

Evans Review No. 4: Functional genomics in chickpea: an emerging frontier for molecular-assisted breeding
Tristan E. Coram A C D , Nitin L. Mantri A , Rebecca Ford B and Edwin C. K. Pang AA RMIT University, School of Applied Sciences, Biotechnology and Environmental Biology, Building 223, Level 1, Plenty Road, Bundoora, Victoria 3083, Australia.
B BioMarka, Faculty of Land and Food Resources, The University of Melbourne, Victoria 3010, Australia.
C Present address: US Department of Agriculture, Agricultural Research Service, Wheat Genetics, Quality, Physiology and Disease Research Unit and Department of Plant Pathology, Washington State University, Pullman, WA 99164-6430, USA.
D Corresponding author. Email: tcoram@mail.wsu.edu
Functional Plant Biology 34(10) 861-873 https://doi.org/10.1071/FP07169
Submitted: 2 July 2007 Accepted: 8 August 2007 Published: 13 September 2007
Abstract
Chickpea is a valuable and important agricultural crop, but yield potential is limited by a series of biotic and abiotic stresses, including Ascochyta blight, Fusarium wilt, drought, cold and salinity. To accelerate molecular breeding efforts for the discovery and introgression of stress tolerance genes into cultivated chickpea, functional genomics approaches are rapidly growing. Recently a series of genetic tools for chickpea have become available that have allowed high-powered functional genomics studies to proceed, including a dense genetic map, large insert genome libraries, expressed sequence tag libraries, microarrays, serial analysis of gene expression, transgenics and reverse genetics. This review summarises the development of these genomic tools and the achievements made in initial and emerging functional genomics studies. Much of the initial research focused on Ascochyta blight resistance, and a resistance model has been synthesised based on the results of various studies. Use of the rich comparative genomics resources from the model legumes Medicago truncatula and Lotus japonicus is also discussed. Finally, perspectives on the future directions for chickpea functional genomics, with the goal of developing elite chickpea cultivars, are discussed.
Additional keywords: abiotic stress, biotic stress, defence, transcriptomics, resistance.
Agricultural value and production constraints
Cultivated chickpea, Cicer arietinum L., is a self-pollinated, diploid (2n = 2x = 16) annual pulse crop with a genome size of 740 Mbp (Arumuganathan and Earle 1991). Chickpea originated in South-Eastern Anatolia (Turkey) (Ladizinsky 1975) and was traditionally cultivated in Asia, the Mediterranean, the Middle East and northern Africa. In contemporary times, chickpea has also become popular throughout the temperate regions of the world, in countries such as Mexico, Canada and Australia (Duke 1981). Chickpea is valued for its nutritious seeds, which contain 20–30% protein, ~40% carbohydrate and only 3–6% oil (Gil et al. 1996). Chickpea is mainly grown for human consumption, providing an important source of protein, especially for people in developing countries. Chickpea is often grown as a disease break in rotation with other crops and contributes to the maintenance of soil fertility through the fixation of atmospheric nitrogen (Singh 1997). In Australia, chickpea is commonly grown in rotation with wheat for the benefits of increased grain yield and grain protein concentration attributable to increased nitrogen supply and improved water-use efficiency (Dalal et al. 1998; Marcellos et al. 1998).
Chickpea is the third most important pulse crop in the world behind dry bean (Phaseolus vulgaris L.) and field pea (Pisum sativum L.), and Australia is currently the largest exporter (FAOSTAT 2005). World production of chickpea in 2005 was ~9.2 million metric tonnes from ~11.2 million hectares, constituting ~15% of the world pulse production from ~15% of the total global area used to grow pulses (FAOSTAT 2005). Despite a proposed yield potential of 6 metric tonnes ha–1 (Singh 1987), actual yields have remained low compared with other pulses (world average ~0.8 metric tonnes ha–1; FAOSTAT 2005), mainly because of biotic and abiotic stresses that reduce yield and yield stability. The necrotrophic foliar fungal disease Ascochyta blight (Ascochyta rabiei (Pass.) Labrousse) and the soil-borne necrotrophic fungal disease Fusarium wilt (Fusarium oxysporum f. sp. ciceris) are considered the most serious biotic stresses. Although winter sowing improves yields, it is rarely adopted in Mediterranean regions because the cool and wet conditions also favour Ascochyta blight. Other minor diseases of chickpea are more geographically localised and include pod borer (Helicoverpa armigera) in Australia and India (Nene and Reddy 1987), botrytis grey mould in areas that favour overgrowth and dense canopy (Kaiser et al. 2000), root rots in the tropics and sub-tropics (Kraft et al. 2000), rust in high-altitude regions (Nene and Reddy 1987) and broomrape in winter-sown areas (Rubiales et al. 1999).
