

Seed bacterial microbiota in post-submergence tolerant and sensitive barley genotypes
Eva María Gómez-Álvarez A , Monique Salardi-Jost B , Germán Darío Ahumada A , Pierdomenico Perata A , Matteo Dell’Acqua B and Chiara Pucciariello A *
A , Monique Salardi-Jost B , Germán Darío Ahumada A , Pierdomenico Perata A , Matteo Dell’Acqua B and Chiara Pucciariello A *
A
B
Abstract
Flooding is a predominant abiotic stress for cultivated plants, including barley. This cereal crop shows a large adaptability to different environmental conditions, suggesting the presence of key traits to tolerate adverse conditions. During germination, genetic variations account for dissimilarities in flooding tolerance. However, differences in the seed microbiota may also contribute to tolerance/sensitivity during seedling establishment. This work investigated differences in microbiome among the grains of barley accessions. Two barley phenotypes were compared, each either tolerant or sensitive to a short submergence period followed by a recovery. The study used a metataxonomic analysis based on 16S ribosomal RNA gene sequencing and subsequent functional prediction. Our results support the hypothesis that bacterial microbiota inhabiting the barley seeds are different between sensitive and tolerant barley accessions, which harbour specific bacterial phyla and families. Finally, bacteria detected in tolerant barley accessions show a peculiar functional enrichment that suggests a possible connection with successful germination and seedling establishment.
Keywords: abiotic stress, barley, flooding, germination, Hordeum vulgare, hypoxia, microbiota, submergence.
Introduction
Barley (Hordeum vulgare ssp. vulgare) is an important cereal crop, cultivated in a wide range of environments throughout the world (Bustos-Korts et al. 2019). Its plasticity and adaptability to different climatic conditions suggest that it has key traits for responding to abiotic and biotic stresses.
Flooding is one of the predominant abiotic stresses for plants. When a rise of water due to heavy rains in unexpected areas or seasons is associated with the poor capacity of the soil to drain water, this can have a severe impact on sensitive plant species. In fact, flooding consequences include less oxygen (O2) for respiration, less carbon dioxide and poor light for photosynthesis in turbid water (Geigenberger 2003; Voesenek and Bailey-Serres 2015; Loreti and Perata 2020; Cho et al. 2021).
In a previous study, we compared barley accessions characterised by sensitivity or tolerance to submergence stress followed by a recovery period during germination (Gómez-Álvarez et al. 2023). The different phenotypic outcome was associated with the presence of secondary dormancy in sensitive barley accessions, in which a more lignified seed coat was the result of transcriptional regulation during seed development. Sensitive accessions of barley were associated with sequence variation in a locus harbouring a laccase gene, possibly resulting in a lower seed permeability and O2 diffusion in the seed. This, in turn, results in a reduced seed germination during the recovery period (Gómez-Álvarez et al. 2023).
Although plant genetic differences account for variations in flooding tolerance, dissimilarities in the seed microbiota may also contribute to this trait. Seed-borne bacteria can be extremely important for germination and seedling establishment because potential effects might be expressed in the initial developmental stage. Hypoxic conditions might shape the microbiome composition, favouring the presence of facultative anaerobic and strict anaerobic microbes.
The microbiome is an integral part of the resilience to flooding stress in plants, due to its high adaptability to change (Martínez-Arias et al. 2022). In this sense, the release of plant substances from root exudates to the endosphere would seem to attract certain microbes and recruit some of them through a ‘cry for help’ mechanism (Rizaludin et al. 2021; Martínez-Arias et al. 2022). Despite this, during flooding stress, beneficial bacteria in the rhizosphere of spring wheat have been found to decrease, while the abundance of potential detrimental bacteria increases (Francioli et al. 2022).
Our previous results on rice, characterised by either short or long coleoptiles under submergence, identified a very specific microbiome in the variety showing a long coleoptile under submergence (Ahumada et al. 2022). In parallel, the isolation of bacteria species and the analysis of plant growth promoting activities, identified bacteria that possibly support rice tolerance to submergence. In fact, some strains were found to have amylase activity and the capacity to produce indole-related compounds, such as auxins, in vitro (Ahumada et al. 2022). Indeed, rice’s capacity to germinate under submergence depends on the activation of a set of enzymes involved in starch degradation to fuel the growing embryo (Perata et al. 1992; Guglielminetti et al. 1995; Kretzschmar et al. 2015), and auxin contributes to the coleoptile elongation under water (Nghi et al. 2019, 2021).
Barley grains contain a complex microbial population with a genotype-dependent endophytic seed microbiome but also a consistent core (Bziuk et al. 2021). In fact, despite genotype specificity, a core of seed endophytic bacteria was identified, with Enterobacteriaceae members being the dominant group and a high abundance of Curtobacterium, Paenibacillus and Pantoea (Bziuk et al. 2021). An investigation of the active part of barley seed microbiome in modern cultivars, using rRNA based sequencing, confirmed a core microbiome that includes Paenibacillaceae and Enterobacteriaceae (Yang et al. 2017). Paenibaciullus and Pantoea genera, observed in the seed endosphere of barley accessions from different geographical locations, were also shown to have plant growth promoting activities (Rahman et al. 2018).
Extensive studies have been conducted on the bacterial composition of barley and its associations with the genotype. Wild and domesticated barley accessions have a different bacteria root and rhizosphere microbiota composition (Bulgarelli et al. 2015) and a correlation is reported between the rhizosphere microbiota diversity and the host genetic diversity (Alegria Terrazas et al. 2020). The selection of the barley rhizosphere microbiota is likely driven by nitrogen shortage, suggesting a plant-driven shaping in growth-limiting conditions (Alegria Terrazas et al. 2022). Finally, a small number of loci appear to be responsible for the composition of the barley rhizosphere microbes, suggesting new possibilities in crop improvement (Escudero-Martinez et al. 2022).
