

Research Front on Coordination Polymers
Christopher J. SumbySchool of Chemistry and Physics, University of Adelaide, Adelaide, SA 5005, Australia. Email: christopher.sumby@adelaide.edu.au

Associate Professor Christopher Sumby is an ARC Future Fellow at the University of Adelaide where he undertakes research into the synthesis, characterization, and properties of nanomaterials. He has been at the University of Adelaide since 2007, after completing a Ph.D. at the University of Canterbury (2003), a post-doctoral position at the University of Leeds (2003–2006), and briefly holding a NZ Science and Technology Fellowship at the University of Otago. Associate Professor Sumby has been awarded various fellowships and awards, including a South Australian Young Tall Poppy Award, 2009. He is Director of the Bragg Crystallography Facility and Deputy Director of the Centre for Advanced Nanomaterials at the University of Adelaide, the latter a University-funded centre where key research themes include: Chemical and Electrical Energy Storage; Energy Waste Management; Heterogeneous Catalysis; and Nanoporous Materials for Gas Separations. |
Australian Journal of Chemistry 66(4) 397-400 https://doi.org/10.1071/CH13052
Published: 15 April 2013
Background and Introduction
Coordination polymers, which are also widely known as metal-organic frameworks (MOFs),[1] are infinite solid-state networks that can be synthesised from combinations of organic links and metals or metal oxide clusters.[2,3] By careful choice of both precursor metal salt and the bridging organic ligand, materials with varying 1-D, 2-D, and 3-D topologies; tailored pore sizes, pore shapes, and internal pore chemistry; and properties can be synthesised.[2,3] Research into the field of coordination polymers and MOFs appears to know no limits. A Web of Science search of both terms as part of the title of published manuscripts (Fig. 1) shows a continual and notable growth over the past 20 years;[4] first of the term ‘coordination polymer’ and then more recently the term ‘metal-organic framework’ or ‘MOF’. This Research Front in Aust. J. Chem. highlights examples of the current state of research in the field of coordination polymer or MOF chemistry and indicates briefly where some of the future challenges and opportunities in the field may lie.
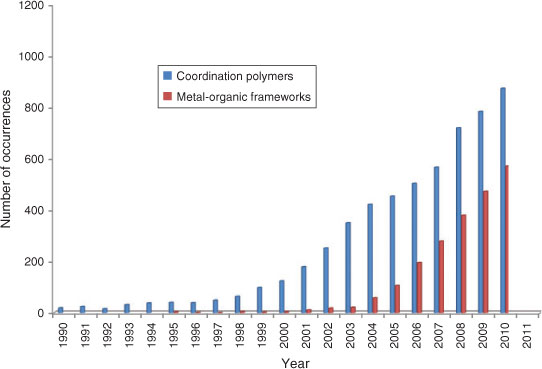
|
Australasia has strong links to the field of coordination polymers with Hoskins and Robson[3a] publishing one of the seminal papers in this area. By combining tetrahedral organic links and tetrahedral CuI metals ions, Hoskins and Robson were able to form a 3-D coordination polymer with a diamond-like cationic framework. This primary contribution outlined a strategy by which judicious choice of both metal ion and organic ligand could lead to the synthesis of coordination polymers with 1-D, 2-D, and 3-D topologies (Scheme 1).[2,3] By modifying the structure metrics of a link or ligand, the pore size and chemistry could be systematically manipulated to generate desirable materials. A flurry of activity in the field has elucidated numerous structurally novel materials from a vast array of metal and ligand combinations, along with the observation of various fundamental phenomenon such as the mechanical interlocking – catenation or interpenetration – of the polymeric structures[2d,5] and the intrinsic physical properties of the frameworks, for example, magnetism and non-linear optics.[6] The appreciation that such materials could be made permanently porous[3c] altered the focus of the field to the properties of coordination polymers that take advantage of the high surfaces areas and large pore volumes; to on-board gas storage[7] initially and hence to separations,[8] catalysis,[9] sensing,[10] and delivery,[11] for example. The identification of a third class of coordination polymers, namely materials with dynamic structures, has added a further focus to the field with the ability to tie changes in the properties of a material to structural changes that occur in the framework.[1b,12]
