

Regulation of alternative splicing in Catharanthus roseus in response to methyl jasmonate modulation during development and stress resilience
Aala A. Abulfaraj A * and Abeer Al-Andal B
A * and Abeer Al-Andal B
A
B
Abstract
Catharanthus roseus has various terpenoid indole alkaloids (TIAs) with adaptive mechanisms to withstand both biotic and abiotic stresses. We investigated the effects of methyl jasmonate (MeJA) on alternative splicing (AS) mechanisms in C. roseus to identify differentially expressed alternatively spliced (DAS) genes following MeJA treatment. We found pairs of co-expressed splicing factors (SFs) and DAS genes and potential roles of co-expressed SFs in the maturation of their respective transcripts. Twenty two clusters encompassing 17 MeJA-responsive DAS genes co-expressed with 10 SF genes. DAS genes, C3H62, WRK41, PIL57, NIP21, and EDL6, exhibited co-expression with the SF genes SR34a, DEAD29, SRC33, DEAH10, and DEAD29, respectively. These gene pairs are implicated in plant developmental processes and/or stress responses. We suggest that MeJA activates the expression of genes encoding SFs that are regulated in tandem with their co-expressed DAS genes and MeJA may enhance the regulatory frameworks that control splicing mechanisms, resulting in the generation of specific mRNA isoforms. This triggers the expression of particular DAS gene variants to allow the plant to effectively respond to environmental stimuli and developmental signals. Our study advances our understanding on how MeJA modulates alternative splicing in C. roseus, potentially influencing various aspects of plant physiology and metabolism. It is recommended that future studies focus on validating the functional relationships between the identified SF/DAS gene pairs and their specific roles in plant development and stress responses, and exploring the potential of manipulating these splicing mechanisms to enhance the production of valuable TIAs in C. roseus.
Keywords: abiotic stress, concordant expression, functional annotation, gene isoform, RNA-Seq, serine-arginine-rich, splicing factor, TIA.
Introduction
Catharanthus roseus, commonly referred to as Madagascar periwinkle, is a perennial herbaceous species within the Apocynaceae family, renowned for its biosynthesis of over 130 terpenoid indole alkaloids (TIAs), notably including the prominent anticancer agents vinblastine and vincristine (Van Der Heijden et al. 2004). Recent genetic studies have revealed that C. roseus possesses a diploid genome (2n = 16) with an estimated size of 738 Mb (Nishanth et al. 2018). The plant’s genome contains approximately 33,829 protein-coding genes, with many involved in specialised metabolite in biosynthesis. Transcriptomic analyses have identified numerous genes involved in TIA biosynthesis, including those encoding key enzymes such as strictosidine synthase (STR) and tryptophan decarboxylase (TDC) (Zhu et al. 2015; Nishanth et al. 2018). In addition to its alkaloidal repertoire, C. roseus synthesises a diverse array of phenolic compounds via the phenylpropanoid biosynthetic pathway (Mustafa and Verpoorte 2007), which are integral to activating multifaceted defence mechanisms against both biotic and abiotic stresses (Demkura et al. 2010).
Recent investigations have found additional stress-responsive mechanisms in this species, mediated by phytohormones such as methyl jasmonate (MeJA) and ethylene (Tabatabaeipour et al. 2024). Studies into regulatory networks under abiotic stress conditions have demonstrated that specific transcription factors (TFs) are induced by biotic and abiotic stresses and co-express with other stress-responsive genes (El-Domyati et al. 2017, Bahieldin et al. 2018). Recent research has demonstrated that MeJA treatment significantly alters the expression of genes involved in TIA biosynthesis in C. roseus. For instance, MeJA has been shown to upregulate the expression of ORCA3, a key TF gene that regulates multiple genes in the TIA pathway (Van Der Fits and Memelink 2001). Our understanding of the splicing factors (SFs) and differentially expressed alternatively spliced (DAS) genes that contributes to a plant’s resilience to stress is limited, particularly in how SFs facilitate the generation of subsequent isoforms.
This study aimed to elucidate the effects of MeJA on alternative splicing mechanisms in C. roseus. Our objectives were to: (1) identify DAS genes in C. roseus following MeJA treatment at 6, 12 and 24 days; (2) investigate co-expression patterns between MeJA-induced DAS genes and SFs; (3) analyse the types and functional implications of alternative splicing events triggered by MeJA; and (4) uncover the regulatory mechanisms by which splicing machinery influences DAS gene expression in response to MeJA. By addressing these objectives, we aimed to gain insights into novel gene regulatory mechanisms that could potentially be applied to enhance crop resilience or manipulate metabolite production in plants.
Materials and methods
RNA sequencing and data processing
RNA sequencing datasets were submitted to NCBI and assigned BioProject number PRJNA386850 (https://www.ncbi.nlm.nih.gov/bioproject/), corresponding to a previously documented experiment investigating methyl jasmonate (MeJA) treatment in Catharanthus roseus (Bahieldin et al. 2018). In this study, 4-week-old seedlings were subjected to MeJA (6 μM), with leaf samples harvested at 6, 12, and 24 days post-treatment. Control seedlings received an equivalent volume of dimethyl sulfoxide (DMSO), and leaf samples from both treatments were collected in duplicate at 0, 6, 12, and 24 days after treatment. Total RNA was extracted from these leaf samples and quantified according to established protocols (Hajrah et al. 2017), subsequently dispatched to BGI, China, for high-throughput sequencing.
Raw RNA sequencing reads were aligned to the C. roseus reference genome ASM94934v1 (Kellner et al. 2015) utilising HISAT2 (Hierarchical Indexing for Spliced Transcript Alignment 2, https://github.com/DaehwanKimLab/hisat2) with default settings (Kim et al. 2014). Tophat (ver. 2.0.10) software (https://github.com/DaehwanKimLab/tophat), which relies on HISAT2 for alignment, was used to detect splice junctions in RNA-Seq data. The Trinity package (https://github.com/trinityrnaseq/trinityrnaseq) was used to enhance assembly quality through genome-guided de novo assembly techniques (Grabherr et al. 2011), utilising these alignments and producing a comprehensive transcriptome for subsequent analyses. Aligned reads underwent processing with RSEM package (https://github.com/deweylab/RSEM) for estimating expression levels for individual isoforms, while FPKM (fragments per kilobase of transcript per million mapped reads) values and differentially expressed genes were computed using Cuffdiff (ver. 2.2.1, https://github.com/cole-trapnell-lab/cufflinks) with default settings (Trapnell et al. 2010; Trapnell et al. 2013). The differential expression of isoforms was assessed using edgeR (Robinson et al. 2010), incorporating the normalised expression levels from RSEM (Li and Dewey 2011) to discern differentially expressed isoforms among experimental conditions.
Analysis of differentially expressed alternatively spliced (DAS) genes
Following differential expression profiling, a focused analysis was performed on DAS genes that exhibited robust and informative expression patterns, harboured multiple regulated isoforms, and potentially contributed to the plant’s response to exogenous MeJA treatment. This analysis also included an exploration of their potential co-expression with splicing factors (SFs). Clusters of DAS/SF gene pairs subjected to rigorous quality control were scrutinised for potential investigation.
Functional annotation and visualisation
The selected differentially expressed isoforms were annotated using the Trinotate package (https://github.com/trinotate/trinotate.github.io) (Lee and Whittaker 2017), which integrates various protein databases and tools such as BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi), Protein Families Database (Pfam, https://pfam.xfam.org/), and Conserved Domain Database (CDD, https://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml) to assign putative functions to the isoforms. The JavaScript-based genome browser JBrowse (https://jbrowse.org/) was employed to visualise alternative splicing events in the selected gene isoforms regulated by MeJA treatment (Skinner et al. 2009). Functional annotation of isoforms exhibiting consistent and informative expression patterns was conducted using InterProScan5, facilitating the identification of protein domains, families, and functional sites associated with the expressed isoforms (Jones et al. 2014). The CDD and Pfam were utilised to identify conserved and functional domains within the isoforms of the selected DAS and SF genes. Results from functional annotation and isoform alignments were visualised with Jalview (https://www.jalview.org/) (Waterhouse et al. 2009), providing a comprehensive overview of multiple sequence alignments and the corresponding functional and conserved domains, thereby aiding in the interpretation of isoform structure and function.