Knowledge of seed microbiota composition, and possibly the in vitro isolation of some microbes with plant growth promoting activities, could lead to innovative solutions based on selected microbial applications at the first stage of plant development. This may enhance food security while using natural resources.
In this study, we conducted a metataxonomic analysis based on 16S ribosomal RNA (rRNA) gene sequencing (Marchesi and Ravel 2015) on barley grains in two barley accessions showing a tolerant phenotype (ability to germinate) versus two barley accessions with a sensitive phenotype (inability to germinate), in relation to a submergence period followed by recovery. The aim was to assess whether bacterial seed endophytes show a dissimilar association with sensitive and tolerant barley accessions, and whether they might help during germination. We thus conducted a metataxonomic analysis by investigating the composition of putative endophytic bacteria immediately after submergence stress. The results of our analysis suggest that tolerant accessions of barley have a different microbiome than that found in sensitive accessions, and that this difference may support tolerance.
Materials and methods
Plant materials
Accessions of barley (Hordeium vulgare L. ssp. vulgare) WB-433, WB-281, WB-050 and WB-352 were sourced from the Whealbi project (https://www.whealbi.eu/). Seeds were derived from lines obtained from two rounds of single seed descent (Bustos-Korts et al. 2019), and subsequently reproduced during the 2020 growing season at Fiorenzuola d’Arda in Northern Italy (44°56′N 9°54′E, 80 m.a.s.l.). The soils in Fiorenzuola d’Arda have the following composition: silt 50%, clay 36%, sand 14%; the soil is slightly basic with pH 7.85 and an organic mass of 3.0% in the upper 30 cm of topsoil. They are classified as fine silty, mixed and mesic Udic Ustochrepts (Rizza et al. 2018).
Accessions were selected in terms of previously identified contrasting phenotypes in relation to the Submergence Tolerance Index (STI, Gómez-Álvarez et al. 2023), i.e. whether they have a capacity to germinate after 2 days of submergence followed by 5 days of recovery. This index is high in WB-281 (Tajikistan) and WB-433 (Montenegro) and low in WB-050 (Greece) and WB-352 (Syria).
Seeds were stored at 4°C in a LAB-MIDI refrigerator (Desmon Scientific, Nusco, Italy). Prior to the submergence treatment, hulled seeds were surface sterilised with 75% ethanol for 2 min, 3.5% sodium hypochlorite for 2 min, and 75% ethanol for 1 min (Bertani et al. 2016). Seeds were washed with sterile water at each step, and at the end of the process.
Sampling and submergence experiment
Sterile seeds were placed in Magenta GA-7 (Merck, 77 mm × 77 mm × 97 mm) boxes. They were germinated in a growing incubator (Percival Scientific, Perry, IA, USA) at 20°C and 50% relative humidity (RH) in the dark. In air, seeds were placed on wet sterile filter paper. In submergence, they were transferred to Magenta GA-7 boxes filled with sterile water. The experiments included four replicas for each barley accession under air and submergence, with 15 seeds for each replicate. After 48 h, 1 g of barley seeds for each replicate was stored at −80°C. DNA extraction, library preparation and sequencing were performed at IGATech Technology Services (Udine, Italy). Possible external contamination of submergence water was checked by incubation on LB (Luria–Bertani) plates at 30°C for 48 h under dark aerobic conditions.
DNA extraction, library preparation and sequencing
DNA was extracted from barley seeds using the kit Dneasy Mericon Food (Qiagen, Venlo, NL) IGATech standard protocol. The hypervariable regions V3 and V4 of the 16S rRNA gene were amplified with the 16S-341F 5′-CCTACGGGNBGCASCAG-3′ and 16S-805R 5′-GACTACNVGGGTATCTAATCC-3′ primer set. An initial peptide nucleic acid (PNA) clamping was applied during the first polymerase chain reaction (PCR) step to block amplification of the host mitochondrial and chloroplast 16S sequences (Lundberg et al. 2013), following the PNA Bio protocol (Newbury Park, CA, USA). The amplification included 25 PCR cycles using locus-specific primers, and a subsequent amplification that integrated the relevant flow-cell binding domains and unique indices (NexteraXT Index Kit, Illumina, San Diego, CA, USA). The sequencing of libraries was performed with the 300 bp paired-end mode using MiSeq (Illumina).
Bioinformatic analysis
The quality of the sequencing process was verified with FastQC (Andrews 2010). The removal of low-quality reads was performed with ERNE-FILTERING (Del Fabbro et al. 2013). After denoising using the QIIME2 ver. 1.9.0 DADA2 plugin (Caporaso et al. 2010), high quality sequences with a minimum length of 275 bp were converted into amplicon sequencing variants (ASVs). Chimeras were removed using the QIIME2 UCHIME plugin. ASVs sequences were aligned to the SILVA (Ribosomal RNA Gene Database Project Data1) database (Quast et al. 2013) (ver. 132), using a 99% sequence identity. The taxonomy matrix and ASVs table were used for subsequent studies.
Statistical analysis
R (R Core Team 2021) was used to perform statistical analyses. Briefly, a phyloseq object was obtained with R/Phyloseq (McMurdie and Holmes 2013). After the removal of reads of chloroplasts, mitochondria and unassigned features, the rarefraction of the ASVs table was performed to the minimum library size. The ‘estimate_richness’ function of R/Phyloseq was used to calculate Chao1 and Shannon indexes related to alpha-diversity. After the Shapiro–Wilk test, Student’s t-test was used to calculate differences between treatments and phenotype.
Cumulative sum scaling (CSS) was used to normalise data with cumNorm() from the R/MetagenomeSeq v.3.8 (Paulson et al. 2013). For the calculation of the beta-diversity, the canonical analysis of principal coordinates (CAPs) feature of the ordinate() function of the R/Phyloseq was utilised, with constraining of the phenotype and treatment variables. Weighted Unifrac, Bray–Curtis and Jaccard distances were used to analyse differences, which were confirmed using PERMANOVA analysis for the three distance matrixes. Venn diagrams were created using the ps_venn() function from the MicEco package (Russel 2023), taking into account the number of ASVs belonging to each group. Pheatmap() function from Pheatmap() package (Kolde and Kolde 2015)was utilised to perform the heatmaps, where the data was transformed to log (1 + x).
