

Synthetic Amino Acids for Applications in Peptide Ligation–Desulfurization Chemistry
Lara R. Malins A and Richard J. Payne A BA School of Chemistry, The University of Sydney, Sydney, NSW 2006, Australia.
B Corresponding author. Email: richard.payne@sydney.edu.au

Lara R. Malins completed her undergraduate studies in chemistry in 2009 as a Trustee Scholar at Boston University, where she worked in the laboratory of Associate Professor Scott Schaus on the synthesis of purine-based natural products. She was awarded an International Postgraduate Research Scholarship in 2010 to undertake her Ph.D. at the University of Sydney, Australia, in the group of Professor Richard Payne. She completed her doctorate in April 2014 and is currently a post-doctoral research associate in the Payne laboratory, focusing on the development of novel ligation methodologies for the synthesis of complex peptide and glycopeptide targets. |

Richard J. Payne graduated from the University of Canterbury, New Zealand, in 2002. In 2003, he was awarded a Gates Scholarship to undertake his Ph.D. within the Department of Chemistry, University of Cambridge, under the supervision of Professor Chris Abell. After completing his Ph.D., Richard moved to the Scripps Research Institute under the auspices of a Lindemann Postdoctoral Fellowship where he worked in the laboratory of Professor Chi-Huey Wong. In 2008, he moved to the University of Sydney as a lecturer within the School of Chemistry, where he is currently Professor of Organic Chemistry and Chemical Biology and ARC Future Fellow. Richard's research focuses on utilising the tools of synthetic chemistry, including peptide and carbohydrate chemistry, to address problems of biochemical and medicinal significance. |
Australian Journal of Chemistry 68(4) 521-537 https://doi.org/10.1071/CH14568
Submitted: 15 September 2014 Accepted: 9 October 2014 Published: 23 January 2015
Abstract
Native chemical ligation is a powerful tool for the convergent assembly of homogeneous peptide and protein targets from unprotected peptide fragments. The method involves the chemoselective coupling of a peptide thioester with a peptide bearing an N-terminal cysteine (Cys) residue and is mediated by the nucleophilic Cys thiol functionality. A widely adopted extension of the technique for the disconnection of protein targets at alanine (Ala) ligation junctions has been the application of post-ligation desulfurization protocols for the mild removal of the Cys thiol moiety. Recently, attention has turned to the construction of synthetic amino acid building blocks bearing suitably positioned β-, γ-, or δ-thiol ligation auxiliaries with a view to expanding the scope of the ligation–desulfurization manifold. To date, several thiol-derived amino acids have been prepared, greatly increasing the generality and flexibility of chemoselective ligation technologies for the chemical synthesis of diverse protein targets. This review will highlight the current synthetic approaches to these important amino acid building blocks.
Introduction
Access to homogeneous peptides and proteins with precisely defined covalent structures is crucial to elucidating the intricate relationship between protein structure and function. In this endeavour, synthetic chemistry has served to complement and extend the role of biological expression techniques, which are generally limited to the incorporation of the genetically encoded, canonical amino acids and do not readily accommodate the programmed installation of non-templated protein modifications, such as the vast array of structurally and functionally important enzyme-mediated post-translational modifications.[1] On the contrary, the tools of chemical synthesis facilitate access to high purity, post-translationally modified proteins and enable the construction of an almost infinite array of unnatural, strategically engineered protein variants for use in biological studies and as potential therapeutics. For example, the development and optimization of solid-phase peptide synthesis (SPPS)[2] has enabled the highly efficient, routine preparation of defined polypeptide sequences (bearing native amino acids as well as unnatural amino acid variants) up to ~50 amino acids in length. The advent of chemoselective ligation methodologies has subsequently enabled entry into the realm of functional protein domains, which are typically much larger than 50 amino acids,[3] through the convergent assembly of smaller peptide fragments. In combination with SPPS as a robust platform for fragment synthesis, such ligation methodologies have redefined the capabilities of modern protein synthesis.
The most widely employed ligation methodology for the convergent construction of peptide and protein targets is native chemical ligation.[4] Developed in 1994 by Kent and co-workers, this technique involves the ligation of completely unprotected peptide fragments, in aqueous denaturing media and under mild reaction conditions, to afford native peptide and protein targets with discrete covalent structures. In particular, the reaction involves the condensation of a peptide bearing a C-terminal thioester with a peptide bearing an N-terminal cysteine (Cys) residue through an initial, reversible transthioesterification step (Scheme 1a). The resulting bridged thioester intermediate then rearranges in an irreversible, intramolecular S-to-N acyl shift to generate the target amide bond and regenerate the ligation site Cys residue. Originally employed in the total chemical synthesis of the cytokine protein IL-8,[4] this powerful methodology has subsequently been utilised in the synthesis of numerous protein targets, including those bearing complex post-translational modifications, in the twenty years since its dissemination.[3,5] Native chemical ligation has likewise inspired the development of related technologies for the construction of peptides and proteins, including expressed protein ligation (EPL),[6] involving the ligation of one or more recombinantly produced peptide fragments, and kinetically controlled ligation,[7] which has facilitated the rapid construction of peptide and protein targets[8] through iterative ligation chemistry.
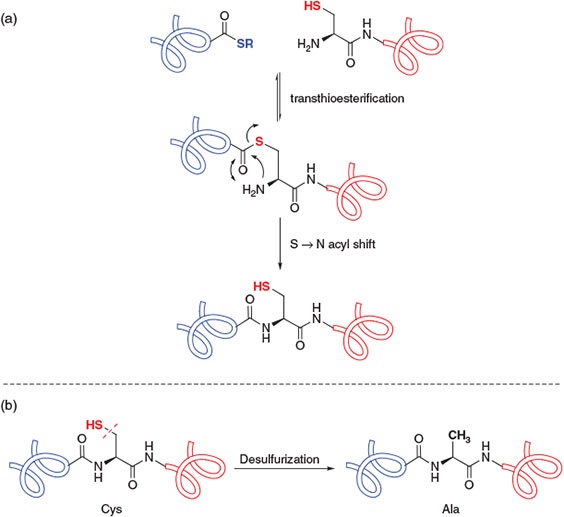
|
The presence of an appropriately placed Cys residue in the target molecule is a prerequisite for the construction of peptides and proteins using native chemical ligation. However, the relative scarcity of Cys in naturally occurring proteins (1.1 %)[9] has prompted intense research efforts to expand the scope of native chemical ligation to include Cys-free protein targets and those bearing Cys residues that are inappropriately positioned for assembly via ligation chemistry. While initial attempts focussed on the use of N-terminal or side-chain thiol auxiliaries[5k,5l,10] and thiol-free methods[11] to overcome this limitation, efforts have recently converged on ligation–desulfurization chemistry[12] using the native chemical ligation concept. This research area was built upon a pioneering study by Yan and Dawson in 2001[13] which demonstrated that Cys residues could be reductively desulfurized, in the presence of a metal catalyst and hydrogen gas, to the corresponding alanine (Ala) residues following the ligation event (Scheme 1b). By demonstrating that ligation at homoCys[14] could also be followed by reductive desulfurization to obtain an α-amino butyric acid (Abu) residue, the authors of this study established the feasibility of the ligation–desulfurization cascade at both β- and γ-thiol ligation auxiliaries. These findings explicitly established the core intellectual framework for the application of fully synthetic amino acid derivatives equipped with suitably positioned β- or γ-thiol auxiliaries for use in ligation–desulfurization chemistry (Scheme 2).[13] Aided by the development of a milder, metal-free radical desulfurization protocol with broad functional group tolerance,[15] this perceptive notion has recently come to fruition, with the preparation of several suitably functionalized amino acid building blocks (Table 1). These compounds have augmented the scope and flexibility of chemoselective ligation methodologies. The rapid development of ligation–desulfurization chemistry, at both Cys and synthetic Cys surrogates,[16] holds great promise for the synthesis of biologically relevant targets and structurally-engineered protein variants.
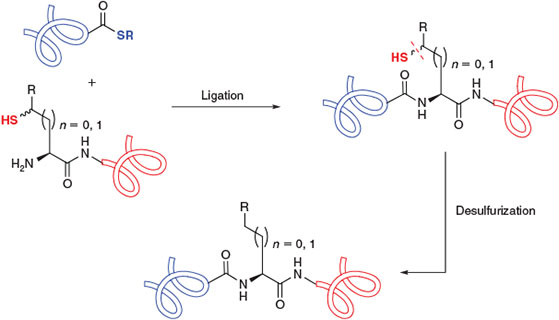
|
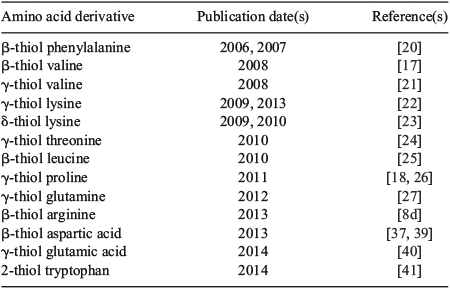
|
The burgeoning success of ligation–desulfurization chemistry may be largely attributed to advances in synthetic methodology that have increased the accessibility of suitably functionalized, thiol-derived amino acids. These building blocks, in turn, serve as enabling tools for the preparation of homogeneous peptides and proteins for biological studies. Ligation–desulfurization chemistry therefore draws upon a long historical precedent for the judicious application of synthetic organic chemistry to the study of important biomolecules. There is perhaps no better exemplification of the systematic construction of small molecules for the purpose of elucidating biological function than in the pioneering work of the late Sir John Cornforth on the intricacies of enzyme catalysis. Indeed, the chemical synthesis of discrete, isotopically labelled compounds played a fundamental role in his Nobel Prize winning work investigating the stereochemical outcome of specific enzymatic transformations. As a tribute to the life and work of Sir John Cornforth, this review will provide an overview of synthetic methods for the construction of thiol-derived amino acids and discuss the utility of these molecules for the preparation of proteins. Specifically, the review will highlight the critical contributions of small-molecule synthesis in the advancement of chemoselective ligation methodologies for the purpose of understanding the structure and function of proteins in the post-genomic era.