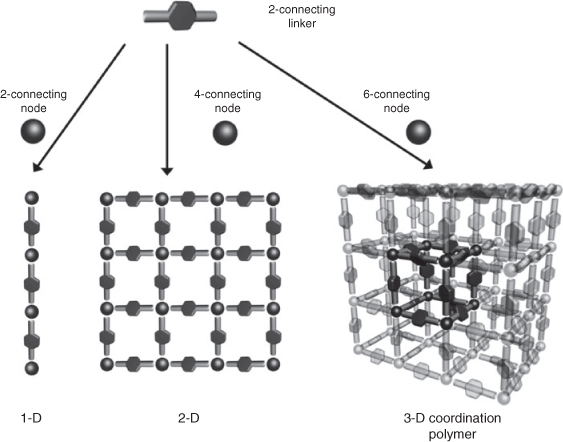
|
As noted, the robust frameworks obtained from combinations of metal ions, or metal-oxide clusters, and organic ligands possess high surfaces areas and large pore volumes that make them suitable for gas storage. Potential for onboard storage of gases such as hydrogen and methane for transportation has been investigated in considerable detail,[7] with control over surface areas and pore volume by extension of the ligands, control of catenation, and generation of open metal sites having been shown to enhance gas uptake.[7a] More recently considerable attention has been directed towards the use of coordination polymers for gas separations, particularly for the separation of CO2 from mixtures of gases that include predominantly methane (natural gas sweetening), hydrogen (pre-combustion CO2 capture), or nitrogen (post-combustion CO2 capture) to counter the increasing levels of CO2 in the atmosphere.[13] Two main strategies have been used to enhance the gas separation performance of MOFs: increasing the affinity of the framework for the gas (e.g. generation of exposed metal sites, post-synthetic metallation, and functionalising pores with Lewis basic sites), or by separating on the basis of different kinetic diameters.[8b,13] Other separations that have been studied included olefin/alkane separations, capture of organosulfur compounds, and enantio-separations.[8a]
The open, crystalline structures of coordination polymers have also led to studies of their potential as catalytic materials, including as shape-selective catalysts, framework catalysts (e.g. Lewis acid catalysis due to structural metal ions), and for heterogenising – embedding – existing homogenous catalysts.[9] Properties of the frameworks such as luminescence, solvatochromism, or vapochromism, and localised Surface Plasmon Resonance have been utilised for sensing,[10] while the large pore volumes have been explored for delivery of pharmaceutical agents.[11] Medicinal applications of coordination polymers themselves have also been explored,[11,14] while their regular crystalline nature makes them the ideal platform to investigate interesting organic reactivity for the linkers or guests.[15]
The ability to unambiguously determine the structure of coordination polymers is essential to the investigation of novel combinations of metal ions and organic ligands and to underpin the exploration of the properties of coordination polymers. In this regard, X-ray crystallography is essential to development of the field. Thus, it is appropriate that 2013 marks 100 years of X-ray crystallography and the first crystal structures reported by William Henry and William Lawrence Bragg.[16] On the 6th December 2012 a symposium was held celebrating the publication of Bragg’s Law and to mark the contributions of William Henry and William Lawrence Bragg to structure determination by X-ray crystallography.[17] Nearly all manuscripts submitted to this particular Research Front contain structures that were determined by X-ray crystallography, underpinning the importance of structure in the investigation of coordination polymers.