Functional prediction based on 16S rRNA gene sequence analysis
Functional prediction based on 16S rRNA gene sequence was performed by using the MicFunPred resource (Mongad et al. 2021).The ASVs table and the representative reads were utilised, maintaining borderline chimeras. The analysis was performed using Pfam and KEGG Orthology (KO) databases (Li et al. 2014; Kanehisa et al. 2016a, 2016b; Keller-Costa et al. 2021; Mistry et al. 2021). The analysis of results was conducted using R (R Core Team 2021). ANOVA analysis (P-value < 0.05) corrected by the False Discovery Rate (FDR) was applied to detect any significant differences among KO groups and Pfam of identified bacteria. The visualisation of MicFunPred results was obtained with the Pheatmap() function, using the viridis colour palette from R/Viridis (Garnier et al. 2021).
Results and discussion
Seed microbiota differs in tolerant and sensitive barley accessions
Two tolerant (WB-281, WB-433) and two sensitive (WB-050, WB-352) barley accessions were previously found to be able or not to germinate after 2 days of submergence followed by 5 days of recovery (Gómez-Álvarez et al. 2023). Seeds belonging to these accessions were collected after 48 h under air and complete submergence before the recovery period (Fig. 1a). In a previous work, we found that a differential lignification during seed development is responsible for the subsequent phenotype observed during germination (Gómez-Álvarez et al. 2023) (Fig. 1a). Our goal was to study whether seed characteristics could be linked to differences in seed microbial communities, which in turn may play a role in the dissimilar responses observed during the recovery period.
(a) The experimental set up, showing the sampling point and differential phenotypes of tolerant and sensitive barley accessions after the recovery period. (b) Alpha-diversity analysis performed by using Shannon and Chao1 indexes (Student t-test) on samples collected before the recovery period. (c) Beta-diversity analysis performed using the canonical analysis of principal coordinates (CAP) constrained for variables on the Bray–Curtis distance (PERMANOVA test). n.s., not significant; sub, submergence.
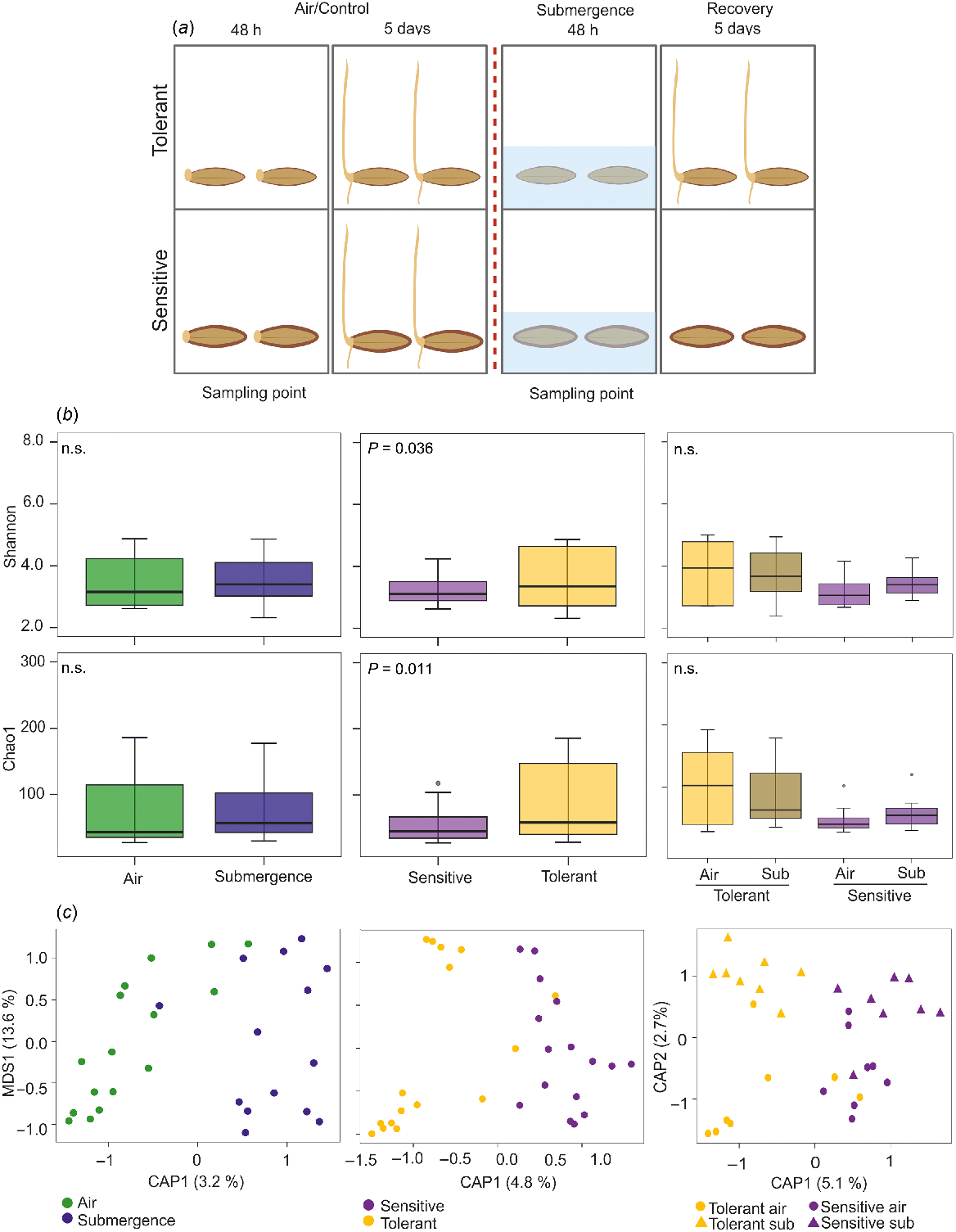
One sampling point was analysed: 48 h in air (control, both tolerant and sensitive barley accessions subsequently germinated in this condition) and 48 h under submergence (treatment, only tolerant varieties subsequently germinated during the recovery period) (Fig. 1a). Our hypothesis is that the differences observed during the recovery period between sensitive and tolerant accessions might be associated with the presence of dissimilar microbial communities after submergence.