Synthesis of Thiol-Derived Amino Acids Through Nucleophilic Displacement
With the exception of β-thiol valine (Val) 1 (more commonly referred to as penicillamine)[17] and 9-fluorenylmethoxycarbonyl (Fmoc)-γ-thiol proline (Pro) 2 (Fig. 1),[18] most thiol-derived amino acids for ligation–desulfurization chemistry are not commercially available. Access to these building blocks therefore requires the design of suitable synthetic pathways. Given the commercial and synthetic availability of protected amino acid derivatives, these molecules have served as efficient starting points for the majority of synthetic routes to thiol-derived amino acids. The application of amino acids as chiral pool precursors also abrogates the need to install the native stereochemical configuration at the α-position, which could considerably complicate the synthesis by requiring the use of chiral ligands or catalysts. Beginning with a generalized amino acid framework, the installation of the key thiol auxiliary therefore becomes the crucial step in the synthetic pathway to thiol-derived amino acids. The earliest employed strategy and by far the most common route to introduce the auxiliary is through displacement of an activated alcohol (e.g. mesylate) derived from a hydroxy-amino acid building block with a suitable thiol nucleophile (e.g. thioacetate). This step is typically followed by several protecting group manipulations, including hydrolysis of the thioacetate moiety, to enable protection of the thiol handle as an asymmetric disulfide or the corresponding S-trityl (Trt) derivative, both of which are compatible with standard conditions employed in Fmoc-strategy SPPS.[19] This overall synthetic strategy has been employed for the synthesis of several thiol-derived amino acids, including β-thiol phenylalanine (Phe),[20] γ-thiol Val,[21] γ-thiol lysine (Lys),[22] δ-thiol Lys,[23] γ-thiol threonine (Thr),[24] β-thiol leucine (Leu),[25] γ-thiol Pro,[26] γ-thiol glutamine (Gln),[27] and β-thiol arginine (Arg).[8d]
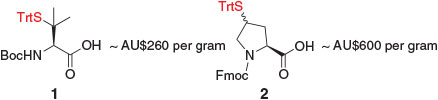
|
In 2007, Crich and Banerjee prepared β-thiol Phe building block 3[20a] for use in ligation–desulfurization chemistry in a 9-step synthesis from l-Phe methyl ester 4 (Scheme 3). Following protection of the free amine as the di-tert-butyloxycarbonyl (Boc) derivative 5, the synthetic pathway relied on selective bromination of the benzylic position of Phe using N-bromosuccinimide[28] to afford 6 as a mixture of diastereomers at the β-position. Subsequent treatment with AgNO3 generated the oxazolidinone intermediate 7 as a 6 : 1 mixture of trans/cis isomers. Chromatographic separation of the diastereomers was followed by treatment of trans-7 with Cs2CO3 to generate the β-hydroxy Phe derivative 8.[29] Activation of the alcohol as the corresponding mesylate and subsequent inversion with thioacetic acid in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) enabled installation of the protected β-thiol functionality. Following hydrolysis of the thioacetate, free thiol 10 was obtained in 55–60 % yield over the three steps. Conversion into the asymmetric disulfide upon treatment with S-ethyl ethanethiosulfinate and saponification of the methyl ester generated Phe building block 3, which was ready for incorporation into model peptides via SPPS. Subsequent application of building block 3 in ligation–desulfurization chemistry served as a general proof of concept for the role of synthetic thiol-derived amino acids in ligation chemistry.
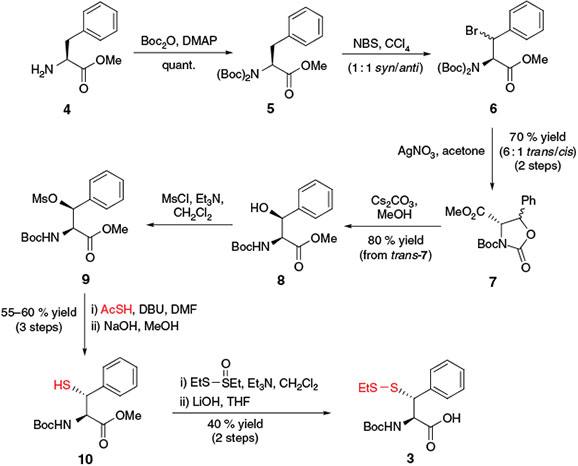
|
A similar synthetic strategy was employed for the synthesis of γ-thiol Val 11, which was accomplished by Danishefsky and co-workers in 2008 (Scheme 4).[21] The 10-step synthesis began with Fmoc-aspartic acid (Asp)-OtBu 12, which was first side-chain protected as the methyl ester 13 and then Fmoc deprotected to allow conversion into the 9-(9-phenylfluorenyl) (PhFl) derivative 14. The PhFl-amine protecting group served to effectively block the α-centre and allow for selective methylation of the β-position using methyl iodide in the presence of a strong base, generating 15 as a 1 : 1 mixture of the β-methyl epimers. Reduction of the methyl ester side-chain of compound 15 to the corresponding γ-alcohol was then accomplished using diisobutylaluminium hydride (DIBAL-H), affording 16 and epi-16, which were readily separated by column chromatography. Compound 16 was activated as the corresponding mesylate 17 and treated, under similar conditions to those employed by Crich and Banerjee for the synthesis of β-thiol Phe,[20a] with thioacetic acid in DBU to install the crucial thiol moiety and provide compound 18 in 73 % yield over the two steps. Protecting group manipulations, including conversion of the S-acetate into the corresponding S-methyl asymmetric disulfide using S-methyl methanethiosulfinate (MMTS) and acidic cleavage of the tBu ester, eventually provided the γ-thiol Val building block 11. Importantly, the β-epimer of 11 was also accessible using an analogous activation–displacement protocol beginning with alcohol epi-16. Both diastereomers were shown to effectively facilitate ligation and could be converted into the native Val residues upon radical desulfurization.[15]
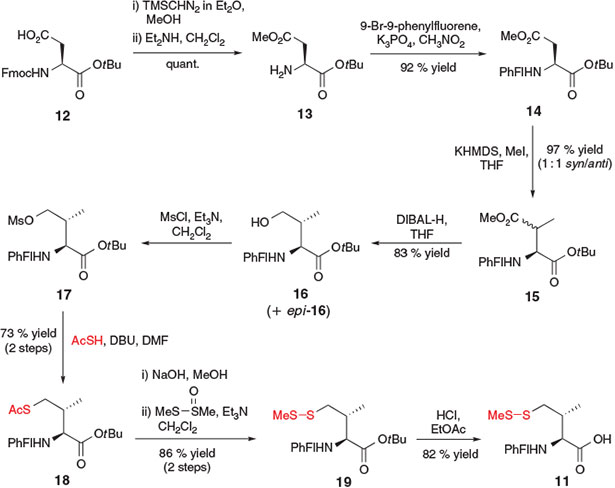
|
In 2009, Liu and co-workers reported the synthesis of a γ-thiol Lys derivative 20 that could effectively mediate ligation at both the α- and ϵ-amino groups of Lys.[22a] Using judicious Cbz-protection of the side-chain ϵ-amino group, γ-thiol Lys derivative 20 was designed to first facilitate ligation at the α-amino group. Subsequent deprotection of the Cbz group would enable modification of the ϵ-amino group in a second ligation step. Removal of the thiol auxiliary using a reductive[13] or radical[15] desulfurization protocol would then afford the doubly functionalized Lys residue. This innovative synthetic strategy is particularly attractive for the synthesis of peptides and proteins bearing post-translational modifications on the Lys side-chain, including acetylation, ubiquitylation, and methylation. Access to the key γ-thiol Lys building block 20 (Scheme 5) was accomplished in 16 steps from Boc-Asp-OtBu 21 by first employing conditions described by Guichard and co-workers[30] for the preparation of 4-hydroxy Lys precursor 22 bearing a protected γ-hydroxy moiety and a free ϵ-hydroxy group. Mesylation and displacement of the primary alcohol of 22 with NaN3 afforded azide 23,[30] which could be reduced to the corresponding amine using catalytic hydrogenation and re-protected in the presence of Cbz-Cl to afford compound 24. At this point, the protected γ-hydroxy moiety was unmasked and activated using mesyl chloride. Displacement of mesylate 26 with potassium thioacetate afforded compound 27, bearing a masked thiol at the γ-position. Conversion into the S-methyl asymmetric disulfide 28 was followed by acidic cleavage of the tBu ester and concomitant loss of the Boc-amine protecting group, which was subsequently reinstalled to afford the final building block 20. After incorporation into model peptides, dual ligations were successfully carried out using this thiol-derived amino acid to generate side-chain ubiquitylated and biotinylated peptide products.[22a]
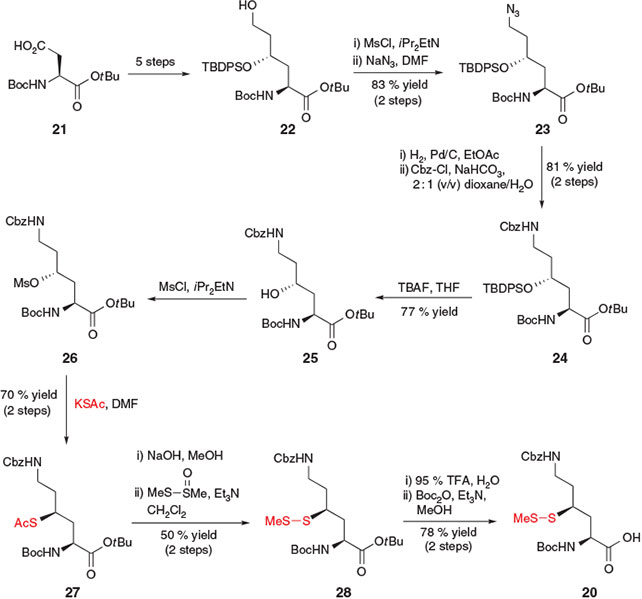
|
Brik and co-workers independently reported a more concise preparation of a δ-thiol Lys derivative 29 for the chemoselective ubiquitylation of peptides (Scheme 6).[23a] This synthetic strategy began with glutamic acid (Glu) 30, which was first converted into aldehyde 31 using conditions previously reported by Martin and co-workers.[31] A subsequent base-catalyzed Henry reaction between aldehyde 31 and MeNO2 in the presence of tetra-n-butylammonium fluoride (TBAF) afforded the nitro alcohol 32 as a 1 : 1 mixture of diastereomers. Rather than installing the key thiol auxiliary at this point through an activation–displacement protocol, the authors opted instead to proceed through the formation of Michael acceptor 33, which was generated in a one-pot acetylation–elimination protocol in the presence of acetic anhydride and 4-dimethylaminopyridine (DMAP). The thiol auxiliary was then installed through Michael addition of lithium t-butylsulfide, generating compound 34 in 85 % yield as a mixture of diastereomers at the δ-position. Reduction of the ϵ-nitro group and subsequent protection with alloxycarbonyl chloride (Alloc-Cl) provided the protected Lys derivative 35. To prevent racemization of the α-centre during base-catalyzed deprotection of the methyl ester, the di-Boc protected α-amine 35 was first converted into the mono-Boc derivative 36. A final saponification step using LiOH then afforded the target δ-thiol Lys derivative 29, which was poised for incorporation into model peptides using Boc-strategy SPPS. Importantly, both δ-thiol epimers mediated ligation at the ϵ-amine, and could be readily removed via radical desulfurization to afford the native Lys residue at the ligation junction.