Results
Overview of transcriptomic analysis
For a cluster analysis of transcriptomic datasets from MeJA-treated at 6, 12, and 24 days after treatment, as well as the untreated control plantlets, see Supplementary Table S1 and Fig. S1. Of 1075 clusters, 216 displayed consistent and informative expression patterns (Table 1). Hierarchical clustering heatmaps were generated for the transcriptomic data of C. roseus plantlets treated and untreated with MeJA (6 μM) at the different time points (Fig. 1 and Fig. S2). The 1D heatmap illustrated the expression levels of individual transcripts, while the 2D heatmap provided a pairwise comparison of overall expression levels across different datasets. Fig. 1 shows that transcriptomes of MeJA-treated and untreated plantlets were closely related at the 12-day and at the 24-day time points. In contrast, transcriptomes at the 6-day time point were completely distinct, indicating that MeJA had a significant impact on transcript levels early in the treatment. This effect of MeJA diminished over time, as evidenced by reduced differential expression between treated and untreated plantlets at 12 and 24 days. The 2D heatmap corroborated these findings, showing high differential expression at the 6-day time point and reduced differential expression at 12 and 24 days (Fig. S2).
| Pattern | Cluster | ||
|---|---|---|---|
| 1 | C0 ↑ | 103, 122, 126, 127, 131, 135, 13, 177, 158, 176, 182, 193, 200, 212, 215, 237, 241, 244, 276, 308, 309, 374, 469, 514, 51, 521, 523, 550, 552, 558, 563, 590, 607, 612, 614, 61, 653, 660, 671, 68, 690, 69, 731, 747, 76, 81, 823, 849, 926, 98, 99 | |
| 2 | C0 ↓ | 109, 124, 179, 198, 224, 269, 275, 322, 336, 367, 380, 392, 399, 400, 443, 447, 489, 493, 555, 56, 5, 600, 611, 640, 644, 699, 6, 733, 78, 82, 91, 927, 931 | |
| 3 | C6 ↑ | 529, 776 | |
| 4 | C6 ↓ | 889 | |
| 5 | C12 ↑ | 138, 171 | |
| 6 | C12 ↓ | 363 | |
| 7 | C24 ↑ | 157, 286, 436 | |
| 8 | C0/6 ↑ | 46, 731 | |
| 9 | C0/6 ↓ | 755 | |
| 10 | C0/24 ↑ | 227, 246, 249, 329, 34, 48, 502, 509, 519, 594, 604, 871 | |
| 11 | C0/24 ↓ | 140, 332, 530, 578, 57, 605, 650 | |
| 12 | 12 ↑ | 10, 11, 123, 12, 163, 184, 190, 196, 205, 20, 22, 341, 366, 36, 80, 897 | |
| 13 | 12 ↓ | 263 | |
| 14 | 24 ↑ | 1064, 110, 125, 147, 216, 243, 251, 258, 264, 334, 402, 421, 459, 480, 488, 630, 636, 813, 846, 883, 938, 955, 977 | |
| 15 | 24 ↓ | 1004, 1007, 189, 266, 324, 384, 449, 598, 746, 766, 821, 838, 83, 941 | |
| 16 | GRDL ↑ | 697 | |
| 17 | M ↑ | 1011, 1060, 132, 156, 209, 253, 267, 268, 272, 28, 335, 441, 460, 474, 499, 500, 507, 513, 571, 589, 621, 627, 715, 73, 789, 832, 833, 891, 901, 967, 971 | |
| 18 | 6M ↑ | 295, 750, 819, 862 | |
| 19 | 6/12M ↑ | 587 | |
| 20 | 12M ↑ | 603 | |
| 21 | 24M ↑ | 174, 194, 204, 210, 349, 742, 949 | |
| 22 | 24M ↓ | 181, 496 | |
Rows highlighted in pink correspond to MeJA-dependent expression patterns; other rows correspond to time-dependent expression patterns. C, control; M = MeJA treatment; ↑, upregulation; ↓, downregulation; GRDL, gradual. Further details are in Table S1.
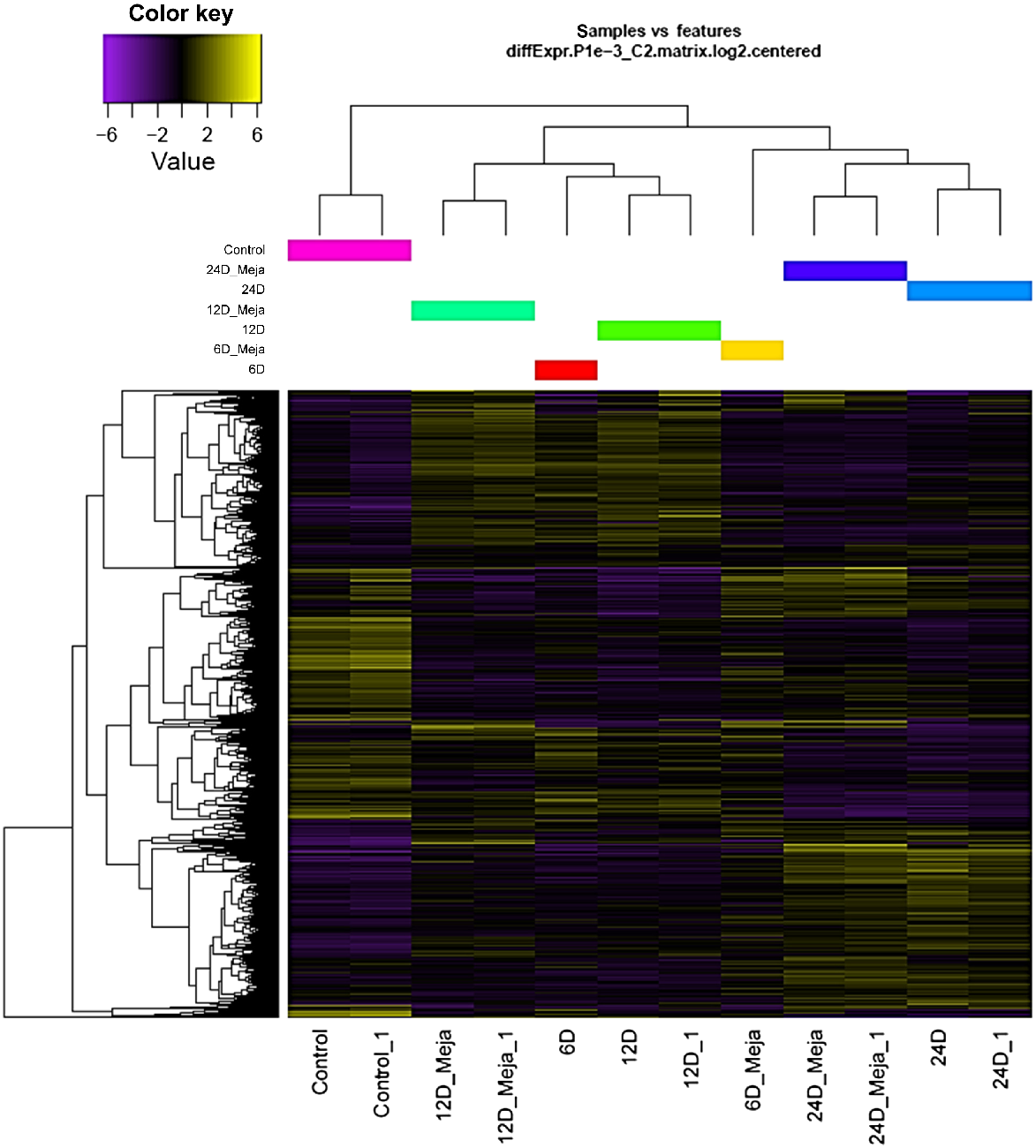
Identification of consistent expression patterns
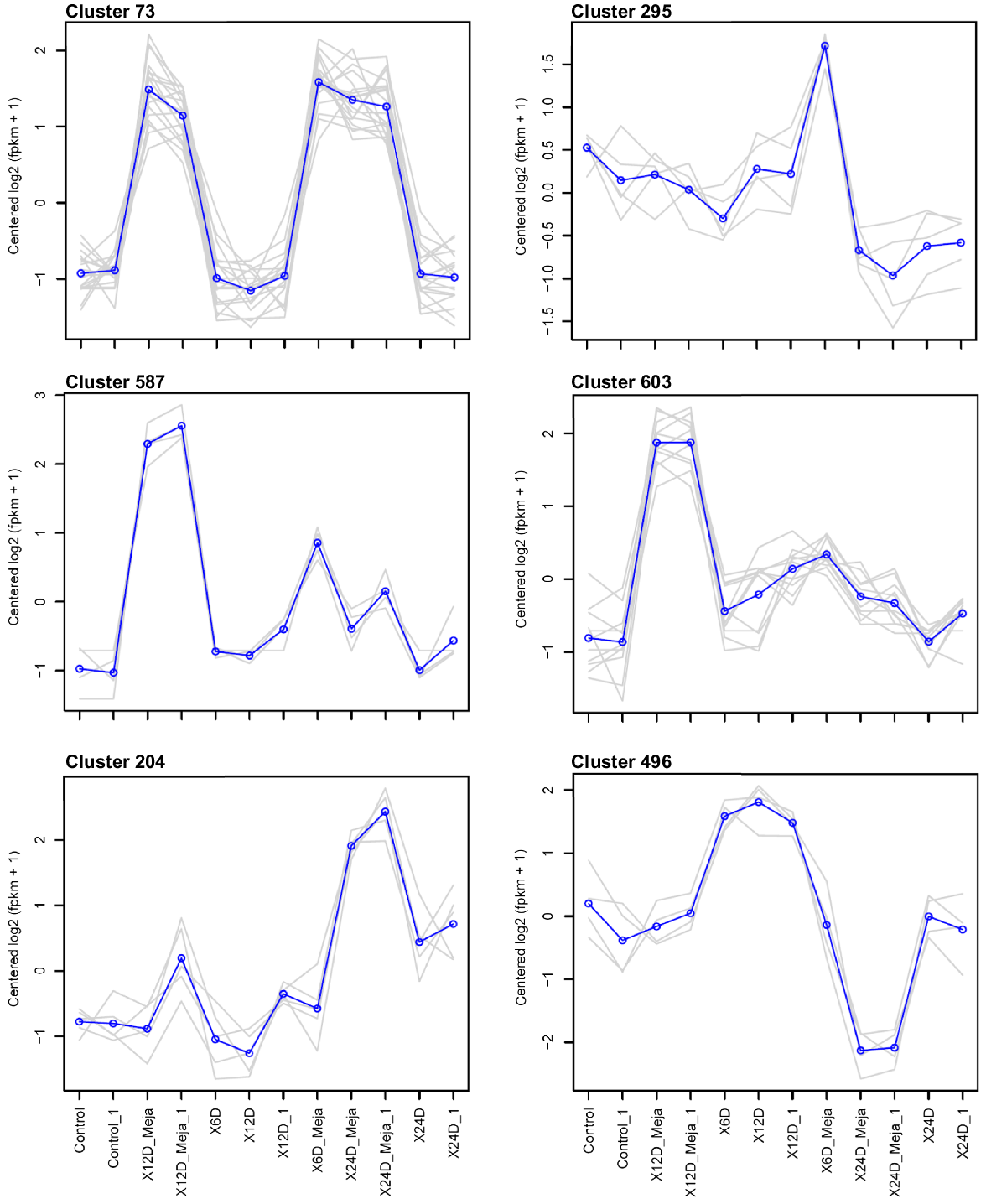
Table 1 shows 22 consistent and informative expression patterns generated from the 216 clusters. Of these, 16 expression patterns displayed time-dependent regulation, while six were specifically associated with MeJA-induced transcripts. Models depicting these expression patterns, which encompass both MeJA-dependent and -independent responses (Figs 2 and S3). The selection criteria for these clusters included meaningful expression changes (either upregulation or downregulation) at one or more time points, and consistent expression patterns across replicates at the same time point within each cluster. Among the six MeJA-dependent expression patterns, one showed upregulated transcripts across all three time points (Fig. 3), while four others displayed upregulation at 6, 12, and 24 days (Figs 4–6, respectively). The sixth pattern involved downregulated transcripts at the 24-day time point of MeJA treatment. Interestingly, clusters corresponding to this downregulated pattern (181 and 496) lacked DAS/SF concordance and were thus not pursued further in the analysis.