The 16S rRNA gene analysis generated 5 016 648 paired-end reads. After in silico removal of chimeras and unassigned sequences and querying the SILVA database (99% sequence similarity), 2010 ASVs were assigned. Alpha-diversity was estimated using the Shannon and Chao1 indexes, comparing the different treatments (air and submergence), the different phenotypes (sensitive and tolerant) and the interaction between them.
The Shannon index of diversity revealed a high range of variability among samples in both air and submergence, whose difference was not significant (Fig. 1b). Although the Shannon index distribution among samples was large in both sensitive and tolerant barley accessions, the difference between phenotypes was significant (Student’s t-test, P < 0.05). This revealed that tolerant accessions have a higher diversity than sensitive accessions regardless of the growth conditions (Fig. 1b). The Chao1 index of richness also showed a large range of variation and the difference between air and submergence conditions was not significant (Fig. 1b). On the contrary, the Chao1 index was significantly different between sensitive and tolerant accessions, although there was a large variation in the tolerant samples (Fig. 1b). For both the indexes, the interaction between treatments and phenotypes was not significant.
Beta-diversity analysis of community composition was performed. Visualisation of the data distribution was computed using the canonical analysis of principal components (CAP) with a constraint for the treatment condition and phenotype (Fig. 1c). The Bray–Curtis CAP was followed by the permutation analysis of variance (PERMANOVA), which found an effect for phenotypes (P < 0.05) but not for treatments and phenotype-by-treatment interactions. The use of weighted Unifrac and Jaccard distances showed the same pattern, albeit not significant (Supplementary Table S1).
These results support the hypothesis of a putative endophytic bacterial microbiota difference between sensitive and tolerant barley accessions, irrespective of the air/submergence conditions. This conclusion differs with the result previously found for rice under submergence (Ahumada et al. 2022), where the tolerant rice accessions under submergence were part of a separate cluster, suggesting a specific bacterial community. In fact, rice and barley differ in their response to submergence in that rice is able to germinate under water whereas barley is not. The Arborio rice tolerant variety is able to elongate much of the coleoptile toward the water surface where it acts as a snorkel in order to allow O2 diffusion to underwater organs (Narsai et al. 2015; Shiono et al. 2022). This particular characteristic may be associated with a different bacterial community than the one in the variety that elongates the coleoptile less. However, this does not happen with barley, where none of the accessions germinate under water.
Sensitive and tolerant barley accessions show specific bacteria phyla and families
A Venn diagram was used to highlight the distribution of the ASVs in barley accessions characterised by different phenotypes, showing significant differences in alpha and beta-diversity indexes. The results show that sensitive and tolerant accessions harbour unique communities with only 117 ASVs in common. Tolerant barley accessions had double the amount of ASVs relative to sensitive accessions (Fig. 2a), reflecting the results obtained with the Chao1 index (Fig. 1b).
(a) Venn diagram for the variable phenotype representing the total number of ASVs identified as described in the ‘Bioinformatic Analysis’ section. (b–e) Heatmap of the relative abundance for the 10 most abundant unique ASVs belonging to sensitive and tolerant varieties grouped by phylum (b, c) and family (d, e). Unique ASVs for sensitive and tolerant accessions are in bold. (f) Boxplots of the significant genera depending on the phenotype variable (FDR < 0.05) and showing differences between air and submergence samples. Relative abundance is calculated as −log(1 + X) of number of counts for given ASVs. m., members.
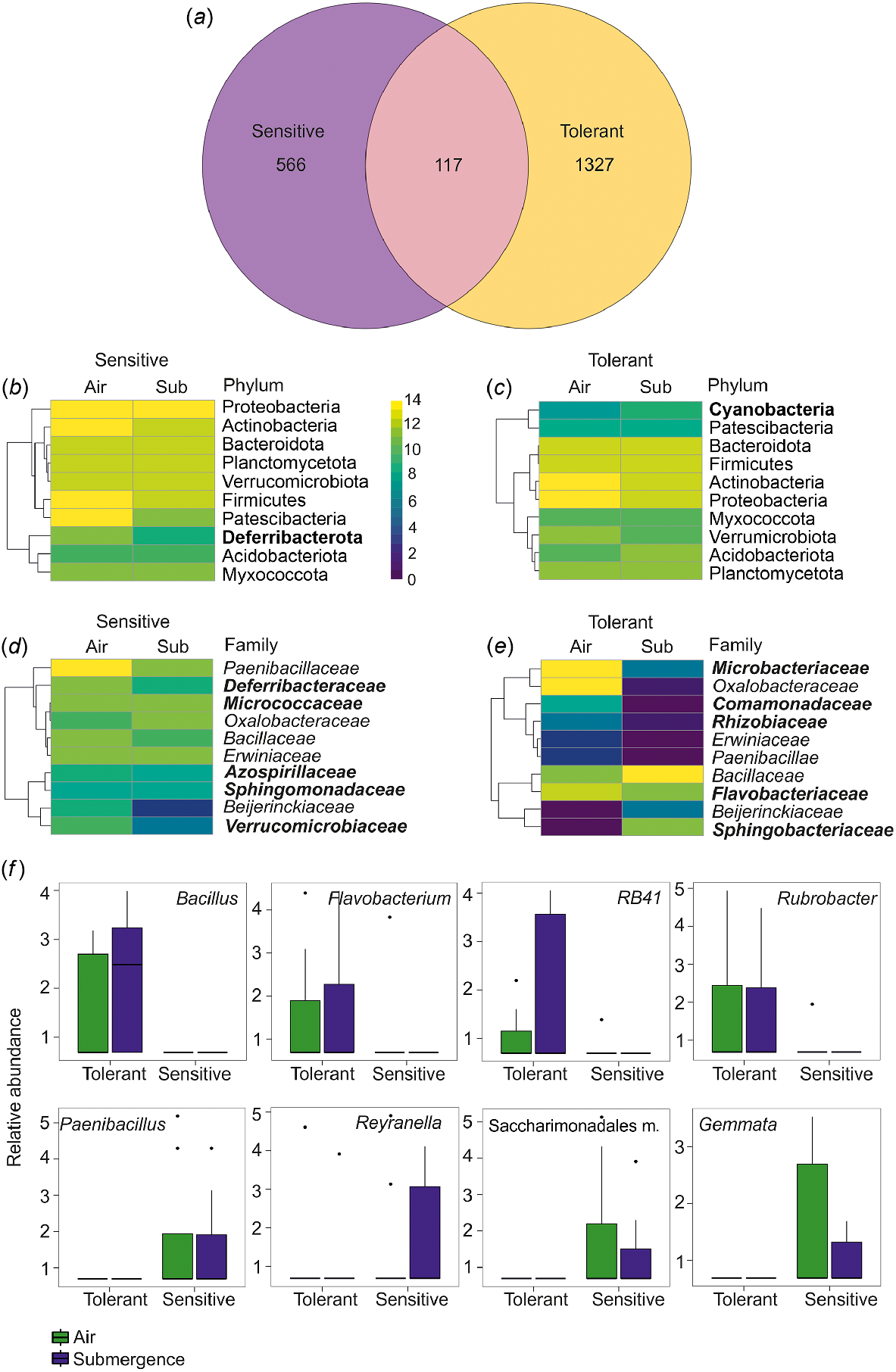