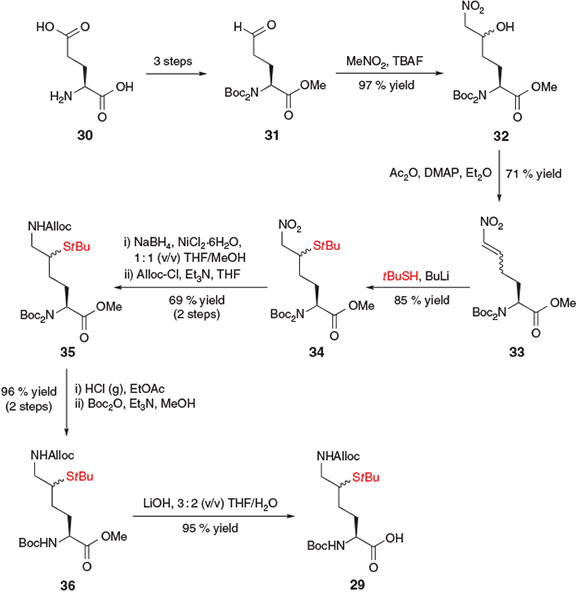
|
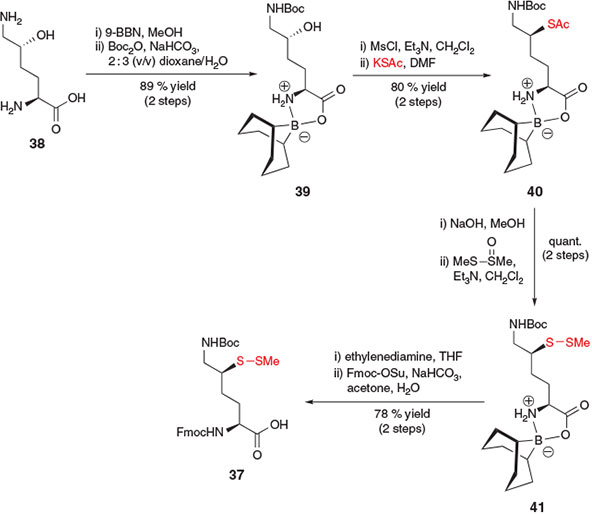
Further interest in the application of δ-thiol Lys in the study of ubiquitylation prompted the development of a subsequent synthetic route to the stereochemically pure δ-thiol Lys derivative 37 by Ovaa and co-workers (Scheme 7).[23b] Beginning with commercially available δ-hydroxy Lys derivative 38, the authors first employed 9-borabicyclo[3.3.1]nonane (9-BBN) as a regioselective protecting group for the α-amino acid functionality[32] and subsequently protected the ϵ-amino group with Boc anhydride to afford compound 39. Activation of the alcohol followed by SN2 displacement with potassium thioacetate facilitated installation of the key thiol auxiliary, affording compound 40 in 80 % yield over the two steps. Hydrolysis of the S-acetate and re-protection as the S-methyl asymmetric disulfide provided compound 41. Removal of the 9-BBN protecting group using ethylenediamine was then followed by Fmoc protection of the α-amine to afford the target δ-thiol Lys building block 37 in a total of eight steps from hydroxy Lys derivative 38.[23b] The same group also reported a modified protocol in 2013 to access a γ-thiol Lys derivative in 8 steps from l-Lys.[22b] This synthetic strategy relied on a radical chlorination reaction to directly activate the γ-position of Lys. Protection of the α-amino acid of the resulting γ-chloro-Lys with 9-BBN enabled subsequent nucleophilic displacement of the γ-chloro functionality with potassium thioacetate. Several protecting group manipulations then afforded a suitable building block for direct incorporation into peptides using Fmoc-SPPS.[22b]
|
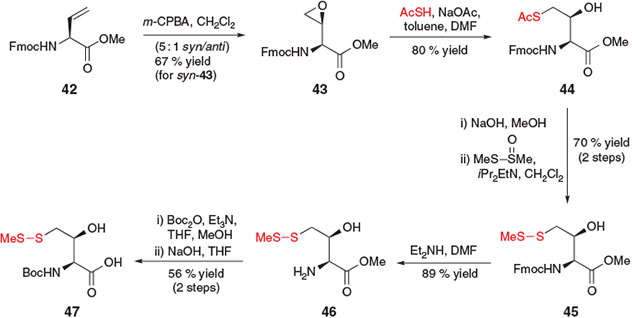
In 2010, Danishefsky and co-workers reported the construction of a γ-thiol Thr derivative for use in ligation–desulfurization chemistry.[24] This synthetic strategy employed a slight variation on the activation–displacement pathway for the installation of the crucial thiol moiety (Scheme 8). Beginning with protected vinylglycine derivative 42, treatment with meta-chloroperbenzoic acid (m-CPBA) first afforded epoxide 43 as a 5 : 1 mixture of syn/anti diastereomers, which were separated by chromatography. Proceeding with the major diastereomer, syn-43, the thiol moiety was installed at the γ-position through opening of the epoxide with sodium thioacetate, liberating the Thr side-chain hydroxyl moiety and affording compound 44 in 80 % yield as a single diastereomer. Conversion into the S-methyl asymmetric disulfide 45 was achieved using standard conditions, and protecting group manipulation enabled straight-forward conversion into the desired γ-thiol Thr building block 47 in seven steps from vinylglycine 42.