Manuscripts in the Research Front
In this Research Front of Aust. J. Chem. a series of papers outline contemporary research in the synthesis and investigation of coordination polymers. In their full paper, Hawes and Kruger[18] report the structural diversity that can be observed when flexible linkers are employed. Their exploration of the coordination chemistry of the bis-bidentate ligand 4,4′-methylenebis(1-(2-pyridyl)-3,5-dimethylpyrazole) unearthed several discrete and polymeric Cuii coordination complexes but also unanticipated reactivity due to the use of solvothermal reaction conditions. For copper(ii) acetate a single stranded unsaturated helical species that forms a highly connected 3-D hydrogen-bonding network was obtained, whereas copper(ii) nitrate provided a coordination polymer derived from [Cu2L] fragments linked together via bridging nitrate anions with undulating 2-D sheets and a (6,3)-topology. By careful control of the solvothermal reaction conditions, three further distinct complexes were obtained which contain a co-ligand formed by either decomposition of the solvent or ligand. These structures were definitively identified by a combination of spectroscopic and structural techniques – including X-ray crystallography – and highlight the need for intimate control and careful adjustment of the reaction conditions (reactant stoichiometry, concentration, and solvothermal reaction temperature) to produce the desired and phase pure products.
As part of a series of papers on silver(i) coordination networks,[19–21] Mak and coworkers report the synthesis of organosilver(i) framework based on underexplored multinuclear heteroaryl ethynide supramolecular synthons RC≡CAgn (n = 4, 5).[19] In their manuscript, Mak and coworkers show by single-crystal X-ray analysis that the position of an ethynide substituent, with respect to the nitrogen donor atom on a quinolinyl or pyridyl nucleus, serves as the dominant factor in directing the assembly of multi-dimensional organosilver(i) networks. Six silver(i) trifluoroacetate complexes of the quinolinyl or pyridyl ethynide ligands were prepared and these show 1-D, 2-D, and 3-D silver-organic networks. These networks were commonly observed to be consolidated by weaker supramolecular interactions.
Steel and Fitchett also report on the positional effects of donors in the synthesis of silver(i) coordination polymers formed from multi-armed pyridylmethyleneoxy ligands.[20] In another contribution to the Research Front that utilises flexible ligands, five isomeric bis(pyridylmethyleneoxy)benzenes, differing in the position of substitution on the benzene and pyridine rings, and three isomeric 1,3,5-tris(pyridylmethyleneoxy)benzenes, differing in the position of substitution on the pyridine ring, were used to form discrete and polymeric silver(i) compounds. The structures of six such complexes were characterised using X-ray crystallography, showing the formation coordination polymers for 3-pyridyl and 4-pyridyl armed ligands and discrete complexes for 2-pyridyl armed ligands.
Biradha and Roy also report a coordination polymer formed from a flexible ligand, 1,3,5-tri(4-cyanophenoxy)benzene and silver(i).[21] In this case the donors are cyano groups as opposed to the pyridyl, quinolyl, or ethynide donors used by Steel and Fitchett[20] or Mak et al.,[19] respectively. Reaction of the flexible tritopic ligand with silver(i) tetrafluoroborate in dichloromethane and different aromatic solvents (benzene and toluene) gave two isostructural 3-D coordination polymers containing channels which are occupied by dichloromethane and the aromatic guest molecules. Based on a particular interest in new luminescent materials for potential application as light-emitting diodes, the solid-state luminescence of the coordination polymer was investigated to reveal similar features to the free ligand.
In a full paper looking to develop MOFs as heterogeneous catalyst platforms, Burgun, Doonan, and Sumby report the first examples of triazolium-containing MOFs whereby the triazolium is integrated into the linker.[22] The choice to integrate the triazolium moiety into the link confers a bridging angle that closely mimics that seen in zeolites, although the work reveals that in a relatively extended ligand the structure can compensate and still generate a framework with a topology that is commonly seen for linear links. Two catenation isomers were isolated from a single reaction of the triazolium link with Cu(NO3)2·3H2O; the α-form, a close-packed three-fold interpenetrated structure, was obtained from reactions undertaken in the presence of nitric acid or at lower temperatures, while undertaking the reaction at higher temperatures leads to a predominance of the two-fold interpenetrated and potentially porous β-form. This work continues to highlight the use of reaction conditions to control interpenetration and provides evidence that charge on structurally similar ligands can drastically alter the types of structures that are accessible due to the requirements for charge balance in the final product.