The heatmap of the ten most abundant phyla (representing 96% of the community) and the most abundant families (representing 46% of the community) for tolerant and sensitive accessions (Fig. 2b–e) showed the predominance of the phyla Proteobacteria and Actinobacteria (alt. Actinomicetota) in both sensitive and tolerant accessions. Both phyla were previously found to be abundant in the rhizosphere of H. vulgare ssp. vulgare elite cultivars (Escudero-Martinez et al. 2022). Proteobacteria and Actinobacteria were also found to be predominant in the rhizosphere and root of the H. vulgare ssp. vulgare landrace, in modern cultivars and in H. vulgare ssp. spontaneum (Bulgarelli et al. 2015) and in the barley seed microbiome of different genotypes (Bziuk et al. 2021). The phylum of Proteobacteria remained high under submergence only in sensitive accessions.
The phylum Cyanobacteria was abundant only in tolerant barley accessions (Fig. 2c). Cyanobacteria have been found in association with terrestrial plants, and their capacity to fix CO2 and atmospheric nitrogen can have a large impact on the host plant, with a growing interest when available in the rice field (Álvarez et al. 2020). On the other hand, the phylum Deferribacterota (alt. Deferribacteres) was abundant only in the list of the sensitive accessions (Fig. 2b). Deferribacterota are halotolerant and frequently found in marine niches (Alauzet and Jumas-Bilak 2014). Interestingly, they are preferentially anaerobes and were found mostly in sensitive seeds, which are likely hypoxic (Gómez-Álvarez et al. 2023).
Among the families, Paenibacillaceae, Oxalobacteraceae, Bacillaceae, Erwiniaceae and Beijerinckiaceae were common to both tolerant and sensitive barley accessions (Fig. 2d, e). Paenibacillaceae and Oxalobacteraceae were among the most abundant in barley seeds also in previous works (Yang et al. 2017; Bziuk et al. 2021), whereas the other families seem to be specific to the accessions that we analysed. In the sensitive barley accessions, the Paenibacillaceae family was the most abundant in air and was reduced under submergence (Fig. 2d).
The unique ASVs of sensitive barley accessions belong to the Deferribacteraceae, Micrococcaceae, Azospirillaceae, Sphingomonadaceae and Verrucomicrobiaceae, which are absent from the list of predominant families in tolerant accessions (Fig. 2d–e). At the family level, in tolerant accessions there is an enrichment of ASVs from Microbacteriaceae, Comamonadaceae, Rhizobiaceae, Flavobacteriaceae and Sphingobacteriaceae, not observed in the 10 most represented groups of sensitive barley accessions (Fig. 2d–e). Of these, Microbacteriaceae, Comamonadaceae and Flavobacteriaceae have been found to be abundant in barley root (Bulgarelli et al. 2015). Sphingobacteriaceae are known to be endophytes of maize (Kämpfer et al. 2016). Microbacteriaceae and Oxalobacteriaceae are reduced under submergence, whereas there were more Bacillaceae (Fig. 2e).
Air and submergence samples show a similar amount of unique ASVs (Fig. S1), reflecting the result of the alpha and beta-diversity analyses. The ASVs that were only present in the submergence samples belong to Chloroflexi, whereas those present only in air are Patescibacteria, which have been found to dominate in the rhizosphere and roots of spring wheat microbiota respectively (Francioli et al. 2021).
At the family level, Sphingobacteriaceae, Xanthobacteriaceae, Bacteroidaceae Vicinamibacteraceae and Pseudomonadaceae are specifically present in submergence (Fig. S1). These families are different from what has been found in spring wheat microbiota under flooding conditions at different growing stages, suggesting a specific response of these plant species (Francioli et al. 2021, 2022).
The most significant genus difference (FDR, for all the single comparisons P-value < 0.05) between sensitive and tolerant barley accessions were analysed (Fig. 2f). Bacillus, Flavobacterium, RB41, and Rubrobacter were found only in tolerant samples both in air and in submergence conditions. Bacillus is one of the twenty most abundant genera in seed and rhizosphere microbiome in barley (Bziuk et al. 2021). Bacillus species with an endophytic lifestyle can provide plants with protection from phytopathogens, increased resistance to abiotic stresses, and plant growth promotion (Lopes et al. 2018). Flavobacterium has been described in Norwegian barley germinated seeds (3–4 days) (Jiao et al. 2022). RB41 has also been previously observed in barley (Lewin et al. 2021).