|
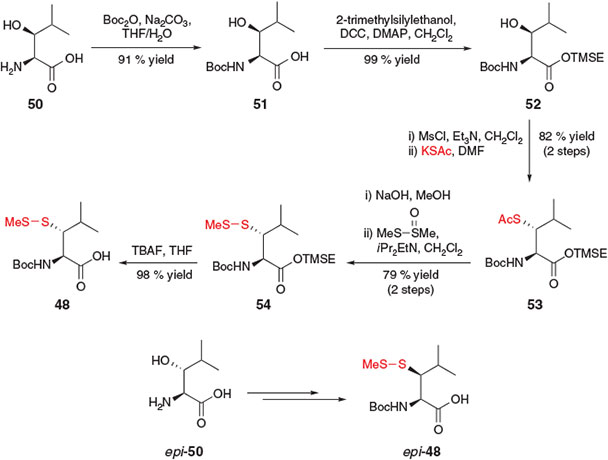
Suitably protected β-thiol Leu derivatives 48 and 49 were independently reported in 2010 by Danishefsky and co-workers[25a] and Brik and co-workers,[25b] respectively. Both groups employed a 7-step protocol beginning with the commercially available β-hydroxy-l-Leu 50 or the corresponding β-epimer, epi-50. Danishefsky and co-workers utilised a similar mesylation, SN2 displacement protocol to those previously described (see above) to install the requisite masked thiol functionality and access the desired β-thiol building block 48 (Scheme 9).[25a] Specifically, an initial Boc-protection of 50 afforded compound 51, which was then treated with 2-trimethylsilylethanol (TMSE-OH) in the presence of N,N′-dicyclohexylcarbodiimide (DCC) and DMAP to provide the corresponding TMSE ester 52. Mesylation of the β-hydroxy moiety followed by nucleophilic displacement with potassium thioacetate afforded thioacetate 53 in 82 % yield over the two steps. Hydrolysis of the thioester and subsequent protection as the asymmetric disulfide 54 was followed by a final C-terminal deprotection step in the presence of TBAF, furnishing the target β-thiol Leu derivative 48. The authors also successfully prepared the corresponding diastereomer, epi-48, by beginning the synthesis with the threo-β-hydroxy-l-Leu derivative, epi-50. Interestingly, a comparison of the rates of ligation facilitated by these diastereomeric building blocks indicated a vast difference between the efficiency of the two diastereomers, with compound 48 mediating more rapid ligation than the corresponding β-epimer, epi-48. The difference in rate was attributed to a steric interaction between the C-terminal peptide chain and the β-isopropyl group of the β-thiol Leu residue imposed by the putative five-membered ring transition state in the S-to-N acyl shift (see Scheme 1a). It was postulated that peptides incorporating building block 48 enabled acyl migration to proceed with a more favourable trans relationship between the two substituents, while the incorporation of epi-48 placed substituents in an unfavourable cis orientation in the five-membered ring transition state.[25a]
|
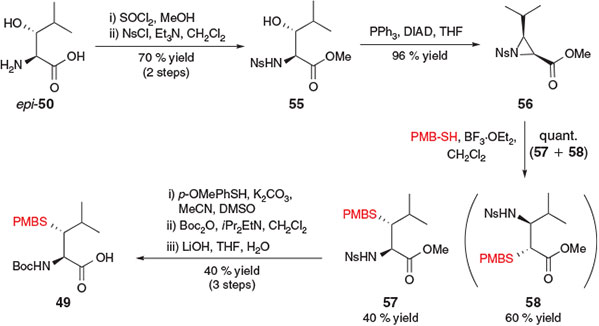
Brik and co-workers also envisaged that the synthesis of β-thiol Leu could be achieved from the corresponding β-hydroxy precursor through mesylation followed by nucleophilic substitution with an appropriate thiol.[25b] However, a competing β-elimination pathway observed upon treatment of the activated β-hydroxy Leu derivative with a nucleophilic thiol prompted the exploration of an alternative synthetic approach (Scheme 10). As such, the strategy adopted by Brik and co-workers for the synthesis of β-thiol Leu derivative 49 began with the conversion of threo-β-hydroxy-l-Leu epi-50 into the corresponding methyl ester and protection of the α-amino group with p-nitrophenylsulfonyl chloride (NsCl) to afford p-nitrosulfonamide 55. Intramolecular ring closure using Mitsunobu conditions then afforded protected aziridine 56 in 96 % yield. Ring opening with p-methoxybenzyl (PMB) thiol in the presence of BF3·OEt2 occurred in quantitative yield in a 4 : 6 ratio of regioisomers 57 : 58, which were separated by flash column chromatography. The desired regioisomer 57, resulting from the attack of PMB-SH onto the β-carbon of aziridine 56, was then converted into the target building block 49 following protecting group manipulations.[25b]
|
Shortly after a report by Danishefsky and co-workers describing ligation–desulfurization chemistry mediated by a commercially available Fmoc-protected trans-γ-thiol Pro derivative 2,[18,33] Otaka and co-workers reported a synthetic approach to the Boc-protected γ-thiol Pro derivative 59 and the corresponding cis diastereomer (Scheme 11).[26] The synthetic pathway to the trans-isomer began with trans-β-hydroxy Pro derivative 60. Intramolecular cyclization using Mitsunobu conditions afforded bridged intermediate 61, which was converted into compound 62, bearing a cis-hydroxy group, following saponification of the lactone in the presence of NaOH and benzyl protection of the resulting carboxylic acid. Installation of the thiol handle was accomplished through activation of alcohol 62 as the corresponding mesylate and subsequent inversion with potassium thioacetate to generate 63 in 87 % yield over the two steps. Simultaneous hydrolysis of the thioacetate and the benzyl ester moiety followed by trityl protection of the unmasked thiol then afforded trans-γ-thiol derivative 59. Preparation of the corresponding cis-isomer was accomplished using a direct activation–displacement protocol beginning with compound 60.[26]

|
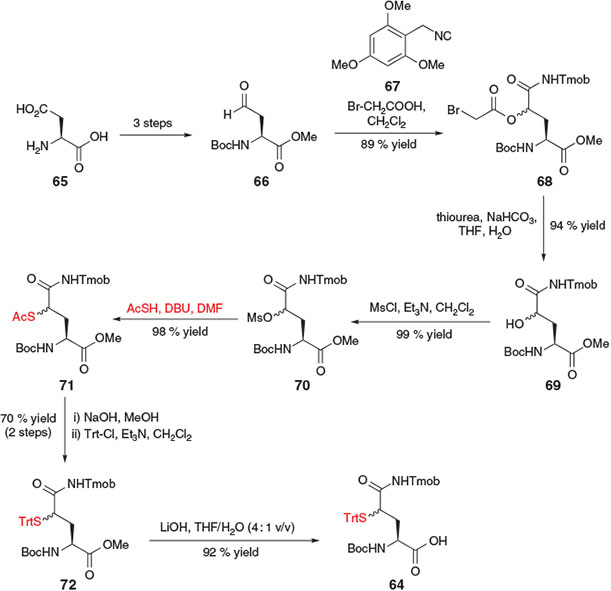
In 2012, Brik and co-workers reported ligation–desulfurization chemistry at γ-thiol Gln via the synthesis of suitably protected amino acid derivative 64 (Scheme 12).[27] The 10-step synthesis of the requisite building block began with l-Asp 65, which was readily converted into the side-chain aldehyde 66 in three steps. A Passerini three component reaction between aldehyde 66, 2,4,6-trimethoxybenzyl isocyanide (Tmob-NC) 67, and bromoacetic acid then afforded a diastereomeric mixture of the γ-acyloxy Gln derivative 68 in 89 % yield. Treatment of 68 with thiourea in basic media provided the γ-hydroxy derivative 69, which was converted into mesylate 70 upon activation with mesyl chloride and triethylamine. Nucleophilic displacement of the mesylate with thioacetic acid in the presence of DBU introduced the crucial masked thiol functionality, generating compound 71 in 98 % yield. Protecting group manipulations, including saponification of the thioester and subsequent conversion to the S-Trt derivative 72 as well as hydrolysis of the α-carboxy methyl ester, afforded the target γ-thiol Gln building block 64 as a mixture of diastereomers.[27]
|
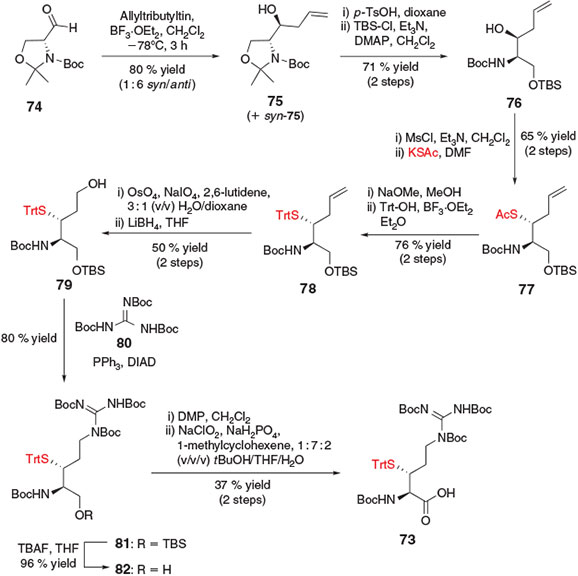
In an effort to explore a common starting material for the synthesis of thiol-derived amino acids, Payne and co-workers devised a strategy for the synthesis of β-thiol Arg 73[8d] from commercially available R-Garner’s aldehyde 74,[34] a configurationally stable, α-amino aldehyde derived from d-serine (Ser) (Scheme 13). It was envisaged that 74 could serve as a common starting material for the synthesis of β-thiol building blocks through the introduction of various proteinogenic amino acid side chains via strategic application of the diverse array of carbon–carbon bond-forming reactions available for the modification of the aldehyde carbonyl carbon. Nucleophilic addition of the desired amino acid side-chain (or side-chain precursor) to the starting aldehyde would have the added benefit of directly generating a β-hydroxy moiety, which could serve as a convenient handle for the introduction of the key thiol functionality through an activation–displacement reaction pathway. As such, the synthesis of β-thiol Arg 73 began with the addition of allyltributyl tin to Garner’s aldehyde 74, affording the corresponding allyl alcohol 75 in 80 % yield as an inseparable, 1 : 6 mixture of syn/anti diastereomers. Deprotection of the hemiaminal ether using p-TsOH was then followed by selective protection of the primary alcohol with tert-butyldimethylsilyl (TBS) chloride to afford compound 76. Activation of the allyl alcohol with mesyl chloride and subsequent inversion with potassium thioacetate occurred in 65 % yield over the two steps to afford thioacetate 77. Facile conversion into the S-trityl derivative 78 was followed by oxidative cleavage of the allyl group in the presence of OsO4 and NaIO4 to generate the corresponding aldehyde. Immediate reduction with LiBH4 afforded primary alcohol 79 predominately as the threo-diastereomer, which was separated at this point from the minor erythro-isomer. Installation of the protected guanidine side-chain of Arg was then accomplished using Mitsunobu conditions in the presence of N,N′,N″-tri-Boc-guanidine 80 to afford compound 81. Subsequent removal of the TBS protecting group afforded primary alcohol 82 which was converted using a two-step oxidation protocol into the target β-thiol Arg derivative 73. Notably, the overall synthetic logic of Garner’s aldehyde as a common starting point for the synthesis of functionalized amino acids for use in ligation chemistry has also been exploited for the preparation of a β-selenol derivative of phenylalanine.[35] This building block was successfully utilised in the construction of peptides using chemoselective ligation–deselenization chemistry,[36] a related field that has been the subject of recent reviews.[5o,12b]
|
Synthesis of Thiol-Derived Amino Acids via Electrophilic Substitution
As a general synthetic strategy for the preparation of thiol-derived amino acids, the introduction of auxiliaries through nucleophilic substitution of activated hydroxyl groups has been highly successful.[16] Recently, several synthetic approaches to thiol-derived amino acids have also employed electrophilic sulfur reagents for the direct functionalization of amino acids. These strategies have tended to exploit the innate reactivity of protected amino acid derivatives bearing side-chain esters, such as Asp and Glu, which can be readily functionalized adjacent to the side-chain carbonyl carbon through base-mediated enolization followed by reaction with a suitable electrophile. As a more direct means of installing the thiol ligation auxiliary than the activation–displacement protocol (see above), these synthetic strategies also tend to be very concise, minimizing functional group and protecting group interconversions and facilitating rapid access to the target thiol-derived amino acids.