Six models of clusters showing the most informative MeJA-dependent expression patterns constructed using transcriptome data from 4-week-old Catharanthus roseus seedlings exposed to MeJA (6 μM) for 6, 12, and 24 days. M, MeJA; ↑, upregulation; ↓, downregulation; Cluster 73, M ↑; Cluster 295, 6M ↑; Cluster 587, 6/12M ↑; Cluster 603, 12M ↑; Cluster 204, 24M ↑; Cluster 496, 24M ↓. Note: Cluster 496 includes no DAS/SF concordant expression. More details are in Tables 1 and S1.
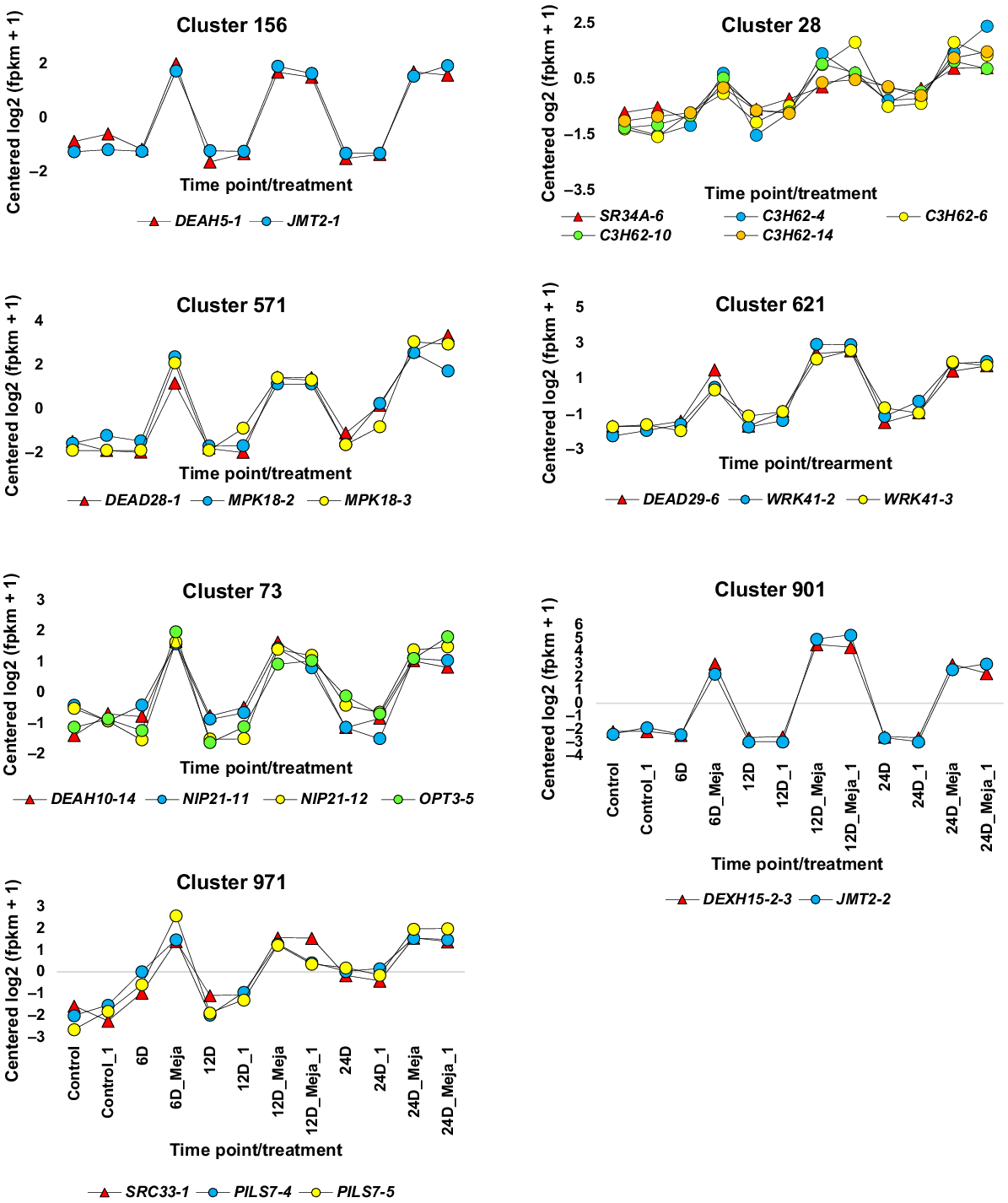
Expression profiling of differentially expressed alternatively spliced (DAS) genes that have two or more isoforms containing active and/or conserved domains and are concordantly expressed with genes encoding splicing factors (SFs). These genes were upregulated at three time points (6, 12, and 24 days) of MeJA (6 μM) treatment in the leaves of 4-week-old Catharanthus roseus seedlings. These seedlings were compared to the control untreated seedlings harvested at 0, 6, 12, and 24 days. Isoforms of these genes exist in clusters that represent the most informative MeJA-dependent expression patterns. The genes encoding the SFs DEAD5-1 (Cluster 156) and SRC33-1 (Cluster 971) each have only one regulated isoform, so they did not undergo the comparative protein domain analysis. Additionally, the SFs SR45A is concordantly expressed with the DAS gene isoform BRM-25-2, which lacks conserved or active protein domains, leading to no further analysis of this DAS/SF gene pair. More details are in Figs S1, S6–S16, and Tables 2, S1, S3, S4.
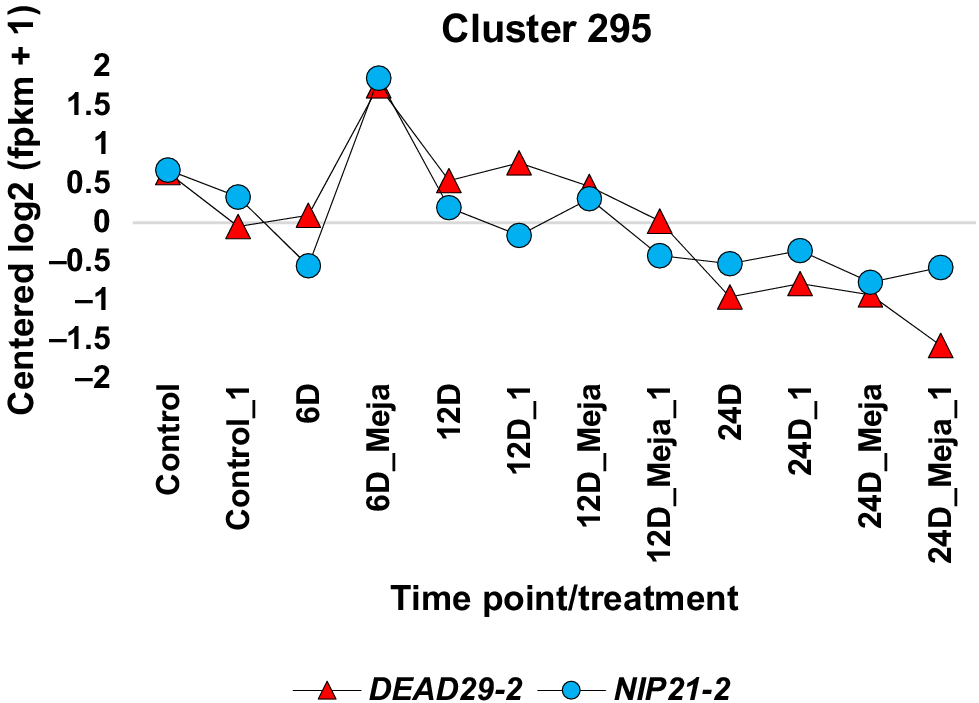
Expression profiling of a differentially expressed alternatively spliced (DAS) gene that has two or more isoforms containing active and/or conserved domains and is concordantly expressed with a gene encoding a splicing factor (SF). These genes were upregulated at 6-day time point of MeJA (6 μM) treatment in the leaves of 4-week-old Catharanthus roseus seedlings. The analysis also included seedlings harvested at 6, 12, and 24 days, with results compared to those from control untreated seedlings collected at 0, 6, 12, and 24 days. This DAS/SF gene pair exist in a cluster that represents one of the most informative MeJA-dependent expression patterns. More details are in Figs S1, S6–S16, and Tables 2, S1, S3, S4.