Neville et al. report the magnetic properties of a family of 3-D coordination polymers based on the three-connecting ligands 2,4,6 tris(3-pyridyl)-1,3,5-triazine (3-tpt) or 2,4,6 tris(4-pyridyl)-1,3,5-triazine (3-tpt).[23] To generate highly cooperative spin crossover (SCO) materials, the authors endeavoured to prepare 3-D networks based around a targeted the [MII(NCS)2(py)4] (MII = Fe, Co, py = 3-tpt and 4-tpt) secondary building unit. By judicious combination of different MII salts and ligands, along with altered crystallisation solvents, a set of 3-D frameworks were obtained, some containing large areas of solvent accessible void volume (~80 %). Despite the presence of metal coordination environments for which SCO has previously been observed, magnetic susceptibilities of this family of materials reveal a high spin nature over the temperature range of the experiments.
Investigations of soft 2-D layered porous coordination polymers synthesised from 1,2-di(4-dipyridyl)ethane, zinc(ii) ions, and bridging dicarboxylates are the focus of a contribution by Horike et al.[24] In their work Horike and coworkers demonstrate that the flexibility of these porous doubly interpenetrated 2-D layer structures is dependent on the substituent group of the dicarboxylate linker. Distinct gating behaviour – opening and closing of the framework during the adsorption and desorption processes – for the 2-D layered porous coordination polymer was observed for CO2, CH4, C2H4, and C2H6 at 195 K and 273 K.
Finally, D’Alessandro and coworkers present a comprehensive investigation of the structures and redox properties of a family of squarate link-based coordination polymers.[25] Structural investigations of the squarate-based frameworks, [MII(C4O4)(H2O)2] (MII = MnII, FeII, CoII, NiII, ZnII, CdII), indicate that all members excepting the CdII analogue exhibit a cubic structure. In the cubic phase the squarate ligands are configured in an ‘eclipsed’ arrangement and are characterised by a positive coefficient of thermal expansion. Meanwhile the CdII analogue exhibits a trigonal structure with a ‘staggered’ orientation of the ligands and displays zero thermal expansion. Unfortunately, ultraviolet-visible-near infrared spectra and electrochemical measurements indicate that electron delocalisation across the dianionic squarate bridge is absent.
Challenges in the Field
In addition to the existing promise for coordination polymers or MOFs in applications as diverse as separations, catalysis, sensing, and delivery, these robust, crystalline frameworks offer substantial opportunity as platforms to allow the exploration of fundamental reaction chemistry. The crystalline nature of coordination polymers, coupled with the vast number of structures and knowledge of synthetic conditions allows the near unrivalled ability to control the organisation of a species in chemical space. Thus, with this intimate control, a MOF can be treated as a matrix in which to observe an organic transformation, to stabilise a reactive catalytic moiety for detailed investigations or to organise a secondary species, for example. The scope of these investigations and the unearthing of new applications for coordination polymers is only limited by our ability to generate and characterise the necessary materials.
References
[1] (a) K. Biradha, A. Ramanan, J. J. Vittal, Cryst. Growth Des. 2009, 9, 2969.| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXmvV2kt7Y%3D&md5=57a6ae6d2431a62be92c6a58d6ba95e6CAS |
(b) S. R. Batten, N. R. Champness, X.-M. Chen, J. Garcia-Martinez, S. Kitagawa, L. Öhrström, M. O’Keeffe, M. P. Suh, J. Reedijk, CrystEngComm 2012, 14, 3001.
| Crossref | GoogleScholarGoogle Scholar |
[2] (a) R. Robson, Dalton Trans. 2008, 5113.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtFCku7%2FJ&md5=ae3bb4dfc6c159b24fdb3da74150acedCAS | 18813362PubMed |
(b) S. Kitagawa, R. Kitaura, S.-I. Noro, Angew. Chem. Int. Ed. 2004, 43, 2334.
| Crossref | GoogleScholarGoogle Scholar |
(c) G. Férey, Chem. Soc. Rev. 2008, 37, 191.
| Crossref | GoogleScholarGoogle Scholar |
(d) S. R. Batten, S. M. Neville, D. R. Turner, Coordination Polymers: Design, Analysis and Application 2009, p. 471 (Royal Society of Chemistry: Cambridge).