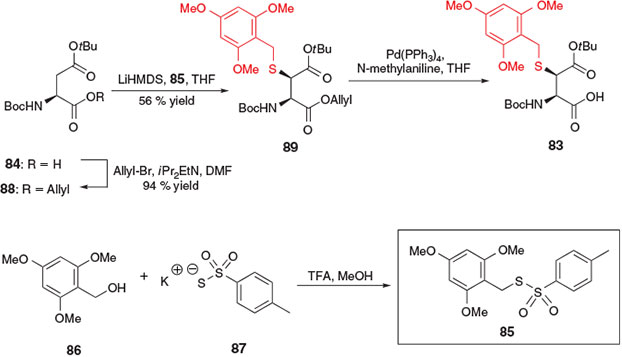
The first application of electrophilic sulfenylation chemistry for the synthesis of a thiol-derived amino acid for ligation–desulfurization chemistry was reported by Thompson et al. in 2013 for the preparation of β-thiol Asp derivative 83 (Scheme 14).[37] The synthesis of this building block was accomplished in three steps from Boc-Asp(OtBu)-OH 84 and employed the novel sulfenylating agent 85, prepared in one step from 2,4,6-trimethoxybenzyl (Tmob) alcohol 86 and potassium toluene thiosulfonate 87.[38] First, protection of the α-carboxylic acid of compound 84 as the allyl ester was accomplished using allyl bromide in the presence of base to afford the fully protected Asp derivative 88. In the presence of 2 equiv. of lithium hexamethyldisilazide (LiHMDS) at –78°C, abstraction of the α-NH proton and enolization of the side-chain ester led to the corresponding dianion of compound 88, which was then treated with the electrophilic sulfenylating reagent 85 to provide the Tmob-protected β-thiol derivative 89 as a 9 : 1 mixture of erythro/threo diastereomers. After separation of the diastereomers by column chromatography, the major erythro isomer was treated with Pd(PPh3)4 and N-methylaniline to remove the allyl ester and afford the target β-thiol Asp building block 83.[37]
|
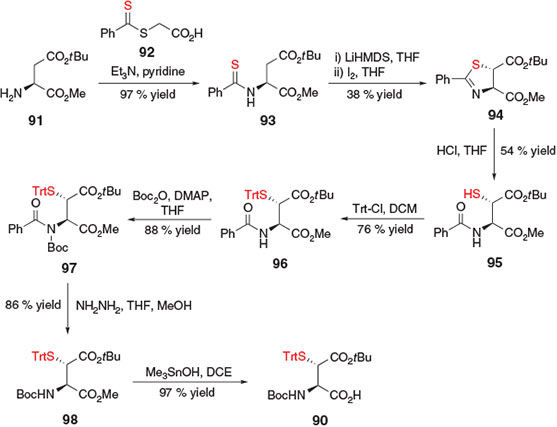
Shortly after the initial report of building block 83,[37] an alternative preparation of β-thiol Asp derivative 90 was reported by Tan and co-workers in eight steps from H-Asp(OtBu)-OMe 91 (Scheme 15).[39] Although conceptually distinct from the sulfenylation protocol employed by Thompson et al.[37] this preparation also utilised a base-mediated enolization of the protected Asp side-chain ester to facilitate installation of the crucial β-thiol auxiliary.[39] Initial condensation of 91 with S-(thiobenzoyl)thioglycolic acid 92 in the presence of Et3N and pyridine afforded thiobenzamidomalonic ester 93. Double deprotonation with LiHMDS to generate the corresponding dianion was followed by treatment with iodine to promote an intramolecular iodocyclization to the trans-thiazoline 94. Selective hydrolysis of the thiazoline ring subsequently provided thiol 95, which was protected as the corresponding S-Trt derivative 96. Boc protection and removal of the benzoyl group provided compound 98, which was treated with trimethyltin hydroxide to facilitate mild cleavage of the C-terminal methyl ester protecting group, affording the final building block 90 for application in ligation-desulfurization chemistry.[39]
|
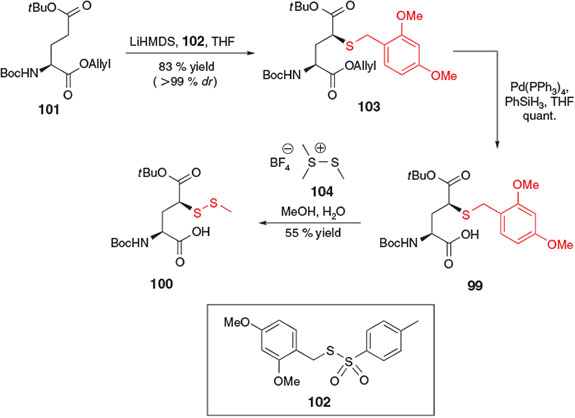
In 2014, the electrophilic sulfenylation pathway employed for the synthesis of β-thiol Asp derivative 83[37] was similarly applied to the preparation of γ-thiol Glu derivatives 99 and 100 by Cergol et al. (Scheme 16).[40] Beginning with Boc-Glu(OtBu)-OAllyl 101, sulfenylation in the presence of 2 equiv. of LiHMDS and the 2,4-dimethoxybenzyl (Dmb)-derived sulfenylating reagent 102 afforded the Dmb-protected γ-thiol Glu derivative 103 in 83 % yield and as a single diastereomer. Deallylation in the presence of catalytic palladium(0) then furnished protected building block 99, which could be directly incorporated into model peptides. Interestingly, when coupled to the N-terminus of a peptide using SPPS, building block 99 was found to be incompatible with the standard acidic conditions employed to facilitate cleavage of the peptide from the resin. The deprotected γ-thiol moiety was shown to facilitate cleavage of the terminal Glu derivative through thiolactone formation under acidic conditions. As such, the alternative building block 100, bearing an S-methyl asymmetric disulfide protecting group at the γ-position, was prepared by direct treatment of Dmb-derivative 99 with dimethyl(methylthio) sulfonium tetrafluoroborate 104. The disulfide protecting group was orthogonal to the acidic conditions employed for cleavage of the resin-bound peptide, and as such, thiolactonization was not observed with building block 100, enabling incorporation into model peptides for use in ligation–desulfurization chemistry.[40]
|
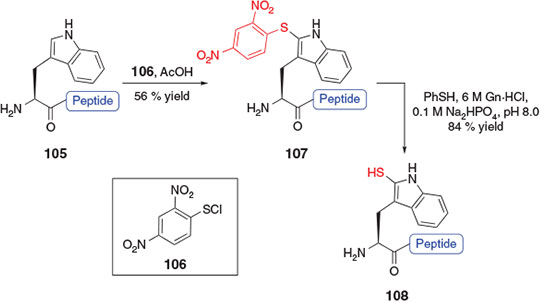
Another application of sulfenylation chemistry for the rapid preparation of a thiol-derived amino acid was reported in 2014 by Payne and co-workers.[41] By employing a chemoselective sulfenylation protocol first described in the late 1960s for the selective modification of tryptophan (Trp) residues in unprotected peptides,[42] this study devised a strategy for ligation–desulfurization chemistry at Trp that avoided the need to prepare a preformed amino acid building block. Specifically, treatment of a fully deprotected peptide bearing an N-terminal Trp residue (e.g. compound 105, Scheme 17) with 2,4-dinitrophenylsulfenyl chloride (DNPS-Cl) 106 in the presence of acetic acid allowed for the selective modification of the 2-position of the indole ring, generating the 2-DNPS adduct 107. Thiolysis of the indole 2-thioether[43] upon treatment with thiophenol unmasked the thiol auxiliary at the 2-position to afford the 2-thiol Trp functionalized peptide 108. The sulfenylation protocol was also adopted for the on-resin modification of Trp-containing peptides, minimizing the number of intermediary purification steps and facilitating the rapid installation of the thiol ligation handle into synthetic peptides. The 2-thiol auxiliary was shown to effectively promote peptide ligation with a variety of C-terminal peptide thioesters and could be removed upon reductive desulfurization in the presence of Pd on Al2O3 following the ligation reaction.[41]
|
The concise synthetic approaches to thiol-derived Asp, Glu and Trp for ligation–desulfurization chemistry have showcased the utility of electrophilic substitution reactions, particularly sulfenylation chemistry, as a facile means of introducing the key thiol auxiliary into thiol-derived amino acid building blocks. It is envisaged that these rapid preparations will facilitate the increasing adoption of such building blocks for the routine construction of peptide and protein targets using ligation–desulfurization chemistry.