Expression profiling of differentially expressed alternatively spliced (DAS) genes that have two or more isoforms containing active and/or conserved domains and are concordantly expressed with a gene encoding a splicing factor (SF). These genes were upregulated at 12-day time point of MeJA (6 μM) treatment in the leaves of 4-week-old Catharanthus roseus seedlings. The analysis also included seedlings harvested at 6, 12, and 24 days, with results compared to those from control untreated seedlings collected at 0, 6, 12, and 24 days. These DAS/SF gene pairs exist in a cluster that represents one of the most informative MeJA-dependent expression patterns. More details are in Figs S1, S6–S16, and Tables 2, S1, S3, S4.
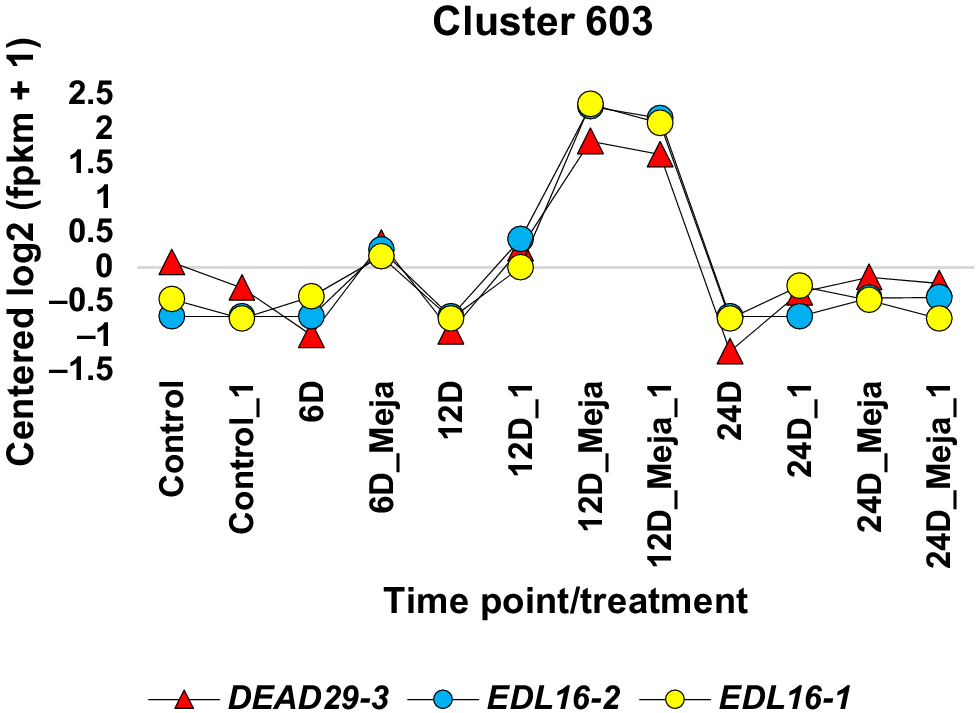
Expression profiling of a differentially expressed alternatively spliced (DAS) gene that has two or more isoforms containing active and/or conserved domains and is concordantly expressed with a gene encoding a splicing factor (SF). These genes were upregulated at 24-day time point of MeJA (6 μM) treatment in the leaves of 4-week-old Catharanthus roseus seedlings. The analysis also included seedlings harvested at 6, 12, and 24 days, with results compared to those from control untreated seedlings collected at 0, 6, 12, and 24 days. This DAS/SF gene pair exist in a cluster that represents one of the most informative MeJA-dependent expression patterns. More details are in Figs S1, S6–S16, and Tables 2, S1, S3, S4.
Selection of DAS genes and splicing factors (SFs)
Clusters corresponding to the five MeJA-dependent expression patterns were extensively analysed to identify DAS genes and SF genes with concordant expression. The primary criterion for selecting candidate DAS genes for further examination was the presence of multiple isoforms for each DAS gene within one or more clusters associated with MeJA-dependent expression profiles. DAS genes that met these criteria are detailed in Table S2. Among these, we focused on DAS genes exhibiting at least two isoforms with consistent and informative expression profiles. Additional criteria for selection included the co-expression of DAS genes with one or more SF genes and the relevance of their functions to MeJA treatment. DAS genes that did not fulfil these criteria were excluded from further analysis. This method resulted in the identification of 17 MeJA-dependent DAS genes that were consistently expressed in conjunction with ten SF genes across 23 clusters (Figs 3–6 and Table S1).
Characterisation of SF genes
Remarkably, seven out of the 10 SF genes exhibited a minimum of two isoforms, each characterised by consistent and informative expression profiles. These SF genes include SR34a (Serine/arginine-rich), DExH15 (DExH-box ATP-dependent RNA helicase), DEAH10 (Pre-mRNA-SF ATP-dependent RNA helicase), DEAD29 (DEAD-box ATP-dependent RNA helicase 29), CRS1 (Chloroplastic group IIA intron splicing facilitator), DExH12 (DExH-box ATP-dependent RNA helicase), and DEAD28 (DEAD-box ATP-dependent RNA helicase). These were designated as SF1, SF2, SF3, SF4, SF7, SF9 and SF10, respectively (Table S2). In contrast, the genes that did not exhibit regulated isoforms include DEAH5 (pre-mRNA-SF ATP-dependent RNA helicase), SR45A (Serine/arginine-rich), and SRC33 (Serine/arginine-rich SC35-like), which were assigned as SF5, SF6 and SF8, respectively. The regulated isoforms of DAS and SF genes are in Figs S4 and S5.
Co-expression analysis and gene pair selection
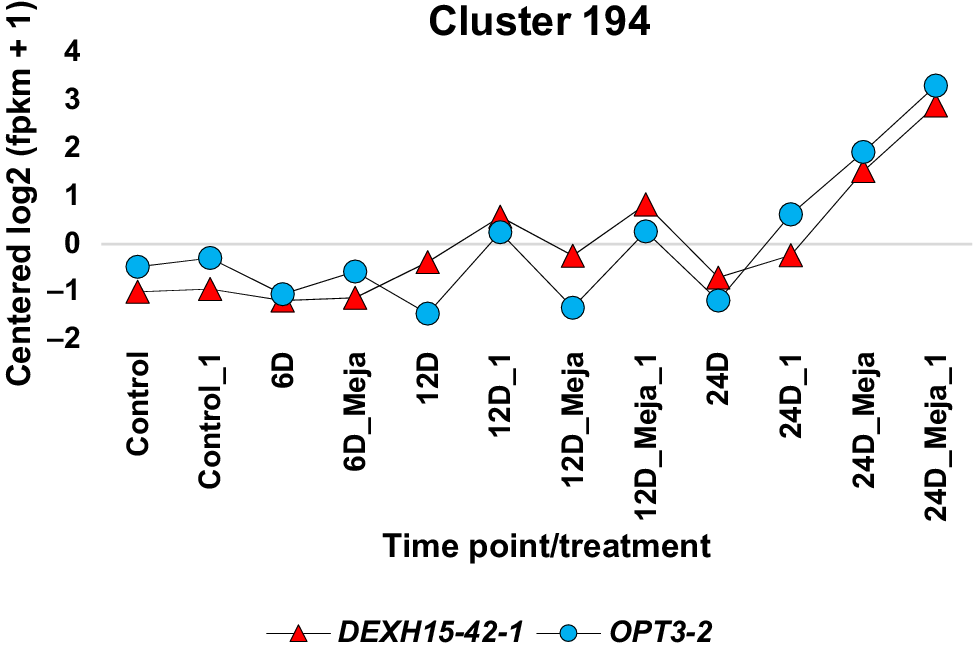
Through comprehensive multiple sequence alignments visualised with Jalview, along with functional annotations derived from the Conserved Domain Database (CDD) and Protein Families Database (Pfam) signature databases, the subset of DAS genes regulated by MeJA was refined to 12. These selected DAS genes demonstrated either conserved or functional domains across their isoforms (Figs S6–S17 and Table S3). The DAS genes omitted from further scrutiny include DAS genes 2, 3, 8, 11 and 17 (Table S2). A model visualisation for DAS gene isoforms lacking conserved or functional domains, exemplified by DAS gene 2, is in Fig. S18. Among the seven SF genes exhibiting MeJA-regulated isoforms, five contained conserved or functional domains, warranting further investigation (Figs S19–S23 and Table S4). The SF genes excluded from subsequent analysis include CRS1 and DExH12. The concordant expression of isoforms for the selected DAS and SF genes intended for further exploration is illustrated in Figs 3–6, while the excluded DAS/SF gene pairs are represented in Figs S24–S26. It is worth noting that the exclusion of a specific DAS gene, such as BRM, typically leads to the corresponding exclusion of its co-expressed SF gene, such as SR45A (Fig. S24), or vice versa, for example, the SF gene CRS1 along with its co-expressed DAS genes KEA3 and LPA1 (Fig. S24), as well as DExH12 and its co-expressed DAS genes SPD1 and PP255 (Fig. S24), unless a consequently excluded DAS or SF gene is co-expressed with other selected genes or isoforms, as seen with one isoform of the DAS gene OPT3 co-expressing with an isoform of the SF gene DExH15, thus analysed further (Fig. 6). Additionally, there are instances where isoforms of a DAS gene co-express with two distinct SFs located in separate clusters. A notable example is the isoforms of the DAS gene JMT2, where one isoform co-expresses with the SF gene DEAH5 from cluster 156, while another isoform co-expresses with the SF gene DExH15 from cluster 901 (Fig. 3). Both clusters demonstrate a similar expression pattern, exhibiting upregulation at all three time points following MeJA treatment.
