 2016, 45, 18931.
| Crossref | GoogleScholarGoogle Scholar | 27841434PubMed |
[25] S. Xie, A. I. Savchenko, M. Kerscher, R. L. Grange, E. H. Krenske, J. R. Harmer, M. J. Bauer, N. Broit, D. J. Watters, G. M. Boyle, P. V. Bernhardt, P. G. Parsons, P. Comba, L. R. Gahan, C. M. Williams, Eur. J. Org. Chem. 2018, 1465.
| Crossref | GoogleScholarGoogle Scholar |
[26] S. Xie, A. I. Savchenko, E. H. Krenske, R. L. Grange, L. R. Gahan, C. M. Williams, Eur. J. Org. Chem. 2018, 3265.
| Crossref | GoogleScholarGoogle Scholar |
[27] B. M. Degnan, C. J. Hawkins, M. F. Lavin, E. J. McCaffrey, D. L. Parry, A. L. Van den Brenk, D. J. Watters, J. Med. Chem. 1989, 32, 1349.
| Crossref | GoogleScholarGoogle Scholar | 2724305PubMed |
[28] C. M. Ireland, A. R. Durso, R. A. Newman, M. P. Hacker, J. Org. Chem. 1982, 47, 1807.
| Crossref | GoogleScholarGoogle Scholar |
[29] F. J. Schmitz, M. B. Ksebati, J. S. Chang, J. L. Wang, M. B. Hossain, D. Van der Helm, M. H. Engel, A. Serban, J. A. Silfer, J. Org. Chem. 1989, 54, 3463.
| Crossref | GoogleScholarGoogle Scholar |
[30] X. Fu, T. Do, F. J. Schmitz, V. Andrusevich, M. H. Engel, J. Nat. Prod. 1998, 61, 1547.
| Crossref | GoogleScholarGoogle Scholar | 9868162PubMed |
[31] M. A. Rashid, K. R. Gustafson, J. H. Cardellina, M. R. Boyd, J. Nat. Prod. 1995, 58, 594.
| Crossref | GoogleScholarGoogle Scholar | 7623037PubMed |
[32] Y. Hamamoto, M. Endo, M. Nakagawa, T. Nakanishi, K. Mizukawa, J. Chem. Soc. Chem. Commun. 1983, 323.
| Crossref | GoogleScholarGoogle Scholar |
[33] T. Ishida, M. Inoue, Y. Hamada, S. Kato, T. Shioiri, J. Chem. Soc. Chem. Commun. 1987, 370.
| Crossref | GoogleScholarGoogle Scholar |
[34] T. Ishida, M. Tanaka, M. Nabae, M. Inoue, S. Kato, Y. Hamada, T. Shioiri, J. Org. Chem. 1988, 53, 107.
| Crossref | GoogleScholarGoogle Scholar |
[35] See, for example, A. Asano, K. Minoura, T. Yamada, M. Doi, J. Pept. Sci. 2016, 22, 156.
| Crossref | GoogleScholarGoogle Scholar | 26856689PubMed |
[36] R. Zhao, S. Gove, J. E. Sundeen, B. C. Chen, Tetrahedron Lett. 2001, 42, 2101.
| Crossref | GoogleScholarGoogle Scholar |
[37] Our group has also published a synthetic route to 2-amino-1,3-thiazoles. See: L. A. Baker, C. M. Williams, J. Heterocycl. Chem. 2003, 40, 353.
| Crossref | GoogleScholarGoogle Scholar |
[38] M. S. Kerr, J. R. de Alaniz, T. Rovis, J. Org. Chem. 2005, 70, 5725.
| Crossref | GoogleScholarGoogle Scholar | 15989360PubMed |
[39] E. A. Merritt, M. C. Bagley, Synthesis 2007, 22, 3535.