Application of Thiol-Derived Building Blocks in the Assembly of Proteins Using Ligation–Desulfurization Chemistry
By far the most widely used ligation–desulfurization method for the synthesis of proteins has been native chemical ligation at Cys followed by post-ligation desulfurization to afford an Ala residue. Indeed, this method has been used for the total chemical synthesis of several complex protein targets.[5q,12] The predominant use of the Cys desulfurization method is mainly owing to the fact that Cys is widely available while the majority of thiol-derived amino acid building blocks have not yet become commercially available. However, recently the power of ligation–desulfurization at synthetic building blocks bearing suitably positioned thiol auxiliaries has been demonstrated through the synthesis of several challenging targets. The following discussion will outline four recent contributions that have employed ligation–desulfurization chemistry at thiol-derived amino acids in the total chemical synthesis of complex protein targets.
An impressive application of the synthetic δ-thiol Lys building block 29[23a] for the study of protein ubiquitylation was reported in 2011 by Brik and co-workers (Scheme 18).[44] The authors of this study prepared the 304-amino acid tetraubiquitin protein 109 using iterative ligation chemistry. Construction of the target compound began with the synthesis of ubiquitin monomer 110, containing a δ-thiol Lys residue at position 48 in the ubiquitin polypeptide chain. Ligation of this fragment with ubiquitin thioester 111, containing a thiazolidine protected δ-thiol Lys residue first afforded a diubiquitin conjugate. Deprotection of the thiazolidine rendered compound 112, which was poised for further functionalization with another equivalent of ubiquitin-derived thioester 111. Thiazolidine deprotection then afforded the ubiquitin trimer 113, which was subjected to a final ligation with ubiquitin thioester 114 containing a native Lys residue at position 48. Removal of the three δ-thiol ligation auxiliaries in a global radical desulfurization protocol provided the target tetraubiquitin protein 109. Analysis of the homogeneous, polyubiquitin protein using circular dichroism and chain disassembly using the deubiquitinating enzyme IsoT confirmed that synthetic protein 109 contained a three-dimensional structure consistent with that of native ubiquitin constructs.[44]

|
Another powerful example of the disconnection of proteins using a combination of ligation–desulfurization at Cys and thiol-derived amino acid building blocks was provided by Danishefsky and co-workers for the preparation of the 84-amino acid, Cys-free human parathyroid hormone (hPTH) 115 (Scheme 19).[45] By employing a convergent ligation–global desulfurization protocol, the authors of this study accessed the target protein 115 through ligation at Cys, β-thiol Leu, and γ-thiol Val from peptide fragments 116–119. First, thiophenyl thioester 116 was utilised in a kinetically controlled ligation reaction[7] with bifunctional peptide 117, containing an N-terminal β-thiol Leu residue and a C-terminal alkyl thioester to generate hPTH(1–38) 120. A γ-thiol Val-mediated ligation reaction between peptides 118 and 119 followed by a thiazolidine deprotection then furnished hPTH (39–84) 121, bearing an unprotected Cys residue at the N-terminus. Convergent assembly of the full-length polypeptide sequence was then accomplished using a native chemical ligation reaction between Cys-peptide 121 and alkyl thioester 120, which proceeded in 63 % yield to afford compound 122. Removal of the Cys, β-thiol Leu, and γ-thiol Val ligation auxiliaries in a global desulfurization protocol subsequently furnished hPTH 115 in 86 % yield. Spontaneous folding of the synthetic protein was confirmed using circular dichroism, and the chemically prepared hPTH was also shown to be of higher purity than the recombinantly produced hPTH used as a reference sample.[45] These results further validate the utility of chemical synthesis, particularly ligation–desulfurization chemistry, for the preparation of well defined protein samples for use in biological assays.

|
Recently, Thompson et al. have described the synthesis of two tick-derived antithrombotic proteins, madanin-1 and chimadanin, using thiol-derived amino acids.[8e] Madanin-1 123 was assembled in the N-to-C direction from peptide fragments 124, 125, and 126 using a three-component, one-pot kinetically controlled ligation–desulfurization protocol (Scheme 20a). Following preparation of the target peptide fragments using Fmoc-SPPS, synthesis began with a kinetically controlled ligation between peptide thioester 124, bearing a reactive C-terminal thioester moiety prepared using trifluoroethanethiol (TFET) 127, and bifunctional peptide 125, containing an N-terminal β-thiol Asp residue[37] and a C-terminal ethyl 3-mercaptopropionate-derived thioester. Importantly, in this kinetically controlled ligation, the electron-withdrawing nature of the trifluoro-substituted TFET alkyl thioester served to enhance the acylating ability of peptide 124 relative to peptide 125, promoting the intermolecular condensation of fragments 124 and 125 over the competing cyclization or oligomerization of bifunctional peptide 125. Following completion of the first ligation, the addition of excess TFET enabled activation of the latent ethyl 3-mercaptopropionate-derived thioester and subsequent ligation with peptide fragment 126, bearing an N-terminal Cys residue, to afford intermediate 128. Without isolation, peptide 128 was directly subjected to radical desulfurization in the presence of tris-(2-carboxyethyl)phosphine (TCEP), the water-soluble radical initiator VA-044,[15] and reduced glutathione[17] to afford the target madanin-1 123 in 42 % isolated yield over the three steps.[8e] This efficient, one-pot ligation–radical desulfurization protocol was facilitated by the use of the alkyl thiol TFET as a novel thiol ligation additive. The innate radical quenching ability of the more commonly employed aryl thiol ligation catalysts (e.g. thiophenol[46] and 4-mercaptophenylacetic acid (MPAA)[47]), generally mandates complete removal of the thiol additive from the ligation mixture before radical desulfurization. In contrast, the alkyl thiol TFET does not interfere with the radical desulfurization transformation and thus enables ligation–desulfurization reactions to be carried out using an operationally simple one-pot protocol.[8e]

|
Application of the TFET-promoted one-pot ligation–desulfurization[8e] has also been demonstrated for the preparation of chimadanin 129 using γ-thiol Glu building block 100 (Scheme 20b).[40] The synthesis of this protein was accomplished through the ligation of three-components, 130, 131, and 132, in the C-to-N direction. Fragment 130, bearing an N-terminal γ-thiol Glu residue was first ligated with peptide 131, bearing a C-terminal thioester and an N-terminal Cys residue masked as the corresponding thiazolidine residue.[48] This reaction was promoted by the presence of excess TFET, which facilitated in situ activation of the ethyl 3-mercaptopropionate peptide thioester 131 to generate the more reactive TFET thioester. Following completion of the ligation, removal of the N-terminal thiazolidine residue was accomplished upon treatment with methoxyamine to afford intermediate 133, bearing a free N-terminal Cys residue. Peptide 133 was subsequently ligated with peptide thioester 132 to afford the full-length polypeptide backbone. Without intermediary purification, a radical desulfurization protocol facilitated direct removal of both the Cys and γ-thiol Glu ligation auxiliaries, affording the native chimadanin 129 in 35 % isolated yield over the four steps.[8e]
Conclusions
The development of ligation–desulfurization chemistry has greatly increased the flexibility of chemoselective ligation technologies for the construction of peptides and proteins. This methodology has extended the scope of native chemical ligation beyond the traditional reliance on the presence of appropriately placed Cys residues within a target sequence. In particular, recent successes in the preparation of thiol-derived amino acids have now enabled the disconnection of target molecules at 11 additional amino acid sites (Phe, Val, Lys, Thr, Leu, Pro, Gln, Arg, Asp, Glu, and Trp). It is predicted that the growing availability of these important synthetic building blocks (e.g. through commercial sources or increasingly concise synthetic routes), together with the very recent development of methods for streamlining the ligation–desulfurization protocol into an efficient, one-pot process,[8e,49] will facilitate the continued adoption of the ligation–desulfurization manifold for the rapid construction of diverse protein targets. Just as the pioneering synthetic work of Sir John Cornforth enabled the careful investigation of the mechanisms of enzyme catalysis, the programmed construction of homogeneous peptide and protein targets using the tools afforded by chemical synthesis will no doubt prove an invaluable resource for understanding the intricate relationship between protein structure and function.
References
[1] (a) C. T. Walsh, S. Garneau-Tsodikova, G. J. Gatto, Angew. Chem. Int. Ed. 2005, 44, 7342.| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXhtlSms77N&md5=4e11c1b7c3c18037db7ff96d347f91b9CAS |
(b) C. Walsh, Posttranslational Modification of Proteins: Expanding Nature’s Inventory 2006 (Roberts and Co. Publishers: Englewood, CO).
[2] (a) R. B. Merrifield, J. Am. Chem. Soc. 1963, 85, 2149.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaF3sXksVajsLg%3D&md5=7222648f6587aad4e9a82b3ec2de3410CAS |
(b) G. Barany, R. B. Merrifield, in The Peptides (Ed. E. G. a. J. Meienhofer) 1979, Vol. 2, pp. 1–284 (Academic Press, New York, NY).