Final selection of DAS and SF genes for alternative splicing analysis
Based on functional annotation outcomes, only eight DAS genes and five SF genes advanced to the analysis of alternative splicing events. The eight DAS genes were JMT2 (Fig. S6), OPT3 (Fig. S8), NIP21 (Fig. S9), C3H62 (Fig. S10), WRK41 (Fig. S11), ELD16 (Fig. S12), MPK18 (Fig. S13), and PILS7 (Fig. S14). The five SF genes were SR34a (Fig. S19), DExH15 (Fig. S20), DEAH10 (Fig. S21), DEAD29 (Fig. S22), and DEAD28 (Fig. S23). The two SF genes DEAD5 and SRC33 showed no isoforms; hence, they were further investigated without the necessity for detecting functional or conserved domains or analysing alternative splicing events. The concordantly expressed pairs of selected DAS and SF genes (Figs 3–6 and Table 2) were investigated for their shared functional roles. This revealed 11 concordantly expressed SF/DAS gene pairs for further analysis (Table 2).
| DAS gene | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| JMT2 | C3H62 | MPK18 | WRK41 | NIP21 B | EDL16 | OPT3 | PIL57 | |||
| SF gene | DEAH5 A | |||||||||
| SR34a | ||||||||||
| DEAD28 A | ||||||||||
| DEAD29 | ||||||||||
| DEAH10 B | ||||||||||
| SRC33 | ||||||||||
| DExH15 | ||||||||||
These SF/DAS gene pairs are grouped into consistent and informative clusters corresponding to the MeJA-dependent expression patterns. The genes were either upregulated at all three time points (6, 12 and 24 days) or at individual time points (6, 12, and 24 days) following MeJA (6 μM) treatment in the leaves of 4-week-old Catharanthus roseus seedlings. The four expression patterns are illustrated in red, blue, yellow, and green boxes. MeJA-treated seedlings were compared to control untreated seedlings collected at 0, 6, 12, and 24 days. The seven splicing factors are classified into two families that play a role in RNA splicing: (1) the serine/arginine-rich (SR) protein family; and (2) the RNA helicase family (which encompasses DEAD/DEAH box proteins). Pink boxes indicate MeJA-regulated SF/DAS genes that show functional alignment and possible connections.
Analysis of alternative splicing events
An investigation into the types of alternative splicing for the isoforms of the SF and DAS genes revealed a predominance of alternative promoter usage (APU) with ten events, followed by alternative 5′ donor site (ADS) and alternative 3′ acceptor site (AAS), both with eight events each, alternative polyadenylation (APA) with seven events, and exon skipping (ES) with four events.
Discussion
The scope of alternative splicing beyond coding regions
The alternative splicing (AS) machinery extends its influence beyond conventional coding regions, occasionally targeting non-coding sequences while maintaining similarities in the encoded protein sequences (Tian and Manley 2017). This phenomenon can lead to the production of identical proteins expressed in disparate temporal or spatial contexts. Recent findings have illuminated the presence of multiple alternative polyadenylation (APA) sites within the 3′ untranslated region (UTR) (Tian and Manley 2017). For instance, the FGFR2 (Fibroblast Growth Factor Receptor 2) gene, pivotal in development and oncogenesis, exemplifies how AS operates within non-coding regions beyond the standard exon-intron paradigm. The 3′ UTR of FGFR2 encompasses APA sites that dictate the inclusion or exclusion of specific sequences in mature mRNA, thereby influencing the transcript’s stability, localisation, and translational efficiency, ultimately impacting FGFR2 expression and its oncogenic roles (Zingg et al. 2022). Another salient example is the ELAV (Embryonic Lethal Abnormal Visual System) gene in Drosophila, crucial for nervous system development, underscoring how AS within non-coding regions can regulate gene expression in context-specific cellular environments (Zhang et al. 2023).
AS can also target alternative promoters for genes featuring multiple enhancer or promoter regions, permitting the inclusion or exclusion of exons at the 5′ end. This phenomenon has been documented in the Bicoid (Bcd) gene hunchback during Drosophila embryogenesis (Wang et al. 2022). Throughout transcription, promoters interact with specific cis-regulatory elements termed enhancers (Haberle and Stark 2018). Eukaryote genomes harbour alternative promoters that collaborate with particular enhancers to yield diverse isoforms, each possessing distinct functions that influence cellular fate during development (Lu et al. 2020). Under MeJA treatment, specific promoters or enhancers/suppressors may be preferentially utilised, resulting in gene variants more aptly aligned with the plant’s responses to stress or developmental cues. Given MeJA’s involvement in stress signalling, alternative promoter usage affords plants the opportunity to modulate their developmental stages and fine-tune responses to environmental challenges, such as pathogen attacks or abiotic stress. By regulating alternative promoter usage, MeJA can augment protein isoform diversity, which may carry varied functions and regulatory roles in the plant.
Notably, the alternative splicing types known as mutually exclusive exons and intron retention were absent in the MeJA-regulated DAS or SF genes. This aligns with the earlier hypothesis that intron retention in plants mostly occurs when transcripts are subjected to environmental stress (Gao et al. 2019).
Validation of potentially shared functional roles in concordantly expressed SF/DAS gene pairs
In this study, seven SF genes belonging to two distinct families were selected for further analysis based on previous criteria. The first family includes SR34a and SRC33 genes that encodes proteins within the serine/arginine-rich (SR) family. The second family comprises DEAD29, DEAD28, DEAH5, DEAH10, and DExH15 genes that encodes RNA helicases affiliated with the DEAD/DEAH box family. Members of the SR family play an integral role in pre-mRNA splicing regulation and participate in the recruitment of the spliceosome to specific splice sites while recognising splice signals (Jin 2022). These SFs can interact synergistically with one another and with other proteins, forming complexes that enhance the splicing process, where collaboration is essential for improving splicing efficiency and accuracy (De et al. 2016). SR34 and SRC33 significantly influence the regulation of alternative splicing events, enabling a single gene to yield multiple mRNA isoforms, which contributes to protein diversity and functional adaptability in response to various cellular, developmental, and environmental conditions (Lam et al. 2022). Both SR34 and SRC33 can respond to stress signals, including hormonal treatments such as MeJA or abiotic stresses. Under such conditions, their expression levels and activity may fluctuate, influencing the splicing of genes implicated in stress response pathways (Punzo et al. 2020). Evidence suggests functional overlap between SR34 and SRC33, indicating that they can partially compensate for one another’s roles in splicing. This redundancy is crucial for maintaining splicing efficiency amidst fluctuating cellular conditions (Zhang et al. 2020).
The second family of SFs, comprising the DEAD/DEAH box proteins, is similarly involved in regulating pre-mRNA splicing by unwinding RNA secondary structures, which is critical for the spliceosome’s access to splice sites (Jin 2022). These factors interact with spliceosome components, facilitating the assembly and activation of the spliceosomal complex. Their helicase activity is pivotal for the rearrangement of RNA during splicing (De et al. 2016). They play significant roles in alternative splicing, influencing the inclusion or exclusion of specific exons in mature mRNA transcripts, resulting in diverse protein isoforms that enhance the plant’s adaptability to external stimuli (Zhang et al. 2020). The expression and activity of these SFs can be modulated in response to environmental stresses, allowing them to participate in the splicing of genes critical for stress response pathways (Liu et al. 2022). There may exist functional redundancy among these factors, permitting one to compensate for another’s loss. Furthermore, they can collaborate in complexes, enhancing splicing efficiency through synergistic interactions. By influencing splicing patterns, these helicases indirectly regulate gene expression levels and profiles, impacting various physiological processes, including growth, development, and stress responses (Jankowsky et al. 2011).
Synergistic interactions between serine/arginine-rich and DEAD/DEAH-box SFs
The SFs SR34 and SRC33 are likely integrated into broader regulatory networks involving additional SFs and regulatory proteins, with their interactions within these networks significantly impacting gene expression profiles and developmental processes in plants (Ali et al. 2007). The SFs DEAD29, DEAD28, DEAH5, DEAH10, and DExH15 may also coordinate their activities with other SFs and regulatory proteins, integrating signals from diverse pathways to finely tune gene expression (Shang et al. 2017). The interplay between serine/arginine-rich SFs and DEAD/DEAH box SFs is characterised by their collaborative roles in splicing regulation, alternative splicing, and responses to environmental stimuli, contributing to the nuanced control of gene expression in plants (Swift et al. 2022; Verta and Jacobs 2022). In other words, the members of the SR protein family collaborate with the SFs DEAD29, DEAD28, DEAH5, DEAH10, and DExH15 during the splicing process, where their interactions enhance the recruitment and stabilisation of the spliceosome at splice sites (Sahebi et al. 2016; Li et al. 2017). Both groups of SFs play pivotal roles in alternative splicing events, where SR34 and SRC33 regulate splice site selection, while the DEAD/DEAH box proteins facilitate the unwinding of RNA structures critical for these decisions, thereby promoting the inclusion or exclusion of specific exons (Taylor and Sobczak 2020).