(e) N. Stock, S. Biswas, Chem. Rev. 2012, 112, 933.
| Crossref | GoogleScholarGoogle Scholar |
[3] (a) B. F. Hoskins, R. Robson, J. Am. Chem. Soc. 1990, 112, 1546.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3cXovVOjtw%3D%3D&md5=52271add077e4b21cbba9770652a658fCAS |
(b) R. Robson, B. F. Abrahams, S. R. Batten, R. W. Gable, B. F. Hoskins, J. P. Liu, ACS Symp. Ser. 1992, 499, 256.
| Crossref | GoogleScholarGoogle Scholar |
(c) M. Eddaoudi, J. Kim, N. L. Rosi, D. T. Vodak, J. Wachter, M. O’Keeffe, O. M. Yaghi, Science 2002, 295, 469.
| Crossref | GoogleScholarGoogle Scholar |
[4] Source, Thomson ISI, Web of Science. www.webofknowledge.com. (accessed 23 November 2012).
[5] (a) S. R. Batten, R. Robson, Angew. Chem. Int. Ed. 1998, 37, 1460.
| Crossref | GoogleScholarGoogle Scholar |
(b) S. R. Batten, CrystEngComm 2001, 3, 67.
| Crossref | GoogleScholarGoogle Scholar |
[6] (a) M. Kurmoo, Chem. Soc. Rev. 2009, 38, 1353.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXkvVamu7s%3D&md5=773b56724455a9f00d9826c7f5f8cd49CAS | 19384442PubMed |
(b) C. Wang, T. Zhang, W. Lin, Chem. Rev. 2012, 112, 1084.
| Crossref | GoogleScholarGoogle Scholar |
[7] (a) M. P. Suh, H. J. Park, T. K. Prasad, D.-W. Lim, Chem. Rev. 2012, 112, 782.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhs1GlsbjM&md5=37b56dc1e2f07e8d6e3f8468bd533f14CAS | 22191516PubMed |
(b) L. J. Murray, M. Dincă, J. R. Long, Chem. Soc. Rev. 2009, 38, 1294.
| Crossref | GoogleScholarGoogle Scholar |
[8] (a) J.-R. Li, J. Sculley, H.-C. Zhou, Chem. Rev. 2012, 112, 869.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXht1OnsbbL&md5=66e0b0302e11a5248b9aaafb8405e444CAS | 21978134PubMed |
(b) K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae, J. R. Long, Chem. Rev. 2012, 112, 724.
| Crossref | GoogleScholarGoogle Scholar |
[9] (a) A. Corma, H. Garcia, F. X. Llabres, I. Xamena, Chem. Rev. 2010, 110, 4606.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXktFers7Y%3D&md5=595b9c4dd6d0c077547dff1fff30c2d3CAS | 20359232PubMed |
(b) A. U. Czaja, N. Trukhan, U. Müller, Chem. Soc. Rev. 2009, 38, 1284.
| Crossref | GoogleScholarGoogle Scholar |
(c) J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen, J. T. Hupp, Chem. Soc. Rev. 2009, 38, 1450.
| Crossref | GoogleScholarGoogle Scholar |
[10] (a) L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne, J. T. Hupp, Chem. Rev. 2012, 112, 1105.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsVCgtL%2FI&md5=be24127619881736a552e017a480a92cCAS | 22070233PubMed |
(b) Y. Cui, Y. Yue, G. Qian, B. Chen, Chem. Rev. 2012, 112, 1126.