[3] P. E. Dawson, S. B. Kent, Annu. Rev. Biochem. 2000, 69, 923.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXnt1ajurc%3D&md5=3bc0d24699c01bc765239b37c38f5ec6CAS | 10966479PubMed |
[4] P. E. Dawson, T. W. Muir, I. Clark-Lewis, S. B. H. Kent, Science 1994, 266, 776.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXitVGgtrw%3D&md5=4bbff7b4624493187902a4dddb36aad1CAS | 7973629PubMed |
[5] (a) S. Kent, J. Pept. Sci. 2003, 9, 574.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXotF2ntro%3D&md5=9212da64ca505f5538b3818051c75861CAS | 14552420PubMed |
(b) D. S. Y. Yeo, R. Srinivasan, G. Y. J. Chen, S. Q. Yao, Chem. – Eur. J. 2004, 10, 4664.
| Crossref | GoogleScholarGoogle Scholar |
(c) J. P. Tam, J. Xu, K. D. Eom, Biopolymers 2001, 60, 194.
| Crossref | GoogleScholarGoogle Scholar |
(d) G. A. Lemieux, C. R. Bertozzi, Trends Biotechnol. 1998, 16, 506.
| Crossref | GoogleScholarGoogle Scholar |
(e) B. L. Nilsson, M. B. Soellner, R. T. Raines, Annu. Rev. Biophys. Biomol. Struct. 2005, 34, 91.
| Crossref | GoogleScholarGoogle Scholar |
(f) B. G. Davis, Chem. Rev. 2002, 102, 579.
| Crossref | GoogleScholarGoogle Scholar |
(g) D. P. Gamblin, E. M. Scanlan, B. G. Davis, Chem. Rev. 2009, 109, 131.
| Crossref | GoogleScholarGoogle Scholar |
(h) D. Macmillan, Angew. Chem. Int. Ed. 2006, 45, 7668.
| Crossref | GoogleScholarGoogle Scholar |
(i) C. P. R. Hackenberger, D. Schwarzer, Angew. Chem. Int. Ed. 2008, 47, 10030.
| Crossref | GoogleScholarGoogle Scholar |
(j) C. Haase, O. Seitz, Angew. Chem. Int. Ed. 2008, 47, 1553.
| Crossref | GoogleScholarGoogle Scholar |
(k) A. Dirksen, P. E. Dawson, Curr. Opin. Chem. Biol. 2008, 12, 760.
| Crossref | GoogleScholarGoogle Scholar |
(l) R. J. Payne, C. H. Wong, Chem. Commun. 2010, 46, 21.
| Crossref | GoogleScholarGoogle Scholar |
(m) S. B. Kent, Chem. Soc. Rev. 2009, 38, 338.
| Crossref | GoogleScholarGoogle Scholar |
(n) H. P. Hemantha, N. Narendra, V. V. Sureshbabu, Tetrahedron 2012, 68, 9491.
| Crossref | GoogleScholarGoogle Scholar |
(o) L. R. Malins, N. J. Mitchell, R. J. Payne, J. Pept. Sci. 2014, 20, 64.
| Crossref | GoogleScholarGoogle Scholar |
(p) C. Unverzagt, Y. Kajihara, Chem. Soc. Rev. 2013, 42, 4408.
| Crossref | GoogleScholarGoogle Scholar |
(q) L. R. Malins, R. J. Payne, Curr. Opin. Chem. Biol. 2014, 22, 70.
| Crossref | GoogleScholarGoogle Scholar |
[6] (a) T. W. Muir, D. Sondhi, P. A. Cole, Proc. Natl. Acad. Sci. USA 1998, 95, 6705.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXjslyntLg%3D&md5=a28a8b90c43929c64eb68dace1d17164CAS | 9618476PubMed |
(b) K. Severinov, T. W. Muir, J. Biol. Chem. 1998, 273, 16205.
| Crossref | GoogleScholarGoogle Scholar |
(c) T. W. Muir, Annu. Rev. Biochem. 2003, 72, 249.
| Crossref | GoogleScholarGoogle Scholar |
(d) V. Muralidharan, T. W. Muir, Nat. Methods 2006, 3, 429.
| Crossref | GoogleScholarGoogle Scholar |
[7] D. Bang, B. L. Pentelute, S. B. Kent, Angew. Chem. Int. Ed. 2006, 45, 3985.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28Xmt1ars7c%3D&md5=d294d635015d1e51708d38683a00a2ceCAS |
[8] (a) T. Durek, V. Y. Torbeev, S. B. Kent, Proc. Natl. Acad. Sci. USA 2007, 104, 4846.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXjvFCjtro%3D&md5=06522c14b9a9119b83227a2b479e040aCAS | 17360367PubMed |
(b) V. Y. Torbeev, S. B. Kent, Angew. Chem. Int. Ed. 2007, 46, 1667.
| Crossref | GoogleScholarGoogle Scholar |
(c) P. Wang, S. Dong, J. A. Brailsford, K. Iyer, S. D. Townsend, Q. Zhang, R. C. Hendrickson, J. Shieh, M. A. S. Moore, S. J. Danishefsky, Angew. Chem. Int. Ed. 2012, 51, 11576.
| Crossref | GoogleScholarGoogle Scholar |
(d) L. R. Malins, K. M. Cergol, R. J. Payne, ChemBioChem 2013, 14, 559.
| Crossref | GoogleScholarGoogle Scholar |
(e) R. E. Thompson, X. Liu, N. Alonso-García, P. J. B. Pereira, K. A. Jolliffe, R. J. Payne, J. Am. Chem. Soc. 2014, 136, 8161.
| Crossref | GoogleScholarGoogle Scholar |
[9] UniprotKB/TrEMBL Protein Database Release 2014_07 Statistics 2014. Available at: http://www.ebi.ac.uk/uniprot/TrEMBLstats (accessed 1 September 2014).
[10] (a) L. E. Canne, S. J. Bark, S. B. H. Kent, J. Am. Chem. Soc. 1996, 118, 5891.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK28Xjtlygu7o%3D&md5=5a70e87e103048f1a9fbd1006ed8a8fcCAS |
(b) J. Offer, P. E. Dawson, Org. Lett. 2000, 2, 23.
| Crossref | GoogleScholarGoogle Scholar |
(c) J. Offer, C. N. C. Boddy, P. E. Dawson, J. Am. Chem. Soc. 2002, 124, 4642.
| Crossref | GoogleScholarGoogle Scholar |
(d) A. Brik, Y. Y. Yang, S. Ficht, C. H. Wong, J. Am. Chem. Soc. 2006, 128, 5626.
| Crossref | GoogleScholarGoogle Scholar |
(e) S. Ficht, R. J. Payne, A. Brik, C. H. Wong, Angew. Chem. Int. Ed. 2007, 46, 5975.
| Crossref | GoogleScholarGoogle Scholar |
(f) M. Y. Lutsky, N. Nepomniaschiy, A. Brik, Chem. Commun. 2008, 1229.
| Crossref | GoogleScholarGoogle Scholar |
(g) J. Offer, Biopolymers 2010, 94, 530.
| Crossref | GoogleScholarGoogle Scholar |
[11] (a) S. Aimoto, Biopolymers 1999, 51, 247.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXislChtA%3D%3D&md5=a5601441f5e9d1946177df3596d5740cCAS | 10618594PubMed |
(b) G. Chen, Q. Wan, Z. Tan, C. Kan, Z. Hua, K. Ranganathan, S. J. Danishefsky, Angew. Chem. Int. Ed. 2007, 119, 7527.
| Crossref | GoogleScholarGoogle Scholar |
(c) R. J. Payne, S. Ficht, W. A. Greenberg, C. H. Wong, Angew. Chem. Int. Ed. 2008, 47, 4411.
| Crossref | GoogleScholarGoogle Scholar |
(d) G. L. Thomas, R. J. Payne, Chem. Commun. 2009, 4260.
| Crossref | GoogleScholarGoogle Scholar |
[12] (a) H. Rohde, O. Seitz, Biopolymers 2010, 94, 551.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXosF2htLw%3D&md5=f5c9533cec9135f6f678d7ca4dac3c8aCAS | 20593472PubMed |
(b) P. E. Dawson, Isr. J. Chem. 2011, 51, 862.
| Crossref | GoogleScholarGoogle Scholar |
[13] L. Z. Yan, P. E. Dawson, J. Am. Chem. Soc. 2001, 123, 526.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXms1Sr&md5=46e3d6c1ef72c133f90f76fbb7248052CAS | 11456564PubMed |
[14] J. P. Tam, Q. Yu, Biopolymers 1998, 46, 319.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXmtlCltr0%3D&md5=6514875579bd40512aa53c25f5285680CAS | 9754028PubMed |
[15] Q. Wan, S. J. Danishefsky, Angew. Chem. Int. Ed. 2007, 46, 9248.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXitFWhtA%3D%3D&md5=adf53dfd5b125cf0866238d846c42ce7CAS |
[16] C. T. T. Wong, C. L. Tung, X. Li, Mol. Biosyst. 2013, 9, 826.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXltVOls7c%3D&md5=d1a0a1f95f42fcadbda23daaa5fcd0ffCAS |
[17] C. Haase, H. Rohde, O. Seitz, Angew. Chem. Int. Ed. 2008, 47, 6807.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtV2iurvJ&md5=3605ed19f72c70d57c21508d03f7e124CAS |
[18] S. Y. Shang, Z. P. Tan, S. W. Dong, S. J. Danishefsky, J. Am. Chem. Soc. 2011, 133, 10784.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXnvFagtrs%3D&md5=8b168bec24ccf6df3b4ccf08c0efb241CAS |
[19] A. Isidro-Llobet, M. Alvarez, F. Albericio, Chem. Rev. 2009, 109, 2455.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXksVSlur4%3D&md5=6fe711d8630c52fe5cef2654c4961a36CAS | 19364121PubMed |
[20] (a) D. Crich, A. Banerjee, J. Am. Chem. Soc. 2007, 129, 10064.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXnvFGntrg%3D&md5=5444367c211ed923ddc36d3345593dd2CAS | 17658806PubMed |
(b) P. Botti, S. Tchertchian, WO/2006/133962 2006.