The SFs SR34 and SRC33 are likely integrated into broader regulatory networks involving additional SFs and regulatory proteins, with their interactions within these networks significantly impacting gene expression profiles and developmental processes in plants (Ali et al. 2007). The SFs DEAD29, DEAD28, DEAH5, DEAH10, and DExH15 may also coordinate their activities with other SFs and regulatory proteins, integrating signals from diverse pathways to finely tune gene expression (Shang et al. 2017). The interplay between serine/arginine-rich SFs and DEAD/DEAH box SFs is characterised by their collaborative roles in splicing regulation, alternative splicing, and responses to environmental stimuli, contributing to the nuanced control of gene expression in plants (Swift et al. 2022; Verta and Jacobs 2022). In other words, the members of the SR protein family collaborate with the SFs DEAD29, DEAD28, DEAH5, DEAH10, and DExH15 during the splicing process, where their interactions enhance the recruitment and stabilisation of the spliceosome at splice sites (Sahebi et al. 2016; Li et al. 2017). Both groups of SFs play pivotal roles in alternative splicing events, where SR34 and SRC33 regulate splice site selection, while the DEAD/DEAH box proteins facilitate the unwinding of RNA structures critical for these decisions, thereby promoting the inclusion or exclusion of specific exons (Taylor and Sobczak 2020).
Additional specific functions of selected SFs beyond RNA splicing
The expression of the gene encoding SR34a has been predominantly observed during the seedling phase and in metabolically active, dividing cells, such as leaf primordia (Palusa et al. 2007; Stankovic et al. 2016). Palusa et al. (2007) also highlighted that splicing variants of this SF include one that undergoes alternative splicing at the 5′ untranslated region, indicative of alternative promoter usage, which aligns with the present study’s findings (Fig. S19). The gene that encodes SRC33, also referred to as SCL33, is temporally regulated during plant development (Shen et al. 2023). The authors suggest that this gene may interact with components of the Pol II elongation complex in Brachypodium distachyon, thereby influencing transcription rates during the flowering stage. Moreover, Palusa and colleagues (Palusa et al. 2007) noted that the expression of SCL33 splice variants in Arabidopsis thaliana is modulated by abiotic stresses.
In terms of the DEAD/DEAH box family, the SF DEAD29 (or RH29) has been identified as crucial for the development and maturation of gametophytes, and may also participate in various aspects of rRNA biogenesis and translation regulation (Chen et al. 2020). DEAH10 has been linked to several developmental processes, including meristem maintenance and the morphogenesis of leaves and roots (Ohtani et al. 2013). The gene encoding DExH15 is associated with antiviral innate immunity by enhancing mitochondrial antiviral signalling (MAVS) and activating the NLRP6 inflammasome (Mosallanejad et al. 2014), as well as antibacterial innate immunity through promoting Wnt-induced antimicrobial protein expression in Paneth cells (Wang et al. 2021). Nevertheless, there is a paucity of literature regarding the roles of DEAD28 and DEAH5.
Although current research does not directly connect MeJA to these SFs, we hypothesise that MeJA treatment could elevate the expression of genes encoding these SFs, thereby augmenting their involvement in splicing DAS genes under either stress or developmental conditions.
Possible MeJA-dependent roles of DAS genes concordantly expressed with SFs
Examining the DAS gene JMT2, previous investigations have demonstrated that the encoded enzyme, jasmonic acid carboxyl methyltransferase (JMT), catalyses the conversion of jasmonic acid (JA) into its methyl ester using S-adenosyl-L-methionine (SAM), playing a vital role in jasmonate-mediated plant responses (Koeduka et al. 2020). Earlier studies indicate that this enzyme regulates both developmental processes and herbivory-induced defence mechanisms in rice (Oryza sativa) (Qi et al. 2016). Furthermore, the overexpression of the JMT gene has been associated with increased tuber yield and size in potatoes (Solanum tuberosum) (Sohn et al. 2011). The functional profile of one of the JMT gene isoforms does not align with the established role of DExH15 gene, suggesting that the concordant expression of DExH15 and JMT2 genes may be coincidental (Table 2). Nonetheless, it remains plausible that DEAH5 gene could be involved in the alternative splicing of a second JMT2 isoform, leading to the generation of one DAS gene variant (Fig. S6).
The DAS gene MPK18, part of the mitogen-activated protein kinase (MAPK) gene family, is crucial for regulating environmental and developmental processes, as well as phytohormone signalling in plants (Colcombet and Hirt 2008). Evidence supports MPK18’s role in mediating cortical microtubule functions, particularly when its expression coincides with that of the PROPYZAMIDE HYPERSENSITIVE 1 (PHS1) gene (Walia et al. 2009). This function is congruent with MeJA actions; however, comparing it with the SF DEAD28 proves challenging due to insufficient information on function of the latter. Therefore, further investigation is warranted regarding the roles of DEAH5 and DEAD28 in producing variants of the JMT2 gene (Fig. S6) and the two variants of the MPK18 gene (Fig. S13).
The three TF genes of DAS (CH62, WRK41, and PIL57) belong to the C3H and WRKY zinc finger TF, and to the phytochrome-interacting factor (PIF) families. Members of these families contribute to seed germination and are instrumental in managing plant development and various biotic and abiotic stresses (Cheng et al. 2020; Li et al. 2024). C3H62 gene is co-expressed with the SF gene SR34a (Table 2), a member of a family known to respond to abiotic stress signals (Punzo et al. 2020) and developmental processes (Ali et al. 2007). WRK41 gene is co-expressed with DEAD29 gene, which is involved in the development and maturation of gametophytes, while PIL57 gene is co-expressed with SRC33 gene, which is involved in plant development, and participates in the Pol II elongation complex during flowering, and responds to abiotic stresses. Given their established functions, it is reasonable to assume that MeJA regulates expression of the three SF genes, leading to the production of multiple gene isoforms (Figs. S10, S11, and S14) that promote functional diversity in these TFs and subsequently drive the expression of numerous downstream development- and stress-related genes.
The DAS gene NIP21, also referred to as NIP2-1, belongs to the aquaporin water channel family, specifically the Nodulin 26-like Intrinsic Protein (NIP) group, facilitating water transport across cellular membranes (Agre 2006). This DAS gene encodes a lactic acid transporter, promoting plant survival under low oxygen stress, and is predominantly expressed in the vascular tissues of roots, which supports its role in metabolic adaptation to oxygen deficit (Beamer et al. 2021). Three MeJA-regulated isoforms of this DAS gene exist, with two co-expressed alongside the SF gene DEAH10 (Fig. 3) and one isoform co-expressed with DEAD29 gene (Fig. 4). Given the involvement of DEAH10 gene in various developmental processes, including leaf and root morphogenesis, its functional alignment with NIP21 gene seems more likely than that of DEAD29 gene.
The DAS gene OPT3 is implicated in the transport of iron from xylem to phloem and in the transmission of shoot-to-root iron signalling in Arabidopsis (Kurt 2021). This function does not appear to align with that of DEAH10 or DExH15 gene, suggesting that the co-expression of this DAS gene’s isoforms with either SF is coincidental. The DAS gene EDL16, or ERD6-like 16, is part of the sugar porter (SP) gene family, which plays roles in plant development and abiotic stress tolerance (Bavnhøj et al. 2021). This DAS gene is involved in regulating cell elongation and responses to abiotic stresses such as dehydration (Kiyosue et al. 1994). The two isoforms of this DAS gene are co-expressed with the SF gene DEAD29 (Fig. 5), suggesting a potential alignment of their functions; thus, it is conceivable that DEAD29 may play a role in generating the two isoforms of this DAS gene.
To further elucidate the interplay between SFs and DAS genes, it is recommended to conduct targeted functional assays and expression analyses under various stress conditions. This approach will help to clarify the specific contributions of SFs like DEAH10 and DEAD29 in regulating gene isoforms associated with stress responses and developmental processes. This approach will clarify the specific roles of SFs like DEAH10 and DEAD29 in regulating expression of gene isoforms linked to stress responses and developmental processes in C. roseus. Understanding the synchronised expression of SFs and their likely influence on promoting specific DAS gene isoforms could facilitate the enhancement of growth and stress tolerance in economically significant plants through biotechnological approaches.
Understanding the synchronised expression of SFs and their likely influence on promoting specific DAS gene isoforms holds significant promise for advancing the growth and stress tolerance of economically important plants through various biotechnological approaches. By elucidating how these SFs interact and function, researchers can develop targeted strategies to manipulate gene expression patterns, enhancing the plants’ ability to respond to environmental challenges. Such insights could lead to the identification of key SFs that regulate critical pathways related to growth and stress resistance. These findings could then be leveraged to engineer plants with improved resilience to factors such as drought, salinity, and pathogen attacks. Moreover, by employing metabolic engineering techniques, it would be possible to optimise the biosynthesis of essential metabolites, improving not only yield but also the nutritional and commercial value of these crops. Ultimately, more understanding of the molecular mechanisms underlying SF expression and its role in shaping DAS gene isoforms could pave the way for innovative biotechnological solutions, contributing to sustainable agricultural practices and enhancing food security in changing climates.