In order of importance, drought, cold and salinity are the three main abiotic stresses that affect chickpea growth and productivity worldwide (Croser et al. 2003). As 90% of chickpea crops are cultivated under rain-fed conditions, drought is of major concern (Kumar and Abbo 2001). Injury from cold, chilling and freezing may affect plants at all developmental stages, however, when this occurs at flowering and/or pod set the consequences may be zero yield (Croser et al. 2003). Sensitivity to sodicity and salinity also adversely affects germination, biomass and yield (Ahmad et al. 2005). Subsequently, increasing resistance/tolerance to biotic and abiotic stresses to increase yield are the predominant aims of chickpea breeders throughout the world. Recently, a series of novel genetic tools have been developed that enable functional genomics studies in chickpea that are ultimately directed towards faster, more precise and efficient breeding to meet these objectives. Thus, the aims of this review are to: (1) provide an overview of the currently developed and developing functional genomics tools available for chickpea, (2) critically analyse the achievements made to date, and (3) propose future directions for chickpea functional genomics efforts for future molecular-assisted breeding efforts.
Molecular breeding
The chickpea cultigen contains high morphological variation, but narrow overall genetic variation, from which many desirable traits may have been excluded through selection (Abbo et al. 2003). For the desirable but missing traits from advanced breeding programs, such as durable resistance/tolerance to the many major biotic and abiotic stresses, breeders have begun to source germplasm more widely, from landraces and closely related species. To speed up the process of recombining ‘wild’ genes into elite genotypes, molecular tools have been integrated with classical breeding approaches. This has included the generation of molecular markers linked to the genes conditioning desirable traits, for efficient pyramiding of the traits. Molecular markers associated with quantitative trait loci (QTL) for resistance to biotic stresses and some morphological traits have been located on both intraspecific and interspecific linkage maps and, importantly, chickpea genotypes tolerant to most major biotic and abiotic stresses have been identified (see the review by Millan et al. 2006). The use of resistant or tolerant cultivars is considered to be the most efficient and effective means of controlling major stresses. However, a major problem for disease-resistant cultivars is that the resistance is incomplete and/or breaks down against new virulent races of pathogens that arise from mutation and genetic recombination. Wild Cicer species have also been identified as sources for resistance to some stresses (Singh et al. 1981; Collard et al. 2001; Croser et al. 2003) and, although interspecific crosses between wild species and C. arietinum have only been successful for Cicer reticulatum and Cicer echinospermum (Singh and Ocampo 1993; Collard et al. 2003), there still exists much potential for transferring resistance genes from wild Cicer species into cultivated chickpea.
Detailed information regarding the number, nature and diversity of genes controlling resistance/tolerance to biotic and abiotic stresses is essential for successful breeding programs. However, problems in dissecting polygenic traits and accurately measuring the underlying physiological mechanisms controlling tolerance to abiotic stresses make this difficult. As a result, molecular genetic studies have not provided a consistent picture of the genetic basis for biotic stress resistance, especially for resistance to Ascochyta blight (see the review by Millan et al. 2006). The narrow genetic variation in cultivated chickpea has limited the generation of informative molecular markers, while QTL for certain stresses differ with developmental stage, bioassay environmental conditions, the genotypes/fungal isolates used, and classifications for resistance and susceptibility. For example, numerous genetic mechanisms controlling Ascochyta blight resistance have been proposed, including single/multiple genes of dominant/recessive nature with modifiers and additive effects, as well as single/multiple QTL. The use of recombinant inbred line (RIL) populations was identified as a strategy to enable resistance studies to be performed on near homozygous individuals with temporal and spatial replication (Tekeoglu et al. 2000). Recent achievements have been made using RIL populations to study Ascochyta blight and Fusarium wilt resistance (Cobos et al. 2006; Iruela et al. 2007). An important QTL for Ascochyta blight resistance was identified on linkage group 2, which appears to cluster with a major gene for resistance to Fusarium wilt.