| Crossref | GoogleScholarGoogle Scholar |
[11] P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris, C. Serre, Chem. Rev. 2012, 112, 1232.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhs1SrtbzN&md5=cb465d237b46ba6f4ff900266a858561CAS | 22168547PubMed |
[12] G. Férey, C. Serre, Chem. Soc. Rev. 2009, 38, 1380.
| Crossref | GoogleScholarGoogle Scholar | 19384443PubMed |
[13] D. M. D’Alessandro, B. Smit, J. R. Long, Angew. Chem. Int. Ed. 2010, 49, 6058.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtVaqu7fL&md5=0625fefdadbc1f140edc0ffb6ea4a6dfCAS |
[14] For example, see: O. Gordon, T. Vig, Slenters, P. S. Brunetto, A. E. Villaruz, D. E. Sturdevant, M. Otto, R. Landmann, K. M. Fromm, Antimicrob. Agents Chemother. 2010, 54, 4208.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtlegtr%2FN&md5=6fe34a367b40ed3dfba67f0fc55a4e3dCAS | 20660682PubMed |
[15] (a) Y. Inokuma, N. Kojima, T. Arai, M. Fujita, J. Am. Chem. Soc. 2011, 133, 19691.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsVCgtLnM&md5=1368c34d2d955dab5c9b6e945a1774dfCAS | 22070216PubMed |
(b) Y. Inokuma, G.-H. Ning, M. Fujita, Angew. Chem. Int. Ed. 2012, 51, 2379.
| Crossref | GoogleScholarGoogle Scholar |
(c) A. D. Burrows, S. O. Hunter, M. F. Mahon, C. Richardson, Chem. Commun. 2013, 49, 990.
| Crossref | GoogleScholarGoogle Scholar |
[16] (a) W. L. Bragg, Proc. R. Soc. Lond. 1913, A89, 248.
(b) W. H. Bragg, W. L. Bragg, Nature 1913, 91, 557.
| Crossref | GoogleScholarGoogle Scholar |
(c) W. H. Bragg, W. L. Bragg, Proc. R. Soc. Lond. 1913, A89, 277.
[17] The Bragg Symposium was held at the University of Adelaide on the 6th of December 2012 and explored some of the historical context and personal links to the Braggs' work. In addition, presentations on the broader scientific and social impacts of the Braggs' work were given. See: S. W. Wilkins, Acta Crystallogr. 2013, A69, 1.
| 1:CAS:528:DC%2BC38XhvVGmsLfM&md5=87667a82d5594367db18ea736fedd3e8CAS |
[18] C. S. Hawes, P. E. Kruger, Aust. J. Chem. 2013, 66, 401.
| Crossref | GoogleScholarGoogle Scholar |
[19] P.-S. Cheng, S. C. K. Hau, T. C. W. Mak, Aust. J. Chem. 2013, 66, 419.
| Crossref | GoogleScholarGoogle Scholar |
[20] P. J. Steel, C. M. Fitchett, Aust. J. Chem. 2013, 66, 443.
| Crossref | GoogleScholarGoogle Scholar |
[21] S. Roy, K. Biradha, Aust. J. Chem. 2013, 66, 436.
| Crossref | GoogleScholarGoogle Scholar |
[22] A. Burgun, C. J. Doonan, C. J. Sumby, Aust. J. Chem. 2013, 66, 409.
| Crossref | GoogleScholarGoogle Scholar |
[23] S. M. Neville, G. J. Halder, K. S. Murray, B. Moubaraki, C. J. Kepert, Aust. J. Chem. 2013, 66, 452.
| Crossref | GoogleScholarGoogle Scholar |
[24] K. Kishida, S. Horike, K. Kongpatpanich, S. Kitagawa, Aust. J. Chem. 2013, 66, 464.
| Crossref | GoogleScholarGoogle Scholar |
[25] P. M. Usov, T. D. Keene, D. M. D’Alessandro, Aust. J. Chem. 2013, 66, 429.
| Crossref | GoogleScholarGoogle Scholar |