[21] J. Chen, Q. Wan, Y. Yuan, J. L. Zhu, S. J. Danishefsky, Angew. Chem. Int. Ed. 2008, 47, 8521.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtlKlu77J&md5=9c8ee9d09ce408d57443c73c1ffd6947CAS |
[22] (a) R. L. Yang, K. K. Pasunooti, F. P. Li, X. W. Liu, C. F. Liu, J. Am. Chem. Soc. 2009, 131, 13592.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtV2jsbfO&md5=87bbf699a6a5cf1210cda49666ee4824CAS |
(b) R. Merkx, G. de Bruin, A. Kruithof, T. van den Bergh, E. Snip, M. Lutz, F. El Oualid, H. Ovaa, Chem. Sci. 2013, 4, 4494.
| Crossref | GoogleScholarGoogle Scholar |
[23] (a) K. S. Ajish Kumar, M. Haj-Yahya, D. Olschewski, H. A. Lashuel, A. Brik, Angew. Chem. Int. Ed. 2009, 48, 8090.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXht1Kju77N&md5=a8a9d8b0aa59cd35e114970f06c9698eCAS |
(b) F. El Oualid, R. Merkx, R. Ekkebus, D. S. Hameed, J. J. Smit, A. de Jong, H. Hilkmann, T. K. Sixma, H. Ovaa, Angew. Chem. Int. Ed. 2010, 49, 10149.
| Crossref | GoogleScholarGoogle Scholar |
[24] J. Chen, P. Wang, J. L. Zhu, Q. Wan, S. J. Danishefsky, Tetrahedron 2010, 66, 2277.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXjtVWjt70%3D&md5=7cff71540b805cef481c2d0ee3400b99CAS | 20798898PubMed |
[25] (a) Z. P. Tan, S. Y. Shang, S. J. Danishefsky, Angew. Chem. Int. Ed. 2010, 49, 9500.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsFSls7fI&md5=34b039012c47e4f8e6319c57b57ddcb1CAS |
(b) Z. Harpaz, P. Siman, K. S. A. Kumar, A. Brik, ChemBioChem 2010, 11, 1232.
| Crossref | GoogleScholarGoogle Scholar |
[26] H. Ding, A. Shigenaga, K. Sato, K. Morishita, A. Otaka, Org. Lett. 2011, 13, 5588.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtFOmsbrF&md5=4cfca8a5fe5e39904b052044fd57da88CAS | 21916452PubMed |
[27] P. Siman, S. V. Karthikeyan, A. Brik, Org. Lett. 2012, 14, 1520.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XivVOgsbc%3D&md5=1a419225f04dd81113beb474e497a6c5CAS | 22360701PubMed |
[28] (a) C. J. Easton, C. A. Hutton, E. W. Tan, E. R. T. Tiekink, Tetrahedron Lett. 1990, 31, 7059.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3MXhsVeqtLs%3D&md5=61b360505fe1e336649c4ea5d696e341CAS |
(b) C. J. Easton, C. A. Hutton, P. D. Roselt, E. R. T. Tiekink, Tetrahedron 1994, 50, 7327.
| Crossref | GoogleScholarGoogle Scholar |
[29] D. Crich, A. Banerjee, J. Org. Chem. 2006, 71, 7106.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XnvVCltbo%3D&md5=515c907e7f6da60887b39cf5e27ac1d6CAS | 16930077PubMed |
[30] J. Marin, C. Didierjean, A. Aubry, J. R. Casimir, J. P. Briand, G. Guichard, J. Org. Chem. 2004, 69, 130.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXps1ymsLw%3D&md5=34715d571ac12334bfbb583dbe641a11CAS | 14703388PubMed |
[31] J. M. Padrón, G. Kokotos, T. Martín, T. Markidis, W. A. Gibbons, V. S. Martín, Tetrahedron Asymmetry 1998, 9, 3381.
| Crossref | GoogleScholarGoogle Scholar |
[32] B. M. Syed, T. Gustafsson, J. Kihlberg, Tetrahedron 2004, 60, 5571.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXks1Kisbk%3D&md5=feee8c852a19f7d9bb3208dbeb3dc2eaCAS |
[33] (a) S. D. Townsend, Z. Tan, S. Dong, S. Shang, J. A. Brailsford, S. J. Danishefsky, J. Am. Chem. Soc. 2012, 134, 3912.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XitlGjtb8%3D&md5=ea83174d7f254f1914e5f0b136816849CAS | 22332757PubMed |
(b) S. Dong, S. Shang, Z. Tan, S. J. Danishefsky, Isr. J. Chem. 2011, 51, 968.
| Crossref | GoogleScholarGoogle Scholar |
[34] P. Garner, Tetrahedron Lett. 1984, 25, 5855.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2MXhsV2ksbk%3D&md5=ec56fe13dcd4f3f040d8e6d74f349a24CAS |
[35] L. R. Malins, R. J. Payne, Org. Lett. 2012, 14, 3142.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XnsF2gurg%3D&md5=a7b81fcf6babcd97b4b7b4ad1c4544cfCAS | 22642500PubMed |
[36] N. Metanis, E. Keinan, P. E. Dawson, Angew. Chem. Int. Ed. 2010, 49, 7049.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtF2lurbM&md5=d0eefee0fc390468e8e316792410bfa7CAS |
[37] R. E. Thompson, B. Chan, L. Radom, K. A. Jolliffe, R. J. Payne, Angew. Chem. Int. Ed. 2013, 52, 9723.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXhtFOitLnI&md5=7cf80f30b0b4c00e4d11e42b5edd106cCAS |
[38] N. Shibata, J. E. Baldwin, A. Jacobs, M. E. Wood, Tetrahedron 1996, 52, 12839.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK28XlvFKrsb0%3D&md5=446b1705f6b83c9b98b9f72054920dc0CAS |
[39] X. Guan, M. R. Drake, Z. Tan, Org. Lett. 2013, 15, 6128.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXhvVCmurjN&md5=fc14b9d90365f251a4488e64a733d097CAS | 24266801PubMed |
[40] K. M. Cergol, R. E. Thompson, L. R. Malins, P. Turner, R. J. Payne, Org. Lett. 2014, 16, 290.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXhvVKjtrnI&md5=0383733fe3f10e4a4ec45000b7a0edebCAS | 24294973PubMed |
[41] L. R. Malins, K. M. Cergol, R. J. Payne, Chem. Sci. 2014, 5, 260.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXhvVGntLbO&md5=d2790eee224f07a836ff6e76f910d83dCAS |
[42] (a) E. Scoffone, A. Fontana, R. Rocchi, Biochem. Biophys. Res. Commun. 1966, 25, 170.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaF2sXivFSltw%3D%3D&md5=3d81c1e166fc7025608d8a1a27b449d7CAS | 5971763PubMed |
(b) E. Scoffone, A. Fontana, R. Rocchi, Biochemistry 1968, 7, 971.
| Crossref | GoogleScholarGoogle Scholar |
[43] M. Wilchek, T. Miron, Biochem. Biophys. Res. Commun. 1972, 47, 1015.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE38XksVGhsrc%3D&md5=5b5dd9c8dd00bb982a1a1c0b2170c10bCAS | 5029854PubMed |
[44] K. S. Kumar, S. N. Bavikar, L. Spasser, T. Moyal, S. Ohayon, A. Brik, Angew. Chem. Int. Ed. 2011, 50, 6137.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXnvFWjtL0%3D&md5=ba02f22d258dab9ef349a4d6a655019bCAS |
[45] S. Shang, Z. Tan, S. J. Danishefsky, Proc. Natl. Acad. Sci. USA 2011, 108, 5986.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXltVymt7Y%3D&md5=5a111916031f7b6c2a092a7a9aff8e3bCAS | 21444787PubMed |
[46] P. E. Dawson, M. J. Churchill, M. R. Ghadiri, S. B. H. Kent, J. Am. Chem. Soc. 1997, 119, 4325.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXjsVCjsLw%3D&md5=602dde5f747cd3fdd5f364b5fa803525CAS |
[47] E. C. B. Johnson, S. B. H. Kent, J. Am. Chem. Soc. 2006, 128, 6640.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XjvFyitrc%3D&md5=9cc535c80a1a303bfba008fe67ed8081CAS |
[48] D. Bang, S. B. Kent, Angew. Chem. Int. Ed. 2004, 43, 2534.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXkt1ejtr0%3D&md5=c6e5294c03cef809f5ab6df405605f2fCAS |
[49] T. Moyal, H. P. Hemantha, P. Siman, M. Refua, A. Brik, Chem. Sci. 2013, 4, 2496.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXntVyqtLw%3D&md5=f9a613084c674115d78e0c5a86590fb2CAS |