In our study, the genera Gemmata, Paenibacillus, Reyranella and Saccharimonadales members were found only in sensitive varieties. Paenibacillus has been reported as one of the most abundant in barley seeds (Yang et al. 2017; Bziuk et al. 2021) and, when isolated, it is able to produce protease, β-1-3-glucanase, cellulase and indole-3-acetic acid (IAA) (Bziuk et al. 2021). It has also been reported in ungerminated Norwegian barley seeds (Østlie et al. 2021).
Bacteria identified in tolerant barley accessions are functionally enriched
Significantly different functions between bacteria identified in sensitive and tolerant barley accessions were predicted, using the KEGG ORTHOLOGY (KO) database (Fig. 3). Tolerant barley accessions showed a bacterial functional enrichment compared to sensitive accessions in all the categories analysed. Interestingly, these include several categories involved in sugar-related mechanisms, such as the alpha-glucoside transport system, pentose phosphate pathway, maltogenic alpha-amylase, starch and sucrose metabolism, multiple sugar transport system and D-allose transport system (Fig. 3). Such activities might be involved in the use of sugar when the plant germinates after submergence during the recovery period and thus help the plant to establish the seedlings. On the other hand, they may suggest that the availability of sugars from seeds in germination enriches the bacterial community.
Functional analysis with MicFunPred using the KEGG ORTHOLOGY (KO) database depending on the phenotype variable (FDR, P < 0.05). Relative abundance is calculated as −log(1 + X) of number of counts for given ASVs. PTS, phosphotransferase system.

Significantly different functions between air and submergence were also predicted using the KO database (Fig. S2). Interestingly, samples from submerged seeds were enriched in bacteria with putative functions involved in starch and sucrose metabolism, which is crucial in overcoming submergence stress. On the contrary, nitric oxide synthase function is reduced under submergence. Nitric oxide is crucial for seeds to germinate, and submergence stress has been found to induce secondary dormancy in sensitive barley accessions (Gómez-Álvarez et al. 2023), suggesting a potential link with plant phenotype.
Conclusions
The identification of a peculiar microbiome associated with crops that are tolerant to environmental constraints may facilitate an innovative approach in which these bacteria are isolated and applied to sensitive crop species. In this framework, our study aimed to assess possible differences in the microbiota of barley accessions characterised by tolerance/sensitivity to a short submergence stress followed by recovery. Indeed, a further number of accessions, characterised by different relatedness, would help in deciphering whether the shape of microbiota in this context is associated with barley genetics. The results of this study suggest that at the end of submergence, tolerant barley accessions have a different microbiome from the sensitive accessions. This microbiome is present whether the barley is in air or submerged. Tolerant barley accessions are richer and more diverse than sensitive accessions, and have specific communities of amplicon sequence variants. Finally, at the functional level, bacteria associated with tolerant barley accessions are enriched in sugar-related categories. These results, which predict bacteria functions on the basis of a taxonomic representation, suggest a possible connection with a prompt barley germination during the recovery period after submergence.
Declaration of funding
This work was financially supported by Sant’Anna School of Advanced Studies and by the microbING project, Italian Ministry for University and Research (MUR) (PNR, EU regulation 241/2021, PNRR 2021-2026).
Author contributions
CP and PP conceived the work, CP designed and supervised the experiments. MSJ performed the submergence experiment with the support of GDA. EMGA performed bioinformatic, statistical and functional analysis. MDA supervised the bioinformatics analysis. CP and EMGA wrote the article, which was revised and approved by all the authors.
Acknowledgements
The authors thank Alessandro Tondelli from CREA Research Centre for Genomics and Bioinformatics (Fiorenzuola D’Arda, Italy) for kindly providing barley seeds. They are grateful to Iris Bertani from the International Centre for Genetic Engineering and Biotechnology (Trieste, Italy) for critically reading the manuscript.
References
Ahumada GD, Gómez-Álvarez EM, Dell’Acqua M, Bertani I, Venturi V, Perata P, Pucciariello C (2022) Bacterial endophytes contribute to rice seedling establishment under submergence. Frontiers in Plant Science 13, 908349.
| Crossref | Google Scholar |
Alegria Terrazas R, Balbirnie-Cumming K, Morris J, Hedley PE, Russell J, Paterson E, Baggs EM, Fridman E, Bulgarelli D (2020) A footprint of plant eco-geographic adaptation on the composition of the barley rhizosphere bacterial microbiota. Scientific Reports 10, 12916.
| Crossref | Google Scholar | PubMed |
Alegria Terrazas R, Robertson-Albertyn S, Corral AM, Escudero-Martinez C, Kapadia R, Balbirnie-Cumming K, Morris J, Hedley PE, Barret M, Torres-Cortes G, Paterson E, Baggs EM, Abbott J, Bulgarelli D (2022) Defining composition and function of the rhizosphere microbiota of barley genotypes exposed to growth-limiting nitrogen supplies. mSystems 7, e00934-22.
| Crossref | Google Scholar | PubMed |
Álvarez C, Navarro JA, Molina-Heredia FP, Mariscal V (2020) Endophytic colonization of rice (Oryza sativa L.) by the symbiotic strain Nostoc punctiforme PCC 73102. Molecular Plant-Microbe Interactions 33, 1040-1045.
| Crossref | Google Scholar | PubMed |
Andrews S (2010) FastQC: a quality control tool for high throughput sequence data. Available at http://www.bioinformatics.babraham.ac.uk/projects/fastqc [Accessed 2023]
Bertani I, Abbruscato P, Piffanelli P, Subramoni S, Venturi V (2016) Rice bacterial endophytes: isolation of a collection, identification of beneficial strains and microbiome analysis. Environmental Microbiology Reports 8, 388-398.