Conclusions
In conclusion, we propose that exogenous application of MeJA may activate the expression of genes encoding SFs that are regulated in tandem with their coordinately expressed DAS genes. This interaction implies that MeJA could enhance the regulatory frameworks that control splicing mechanisms, potentially resulting in the generation of specific mRNA isoforms. These isoforms may, in turn, trigger the expression of particular DAS gene variants, allowing the plant to effectively respond to environmental stimuli and developmental signals. By modulating the expression of these MeJA-dependent SF/DAS gene pairs, the overall gene expression profiles are likely to be altered, which can lead to increased functional diversity within the plant. This diversification is crucial for the plant’s ability to adapt and thrive under varying conditions, as it enables a more tailored response to stressors, such as pathogen attacks or abiotic challenges. The dynamic interplay between MeJA, SFs, and DAS genes highlights the complex regulatory networks that underpin plant resilience and adaptability, further emphasising the significance of MeJA in plant biology. The insights into the coordinated expression of SFs and their potential role in promoting specific DAS gene isoforms can aid in enhancing the growth and stress resilience of economically valuable plants through metabolic engineering processes.
Data availability
RNA sequencing datasets are available in the NCBI, assigned the BioProject number PRJNA386850. This corresponds to a previously documented study examining the effects of MeJA treatment in Catharanthus roseus.
Author contributions
Conceptualisation, A. Abulfaraj and A. Andal; methodology, A. Abulfaraj and A. Andal; software, A. Abulfaraj and A. Andal; validation, A. Abulfaraj and A. Andal; formal analysis, A. Abulfaraj and A. Andal; investigation, A. Abulfaraj and A. Andal; resources, A. Abulfaraj and A. Andal; data curation, A. Abulfaraj and A. Andal; writing – original draft preparation, A. Abulfaraj and A. Andal; writing – review and editing, A. Abulfaraj and A. Andal; visualization, A. Abulfaraj and A. Andal; supervision, A. Abulfaraj and A. Andal; project administration, A. Abulfaraj and A. Andal.
References
Agre P (2006) The aquaporin water channels. Proceedings of the American Thoracic Society 3(1), 5-13.
| Crossref | Google Scholar | PubMed |
Ali GS, Palusa SG, Golovkin M, Prasad J, Manley JL, Reddy AS (2007) Regulation of plant developmental processes by a novel splicing factor. PLoS ONE 2(5), e471.
| Crossref | Google Scholar | PubMed |
Bahieldin A, Atef A, Edris S, Gadalla NO, Al-Matary M, Al-Kordy MA, Ramadan AM, Bafeel S, Alharbi MG, Al-Quwaie DAH, Sabir JSM, Al-Zahrani HS, Nasr ME, El-Domyati FM (2018) Stepwise response of MeJA-induced genes and pathways in leaves of C. roseus. Comptes Rendus. Biologies 341(9–10), 411-420.
| Crossref | Google Scholar | PubMed |
Bavnhøj L, Paulsen PA, Flores-Canales JC, Schiøtt B, Pedersen BP (2021) Molecular mechanism of sugar transport in plants unveiled by structures of glucose/H+ symporter STP10. Nature Plants 7, 1409-1419.
| Crossref | Google Scholar |
Beamer ZG, Routray P, Choi W-G, Spangler MK, Lokdarshi A, Roberts DM (2021) Aquaporin family lactic acid channel NIP2;1 promotes plant survival under low oxygen stress in Arabidopsis. Plant Physiology 187(4), 2262-2278.
| Crossref | Google Scholar | PubMed |
Chen D, Wang Y, Zhang W, Li N, Dai B, Xie F, Sun Y, Sun M, Peng X (2020) Gametophyte-specific DEAD-box RNA helicase 29 is required for functional maturation of male and female gametophytes in Arabidopsis. Journal of Experimental Botany 71(14), 4083-4092.
| Crossref | Google Scholar | PubMed |
Cheng X, Cao J, Gao C, Gao W, Yan S, Yao H, Xu K, Liu X, Xu D, Pan X, Lu J, Chang C, Zhang H, Ma C (2020) Identification of the wheat C3H gene family and expression analysis of candidates associated with seed dormancy and germination. Plant Physiology and Biochemistry 156, 524-537.
| Crossref | Google Scholar | PubMed |
Colcombet J, Hirt H (2008) Arabidopsis MAPKs: a complex signalling network involved in multiple biological processes. Biochemical Journal 413(2), 217-226.
| Crossref | Google Scholar | PubMed |
De I, Schmitzová J, Pena V (2016) The organization and contribution of helicases to RNA splicing. Wiley Interdisciplinary Reviews: RNA 7(2), 259-274.
| Crossref | Google Scholar | PubMed |
Demkura PV, Abdala G, Baldwin IT, Ballare CL (2010) Jasmonate-dependent and -independent pathways mediate specific effects of solar ultraviolet B radiation on leaf phenolics and antiherbivore defense. Plant Physiology 152(2), 1084-1095.
| Crossref | Google Scholar | PubMed |
El-Domyati FM, Atef A, Al-Ghamdi AKM, Khan TK, Edris S, Gadalla NO, Al-Kordy MA, Ramadan AM, Saad YM, Sabir M, Al-Zahrani HS, Bahieldin A (2017) Identification of organ-specific regulatory frameworks of Catharanthus Roseus with emphasis to the TIA biosynthetic pathway. Pakistan Journal of Botany 49(6), 2243-2254.
| Google Scholar |
Gao H, Li F, Xu Z, Huang C, Xiong C, Jiang C, Xie N, Leng L, Zhang Y, Yousaf Z, Liu X, Sun W (2019) Genome-wide analysis of methyl jasmonate-regulated isoform expression in the medicinal plant Andrographis paniculata. Industrial Crops and Products 135, 39-48.
| Crossref | Google Scholar |
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnology 29, 644-652.
| Crossref | Google Scholar | PubMed |
Haberle V, Stark A (2018) Eukaryotic core promoters and the functional basis of transcription initiation. Nature Reviews Molecular Cell Biology 19, 621-637.
| Crossref | Google Scholar | PubMed |
Hajrah NH, Obaid AY, Atef A, Ramadan AM, Arasappan D, Nelson CA, Edris S, Mutwakil MZ, Alhebshi A, Gadalla NO, Makki RM, Al-Kordy MA, El-Domyati FM, Sabir JSM, Khiyami MA, Hall N, Bahieldin A, Jansen RK (2017) Transcriptomic analysis of salt stress responsive genes in Rhazya stricta. PLoS ONE 12(5), e0177589.
| Crossref | Google Scholar | PubMed |
Jankowsky A, Guenther U-P, Jankowsky E (2011) The RNA helicase database. Nucleic Acids Research 39(1), D338-D341.
| Crossref | Google Scholar | PubMed |
Jin X (2022) Regulatory network of serine/arginine-rich (SR) proteins: the molecular mechanism and physiological function in plants. International Journal of Molecular Sciences 23(11), 10147.
| Crossref | Google Scholar | PubMed |
Jones P, Binns D, Chang H-Y, Fraser M, Li W, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G, Pesseat S, Quinn AF, Sangrador-Vegas A, Scheremetjew M, Yong S-Y, Lopez R, Hunter S (2014) InterProScan 5: genome-scale protein function classification. Bioinformatics 30(9), 1236-1240.
| Crossref | Google Scholar | PubMed |
Kellner F, Kim J, Clavijo BJ, Hamilton JP, Childs KL, Vaillancourt B, Cepela J, Habermann M, Steuernagel B, Clissold L, McLay K, Buell CR, O’Connor SE (2015) Genome-guided investigation of plant natural product biosynthesis. The Plant Journal 82(4), 680-692.
| Crossref | Google Scholar | PubMed |
Kim D, Langmead B, Salzberg SL (2014) HISAT: hierarchical indexing for spliced alignment of transcripts. BioRxiv 012591.
| Crossref | Google Scholar |
Kiyosue T, Yamaguchi-Shinozaki K, Shinozaki K (1994) Cloning of cDNAs for genes that are early-responsive to dehydration stress (ERDs) in Arabidopsis thaliana L.: identification of three ERDs as HSP cognate genes. Plant Molecular Biology 25, 791-798.
| Crossref | Google Scholar | PubMed |
Koeduka T, Suzuki H, Taguchi G, Matsui K (2020) Biochemical characterization of the jasmonic acid methyltransferase gene from wasabi (Eutrema japonicum). Plant Biotechnology 37(3), 389-392.
| Crossref | Google Scholar | PubMed |
Kurt F (2021) An insight into oligopeptide transporter 3 (OPT3) family proteins. Protein & Peptide Letters 28(1), 43-54.
| Crossref | Google Scholar | PubMed |
Lam PY, Wang L, Lo C, Zhu F-Y (2022) Alternative splicing and its roles in plant metabolism. International Journal of Molecular Sciences 23(13), 7355.
| Crossref | Google Scholar | PubMed |
Lee DW, Whittaker GR (2017) Use of AAScatterPlot tool for monitoring the evolution of the hemagglutinin cleavage site in H9 avian influenza viruses. Bioinformatics 33(16), 2431-2435.
| Crossref | Google Scholar | PubMed |
Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323.
| Crossref | Google Scholar |
Li J, Wang Y, Rao X, Wang Y, Feng W, Liang H, Liu Y (2017) Roles of alternative splicing in modulating transcriptional regulation. BMC Systems Biology 11, 89.