[40] Y. Takikawa, K. Shimada, K. Sato, S. Sato, S. Takizawa, Bull. Chem. Soc. Jpn. 1985, 58, 995.
| Crossref | GoogleScholarGoogle Scholar |
[41] P. Comba, L. R. Gahan, G. R. Hanson, M. Westphal, Chem. Commun. 2012, 48, 9364.
| Crossref | GoogleScholarGoogle Scholar |
[42] P. Comba, L. R. Gahan, G. R. Hanson, M. Maeder, M. Westphal, Dalton Trans. 2014, 43, 3144.
| Crossref | GoogleScholarGoogle Scholar | 24326405PubMed |
[43] A. L. van den Brenk, K. A. Byriel, D. P. Fairlie, L. R. Gahan, G. R. Hanson, C. J. Hawkins, A. Jones, C. H. L. Kennard, B. Moubaraki, K. S. Murray, Inorg. Chem. 1994, 33, 3549.
| Crossref | GoogleScholarGoogle Scholar |
[44] (a) MacroModel, version 10.6 2014 (Schrödinger, LLC: New York, NY).
(b) Maestro, version 9.9 2014 (Schrödinger, LLC: New York, NY).
[45] (a) T. A. Halgren, J. Comput. Chem. 1996, 17, 490.
| Crossref | GoogleScholarGoogle Scholar |
(b) T. A. Halgren, J. Comput. Chem. 1996, 17, 520.
| Crossref | GoogleScholarGoogle Scholar |
(c) T. A. Halgren, J. Comput. Chem. 1996, 17, 553.
| Crossref | GoogleScholarGoogle Scholar |
(d) T. A. Halgren, R. B. Nachbar, J. Comput. Chem. 1996, 17, 587.
(e) T. A. Halgren, J. Comput. Chem. 1996, 17, 616.
| Crossref | GoogleScholarGoogle Scholar |
(f) T. A. Halgren, J. Comput. Chem. 1999, 20, 720.
| Crossref | GoogleScholarGoogle Scholar |
(g) T. A. Halgren, J. Comput. Chem. 1999, 20, 730.
| Crossref | GoogleScholarGoogle Scholar |
[46] M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, D. J. Fox, Gaussian 16, Revision D.01 2016 (Gaussian, Inc.: Wallingford, CT).
[47] (a) A. D. Becke, J. Chem. Phys. 1993, 98, 5648.
| Crossref | GoogleScholarGoogle Scholar |
(b) C. Lee, W. Yang, R. G. Parr, Phys. Rev. B Condens. Matter 1988, 37, 785.
| Crossref | GoogleScholarGoogle Scholar |
(c) S. H. Vosko, L. Wilk, M. Nusair, Can. J. Phys. 1980, 58, 1200.
| Crossref | GoogleScholarGoogle Scholar |
(d) P. J. Stephens, F. J. Devlin, C. F. Chabalowski, M. J. Frisch, J. Phys. Chem. 1994, 98, 11623.
| Crossref | GoogleScholarGoogle Scholar |
[48] Y. Zhao, D. G. Truhlar, J. Chem. Phys. 2006, 125, 194101.
| Crossref | GoogleScholarGoogle Scholar | 17129083PubMed |
[49] (a) Y. Zhao, D. G. Truhlar, Theor. Chem. Acc. 2008, 120, 215.
| Crossref | GoogleScholarGoogle Scholar |
(b) A. V. Marenich, C. J. Cramer, D. G. Truhlar, J. Phys. Chem. B 2009, 113, 6378.
| Crossref | GoogleScholarGoogle Scholar |
[50] P. Comba, N. Dovalil, G. R. Hanson, G. Linti, Inorg. Chem. 2011, 50, 5165.
| Crossref | GoogleScholarGoogle Scholar | 21563768PubMed |
[51] L. J. Farrugia, J. Appl. Cryst. 1999, 32, 837.
| Crossref | GoogleScholarGoogle Scholar |
[52] G. M. Sheldrick, Acta Crystallogr. Sect. A 2008, 64, 112.
| Crossref | GoogleScholarGoogle Scholar |
[53] L. J. Farrugia, J. Appl. Cryst. 1997, 30, 565.
| Crossref | GoogleScholarGoogle Scholar |