| Crossref | Google Scholar | PubMed |
Bulgarelli D, Garrido-Oter R, Münch PC, Weiman A, Dröge J, Pan Y, McHardy AC, Schulze-Lefert P (2015) Structure and function of the bacterial root microbiota in wild and domesticated barley. Cell Host & Microbe 17, 392-403.
| Crossref | Google Scholar | PubMed |
Bustos-Korts D, Dawson IK, Russell J, Tondelli A, Guerra D, Ferrandi C, Strozzi F, Nicolazzi EL, Molnar-Lang M, Ozkan H, Megyeri M, Miko P, Çakır E, Yakışır E, Trabanco N, Delbono S, Kyriakidis S, Booth A, Cammarano D, Mascher M, Werner P, Cattivelli L, Rossini L, Stein N, Kilian B, Waugh R, van Eeuwijk FA (2019) Exome sequences and multi-environment field trials elucidate the genetic basis of adaptation in barley. The Plant Journal 99, 1172-1191.
| Crossref | Google Scholar | PubMed |
Bziuk N, Maccario L, Straube B, Wehner G, Sørensen SJ, Schikora A, Smalla K (2021) The treasure inside barley seeds: microbial diversity and plant beneficial bacteria. Environmental Microbiome 16, 20.
| Crossref | Google Scholar | PubMed |
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, Mcdonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R (2010) QIIME allows analysis of high-throughput community sequencing data. Nature Methods 7, 335-336.
| Crossref | Google Scholar | PubMed |
Cho H-Y, Loreti E, Shih M-C, Perata P (2021) Energy and sugar signaling during hypoxia. New Phytologist 229, 57-63.
| Crossref | Google Scholar | PubMed |
Del Fabbro C, Scalabrin S, Morgante M, Giorgi FM (2013) An extensive evaluation of read trimming effects on Illumina NGS data analysis. PLoS ONE 8, e85024.
| Crossref | Google Scholar | PubMed |
Escudero-Martinez C, Coulter M, Alegria Terrazas R, Foito A, Kapadia R, Pietrangelo L, Maver M, Sharma R, Aprile A, Morris J, Hedley PE, Maurer A, Pillen K, Naclerio G, Mimmo T, Barton GJ, Waugh R, Abbott J, Bulgarelli D (2022) Identifying plant genes shaping microbiota composition in the barley rhizosphere. Nature Communications 13, 3443.
| Crossref | Google Scholar | PubMed |
Francioli D, Cid G, Kanukollu S, Ulrich A, Hajirezaei M-R, Kolb S (2021) Flooding causes dramatic compositional shifts and depletion of putative beneficial bacteria on the spring wheat microbiota. Frontiers in Microbiology 12, 773116.
| Crossref | Google Scholar |
Francioli D, Cid G, Hajirezaei M-R, Kolb S (2022) Leaf bacterial microbiota response to flooding is controlled by plant phenology in wheat (Triticum aestivum L.). Scientific Reports 12, 11197.
| Crossref | Google Scholar | PubMed |
Garnier S, Ross N, Rudis B, Sciaini M, Scherer C (2021) viridis: default color maps from ‘matplotlib’. Available at https://cran.r-project.org/web/packages/viridis/index.html
Geigenberger P (2003) Response of plant metabolism to too little oxygen. Current Opinion in Plant Biology 6, 247-256.
| Crossref | Google Scholar | PubMed |
Guglielminetti L, Yamaguchi J, Perata P, Alpi A (1995) Amylolytic activities in cereal seeds under aerobic and anaerobic conditions. Plant Physiology 109, 1069-1076.
| Crossref | Google Scholar | PubMed |
Gómez-Álvarez EM, Tondelli A, Nghi KN, Voloboeva V, Giordano G, Valè G, Perata P, Pucciariello C (2023) The inability of barley to germinate after submergence depends on hypoxia-induced secondary dormancy. Journal of Experimental Botany 74, 4277-4289.
| Crossref | Google Scholar | PubMed |
Jiao N, Song X, Song R, Yin D, Deng X (2022) Diversity and structure of the microbial community in rhizosphere soil of Fritillaria ussuriensis at different health levels. PeerJ 10, e12778.
| Crossref | Google Scholar | PubMed |
Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M (2016a) KEGG as a reference resource for gene and protein annotation. Nucleic Acids Research 44, D457-D462.
| Crossref | Google Scholar | PubMed |
Kanehisa M, Sato Y, Morishima K (2016b) BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. Journal of Molecular Biology 428, 726-731.
| Crossref | Google Scholar | PubMed |
Keller-Costa T, Lago-Lestón A, Saraiva JP, Toscan R, Silva SG, Gonçalves J, Cox CJ, Kyrpides N, Nunes da Rocha U, Costa R (2021) Metagenomic insights into the taxonomy, function, and dysbiosis of prokaryotic communities in octocorals. Microbiome 9, 72.
| Crossref | Google Scholar | PubMed |
Kolde R, Kolde MR (2015) pheatmap: Pretty Heatmaps. R package 1(7), 790. Available at https://CRAN.R-project.org/package=pheatmap
Kretzschmar T, Pelayo MAF, Trijatmiko KR, Gabunada LFM, Alam R, Jimenez R, Mendioro MS, Slamet-Loedin IH, Sreenivasulu N, Bailey-Serres J, Ismail AM, Mackill DJ, Septiningsih EM (2015) A trehalose-6-phosphate phosphatase enhances anaerobic germination tolerance in rice. Nature Plants 1, 15124.
| Crossref | Google Scholar | PubMed |
Kämpfer P, Busse H-J, Kleinhagauer T, McInroy JA, Glaeser SP (2016) Sphingobacterium zeae sp. nov., an endophyte of maize. International Journal of Systematic and Evolutionary Microbiology 66, 2643-2649.
| Crossref | Google Scholar | PubMed |
Lewin S, Francioli D, Ulrich A, Kolb S (2021) Crop host signatures reflected by co-association patterns of keystone Bacteria in the rhizosphere microbiota. Environmental Microbiome 16, 18.