| Crossref | Google Scholar |
Li G, Kazmi A, Feng M, Hou H (2024) Phytochrome-interacting factors (PIFs) regulate phytohormone-mediated plant environmental adaptation. Environmental and Experimental Botany 218, 105610.
| Crossref | Google Scholar |
Liu X-X, Guo Q-H, Xu W-B, Liu P, Yan K (2022) Rapid regulation of alternative splicing in response to environmental stresses. Frontiers in Plant Science 13, 832177.
| Crossref | Google Scholar | PubMed |
Lu D, Sin H-S, Lu C, Fuller MT (2020) Developmental regulation of cell type-specific transcription by novel promoter-proximal sequence elements. Genes & Development 34, 663-677.
| Crossref | Google Scholar | PubMed |
Mosallanejad K, Sekine Y, Ishikura-Kinoshita S, Kumagai K, Nagano T, Matsuzawa A, Takeda K, Naguro I, Ichijo H (2014) The DEAH-box RNA helicase DHX15 activates NF-κB and MAPK signaling downstream of MAVS during antiviral responses. Science Signaling 7(323), ra40.
| Crossref | Google Scholar |
Mustafa NR, Verpoorte R (2007) Phenolic compounds in Catharanthus roseus. Phytochemistry Reviews 6, 243-258.
| Crossref | Google Scholar |
Nishanth MJ, Sheshadri SA, Rathore SS, Srinidhi S, Simon B (2018) Expression analysis of Cell wall invertase under abiotic stress conditions influencing specialized metabolism in Catharanthus roseus. Scientific Reports 8(1), 15059.
| Crossref | Google Scholar | PubMed |
Ohtani M, Demura T, Sugiyama M (2013) Arabidopsis root initiation defective1, a DEAH-box RNA helicase involved in pre-mRNA splicing, is essential for plant development. The Plant Cell 25(6), 2056-2069.
| Crossref | Google Scholar | PubMed |
Palusa SG, Ali GS, Reddy AS (2007) Alternative splicing of pre-mRNAs of Arabidopsis serine/arginine-rich proteins: regulation by hormones and stresses. The Plant Journal 49(6), 1091-1107.
| Crossref | Google Scholar | PubMed |
Punzo P, Grillo S, Batelli G (2020) Alternative splicing in plant abiotic stress responses. Biochemical Society Transactions 48(5), 2117-2126.
| Crossref | Google Scholar | PubMed |
Qi J, Li J, Han X, Li R, Wu J, Yu H, Hu L, Xiao Y, Lu J, Lou Y (2016) Jasmonic acid carboxyl methyltransferase regulates development and herbivory-induced defense response in rice. Journal of Integrative Plant Biology 58(6), 564-576.
| Crossref | Google Scholar | PubMed |
Robinson MD, Mccarthy DJ, Smyth GK (2010) EdgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26(1), 139-140.
| Crossref | Google Scholar | PubMed |
Sahebi M, Hanafi MM, Van Wijnen AJ, Azizi P, Abiri R, Ashkani S, Taheri S (2016) Towards understanding pre-mRNA splicing mechanisms and the role of SR proteins. Gene 587(2), 107-119.
| Crossref | Google Scholar | PubMed |
Shang X, Cao Y, Ma L (2017) Alternative splicing in plant genes: a means of regulating the environmental fitness of plants. International Journal of Molecular Sciences 18(2), 432.
| Crossref | Google Scholar | PubMed |
Shen Y, Qin Z, Ren G, Deng P, Ji W, Jiao C, Wu L (2023) Complexity and regulation of age-dependent alternative splicing in Brachypodium distachyon. Plant Physiology 192(4), 2703-2722.
| Crossref | Google Scholar | PubMed |
Skinner ME, Uzilov AV, Stein LD, Mungall CJ, Holmes IH (2009) JBrowse: a next-generation genome browser. Genome Research 19, 1630-1638.
| Crossref | Google Scholar | PubMed |
Sohn HB, Lee HY, Seo JS, Jung C, Jeon JH, Kim J-H, Lee YW, Lee JS, Cheong J-J, Choi YD (2011) Overexpression of jasmonic acid carboxyl methyltransferase increases tuber yield and size in transgenic potato. Plant Biotechnology Reports 5, 27-34.
| Crossref | Google Scholar |
Stankovic N, Schloesser M, Joris M, Sauvage E, Hanikenne M, Motte P (2016) Dynamic distribution and interaction of the Arabidopsis SRSF1 subfamily splicing factors. Plant Physiology 170(2), 1000-1013.
| Crossref | Google Scholar | PubMed |
Swift J, Greenham K, Ecker JR, Coruzzi GM, Robertson Mcclung C (2022) The biology of time: dynamic responses of cell types to developmental, circadian and environmental cues. The Plant Journal 109, 764-778.
| Crossref | Google Scholar | PubMed |
Tabatabaeipour SN, Shiran B, Ravash R, Niazi A, Ebrahimie E (2024) Comprehensive transcriptomic meta-analysis unveils new responsive genes to methyl jasmonate and ethylene in Catharanthus roseus. Heliyon 10, e27132.
| Crossref | Google Scholar |
Taylor K, Sobczak K (2020) Intrinsic regulatory role of RNA structural arrangement in alternative splicing control. International Journal of Molecular Sciences 21(14), 5161.
| Crossref | Google Scholar | PubMed |
Tian B, Manley JL (2017) Alternative polyadenylation of mRNA precursors. Nature Reviews Molecular Cell Biology 18, 18-30.
| Crossref | Google Scholar | PubMed |
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, Van Baren MJ, Salzberg SL, Wold BJ, Pachter L (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology 28, 511-515.
| Crossref | Google Scholar | PubMed |
Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L (2013) Differential analysis of gene regulation at transcript resolution with RNA-seq. Nature Biotechnology 31, 46-53.
| Crossref | Google Scholar | PubMed |
Van Der Fits L, Memelink J (2001) The jasmonate-inducible AP2/ERF-domain transcription factor ORCA3 activates gene expression via interaction with a jasmonate-responsive promoter element. The Plant Journal 25(1), 43-53.
| Crossref | Google Scholar | PubMed |
Van Der Heijden R, Jacobs DI, Snoeijer W, Hallard D, Verpoorte R (2004) The Catharanthus alkaloids: pharmacognosy and biotechnology. Current Medicinal Chemistry 11(5), 607-628.
| Crossref | Google Scholar |
Verta J-P, Jacobs A (2022) The role of alternative splicing in adaptation and evolution. Trends in Ecology & Evolution 37(4), 299-308.
| Crossref | Google Scholar | PubMed |
Walia A, Lee JS, Wasteneys G, Ellis B (2009) Arabidopsis mitogen-activated protein kinase MPK18 mediates cortical microtubule functions in plant cells. The Plant Journal 59(4), 565-575.
| Crossref | Google Scholar | PubMed |
Wang Y, He K, Sheng B, Lei X, Tao W, Zhu X, Wei Z, Fu R, Wang A, Bai S, Zhang Z, Hong N, Ye C, Tian Y, Wang J, Li M, Zhang K, Li L, Yang H, Li H-B, Flavell RA, Zhu S (2021) The RNA helicase Dhx15 mediates Wnt-induced antimicrobial protein expression in Paneth cells. Proceedings of the National Academy of Sciences of the United States of America 118(4), e2017432118.
| Crossref | Google Scholar | PubMed |
Wang J, Zhang S, Lu H, Xu H (2022) Differential regulation of alternative promoters emerges from unified kinetics of enhancer-promoter interaction. Nature Communications 13, 2714.
| Crossref | Google Scholar | PubMed |
Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ (2009) Jalview version 2 – a multiple sequence alignment editor and analysis workbench. Bioinformatics 25(9), 1189-1191.
| Crossref | Google Scholar |
Zhang D, Chen M-X, Zhu F-Y, Zhang J, Liu Y-G (2020) Emerging functions of plant serine/arginine-rich (SR) proteins: lessons from animals. Critical Reviews in Plant Sciences 39(2), 173-194.
| Crossref | Google Scholar |
Zhang Z, Bae B, Cuddleston WH, Miura P (2023) Coordination of alternative splicing and alternative polyadenylation revealed by targeted long read sequencing. Nature Communications 14, 5506.
| Crossref | Google Scholar | PubMed |
Zhu W, Yang B, Komatsu S, Lu X, Li X, Tian J (2015) Binary stress induces an increase in indole alkaloid biosynthesis in Catharanthus roseus. Frontiers in Plant Science 6, 582.
| Crossref | Google Scholar | PubMed |
Zingg D, Bhin J, Yemelyanenko J, Kas SM, Rolfs F, Lutz C, Lee JK, Klarenbeek S, Silverman IM, Annunziato S, Chan CS, Piersma SR, Eijkman T, Badoux M, Gogola E, Siteur B, Sprengers J, de Klein B, de Goeij-de Haas RR, Riedlinger GM, Ke H, Madison R, Drenth AP, van der Burg E, Schut E, Henneman L, van Miltenburg MH, Proost N, Zhen H, Wientjens E, de Bruijn R, de Ruiter JR, Boon U, de Korte-Grimmerink R, van Gerwen B, Féliz L, Abou-Alfa GK, Ross JS, van de Ven M, Rottenberg S, Cuppen E, Chessex AV, Ali SM, Burn TC, Jimenez CR, Ganesan S, Wessels LFA, Jonkers J (2022) Truncated FGFR2 is a clinically actionable oncogene in multiple cancers. Nature 608, 609-617.
| Crossref | Google Scholar | PubMed |