| Crossref | Google Scholar | PubMed |
Li RW, Giarrizzo JG, Wu S, Li W, Duringer JM, Craig AM (2014) Metagenomic insights into the RDX-Degrading potential of the ovine rumen microbiome. PLoS ONE 9, e110505.
| Crossref | Google Scholar | PubMed |
Lopes R, Tsui S, Gonçalves PJRO, de Queiroz MV (2018) A look into a multifunctional toolbox: endophytic Bacillus species provide broad and underexploited benefits for plants. World Journal of Microbiology and Biotechnology 34, 94.
| Crossref | Google Scholar | PubMed |
Loreti E, Perata P (2020) The many facets of hypoxia in plants. Plants 9, 745.
| Crossref | Google Scholar |
Lundberg DS, Yourstone S, Mieczkowski P, et al. (2013) Practical innovations for high-throughput amplicon sequencing. Nature Methods 10, 999-1002.
| Crossref | Google Scholar | PubMed |
Marchesi JR, Ravel J (2015) The vocabulary of microbiome research: a proposal. Microbiome 3, 31.
| Crossref | Google Scholar | PubMed |
Martínez-Arias C, Witzell J, Solla A, Martin JA, Rodríguez-Calcerrada J (2022) Beneficial and pathogenic plant-microbe interactions during flooding stress. Plant, Cell & Environment 45, 2875-2897.
| Crossref | Google Scholar | PubMed |
McMurdie PJ, Holmes S (2013) phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8, e61217.
| Crossref | Google Scholar | PubMed |
Mistry J, Chuguransky S, Williams L, Qureshi M, Salazar GA, Sonnhammer ELL, Tosatto SCE, Paladin L, Raj S, Richardson LJ, Finn RD, Bateman A (2021) Pfam: the protein families database in 2021. Nucleic Acids Research 49, D412-D419.
| Crossref | Google Scholar | PubMed |
Mongad DS, Chavan NS, Narwade NP, Dixit K, Shouche YS, Dhotre DP (2021) MicFunPred: a conserved approach to predict functional profiles from 16S rRNA gene sequence data. Genomics 113, 3635-3643.
| Crossref | Google Scholar | PubMed |
Narsai R, Edwards JM, Roberts TH, Whelan J, Joss GH, Atwell BJ (2015) Mechanisms of growth and patterns of gene expression in oxygen-deprived rice coleoptiles. The Plant Journal 82, 25-40.
| Crossref | Google Scholar | PubMed |
Nghi KN, Tondelli A, Valè G, Tagliani A, Marè C, Perata P, Pucciariello C (2019) Dissection of coleoptile elongation in japonica rice under submergence through integrated genome-wide association mapping and transcriptional analyses. Plant, Cell & Environment 42, 1832-1846.
| Crossref | Google Scholar | PubMed |
Nghi KN, Tagliani A, Mariotti L, Weits DA, Perata P, Pucciariello C (2021) Auxin is required for the long coleoptile trait in japonica rice under submergence. New Phytologist 229, 85-93.
| Crossref | Google Scholar | PubMed |
Østlie HM, Porcellato D, Kvam G, Wicklund T (2021) Investigation of the microbiota associated with ungerminated and germinated Norwegian barley cultivars with focus on lactic acid bacteria. International Journal of Food Microbiology 341, 109059.
| Crossref | Google Scholar | PubMed |
Paulson J, Talukder H, Pop M, Bravo HC (2013) metagenomeSeq: statistical analysis for sparse high-throughput sequencing. Bioconductor package 1.18.0. Available at http://cbcb.umd.edu/software/metagenomeSeq [Accessed 2023]
Perata P, Pozueta-Romero J, Akazawa T, Yamaguchi J (1992) Effect of anoxia on starch breakdown in rice and wheat seeds. Planta 188, 611-618.
| Crossref | Google Scholar | PubMed |
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO (2013) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Research 41, D590-D596.
| Crossref | Google Scholar | PubMed |
R Core Team (2021) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. Available at https://www.R-project.org/
Rahman MM, Flory E, Koyro H-W, Abideen Z, Schikora A, Suarez C, Schnell S, Cardinale M (2018) Consistent associations with beneficial bacteria in the seed endosphere of barley (Hordeum vulgare L.). Systematic and Applied Microbiology 41, 386-398.
| Crossref | Google Scholar | PubMed |
Rizaludin MS, Stopnisek N, Raaijmakers JM, Garbeva P (2021) The chemistry of stress: understanding the ‘Cry for Help’ of plant roots. Metabolites 11, 357.
| Crossref | Google Scholar |
Rizza F, Vasilescu L, Badeck F-W, Morcia C, Alberici R, Bude A, Alionte E, Petcu E, Baronchelli M, Faccini N, Pagani D, Terzi V (2018) Agrobiodiversity for adaptive and yield traits in Romanian and Italian barley cultivars across four continental environments. Agronomy 8, 79.
| Crossref | Google Scholar |
Russel J (2023) MicEco: various functions for microbial community data. doi:10.5281/zenodo.1169176
Shiono K, Yoshikawa M, Kreszies T, Yamada S, Hojo Y, Matsuura T, Mori IC, Schreiber L, Yoshioka T (2022) Abscisic acid is required for exodermal suberization to form a barrier to radial oxygen loss in the adventitious roots of rice (Oryza sativa). New Phytologist 233, 655-669.
| Crossref | Google Scholar | PubMed |
Voesenek LACJ, Bailey-Serres J (2015) Flood adaptive traits and processes: an overview. New Phytologist 206, 57-73.
| Crossref | Google Scholar | PubMed |
Yang L, Danzberger J, Schöler A, Schröder P, Schloter M, Radl V (2017) Dominant groups of potentially active bacteria shared by barley seeds become less abundant in root associated microbiome. Frontiers in Plant Science 8, 1005.
| Crossref | Google Scholar |