Functional genomics
Specific genes involved in resistance to biotic and abiotic stresses in chickpea have not been characterised using a genetics approach, but an enhanced understanding of the chickpea stress response at the genomic level may enable this. Plant stress responses are complex and diverse, and every gene involved, from recognition to signalling to direct involvement, forms part of a coordinated response network. Until recently, the genes and pathways of gene activation controlling effective stress resistance in chickpea remained unknown. Several approaches, including differential screening of cDNA libraries (Ichinose et al. 2000) and the placement of resistance gene analogues onto existing linkage maps (Rajesh et al. 2002), have identified candidate genes that are involved in Ascochyta blight resistance. Functional genomics provides opportunities for illuminating the mechanisms of chickpea resistance/tolerance to major biotic and abiotic stresses, possibly providing information concerning the molecular pathway(s) used by the plant, as well as the function of the candidate genes involved. Functional genomics incorporates several parallel approaches and tools, such as EST generation, transcript profiling, transgenics and reverse/forward genetics, for high throughput studies of gene function (Table 1). Ultimately the goal is to link the genome to the phenome, but understanding of the functional roles of genes is very limited compared with the knowledge of sequence information. Thus, a major challenge is to analyse and interpret the large-scale gene sequence data being produced to discover and understand the functional roles of underlying genes. Functional genomics has become widely used for studying the stress responses of plants, such as tomato (Gibly et al. 2004), rice (Fujiwara et al. 2004), maize (Baldwin 1998), cassava (Lopez et al. 2005), soybean (Moy et al. 2004) and Arabidopsis thaliana (Huitema et al. 2003) to name a few.

|
Initial expression studies
The first, yet indirect, analyses of the chickpea transcriptome focused on a biochemical investigation of Ascochyta-blight-elicited cell-suspension cultures and plant tissue (Mackenbrock et al. 1993; Barz and Mackenbrock 1994). These studies identified several rapid responses following elicitation, including an oxidative burst, extracellular alkalisation followed by acidification, and a K+ efflux. Although specific transcripts were not identified, the proteins involved in isoflavone metabolism were rapidly and transiently induced. Another biochemical study identified several pathogenesis-related (PR) proteins that were elicited much faster in the tissues of a resistant chickpea genotype than a susceptible one (Hanselle and Barz 2001). In addition, chemical inhibition studies identified serine/threonine protein kinases as important for the above-mentioned elicitor-induced responses (Otte et al. 2001). The first biochemical studies of Fusarium wilt resistance in chickpea roots reported the induction of proteases and numerous PR proteins (chitinase and β-1,3-glucanase), as well as the accumulation of phytoalexins and isoflavanoids (Stevenson et al. 1997; Giri et al. 1998; Saikia et al. 2005). Interestingly, these observations overlapped substantially with the results of early biochemical work for Ascochyta blight resistance. Furthermore, several QTL for Ascochyta blight and Fusarium wilt resistance cluster on linkage group 2 (see the review of Millan et al. 2006); thus, there may be an overlap in the resistance mechanisms against both of these necrotrophic fungal pathogens. The first direct study of mRNA abundance, through suppression subtractive hybridisation (SSH) of cDNA libraries from Ascochyta-blight-infected and non-infected tissue, identified 35 candidate defence-related transcripts, including two GTP-binding proteins thought to be involved in cell wall fortification (Cornels et al. 2000; Ichinose et al. 2000). Overall, these initial functional studies were relatively small in scale and did not provide sufficient coverage of the transcriptome to reveal specific defence-related pathways.
Expressed sequence tags and transcriptomics
A common first step in functional genomics studies is EST generation, which involves large-scale single-pass sequencing of randomly selected clones from cDNA libraries constructed from mRNA isolated at a particular developmental stage and in response to a particular stress. Functional identification of sequenced clones is being made easier by the availability of rapidly growing sequence databases, such as GenBank, and the full sequencing of model species genomes. Despite their disadvantage of not always representing full-length gene sequences, EST analysis has become a popular method for gene discovery and mapping in many organisms. For plants such as rice, maize and A. thaliana, comprehensive sets of EST sequences are available and have been used for the generation of molecular markers (Cato et al. 2001; Yu et al. 2004), the identification of gene families (Epple et al. 1997), single nucleotide polymorphism (SNP) markers (Cho et al. 1999) and the study of gene expression with microarrays (Schenk et al. 2000; Lan et al. 2004). Clustering of EST libraries into tentative consensus sequences (TCs) also allows for in silico (‘electronic northern’) analyses to identify putative genes with differential expression. ESTs may be particularly useful for the generation of molecular markers because: (1) an EST marker genetically associated with a trait is likely to represent a gene that directly affects that trait, and (2) EST markers are derived from highly conserved coding DNA sequences, which is likely to render them highly transportable across pedigrees compared with other markers derived from non-expressed sequences (e.g. simple sequence repeat markers) (Cato et al. 2001). As a result, the use of gene sequences derived from ESTs holds much promise for identifying the actual gene(s) controlling a desired trait. Furthermore, the use of EST-derived markers for the development of high-density (saturated) linkage maps will provide researchers with a greater arsenal of tools for QTL mapping and effective use of marker-assisted selection (MAS) (Collard et al. 2005; Dita et al. 2006).
The first report of large-scale EST generation in chickpea was published in 2005 (Coram and Pang 2005b). The assembly consisted of >500 unigenes that were isolated from the stems and leaves of an Ascochyta-blight-resistant chickpea genotype after pathogen inoculation. As a result, many potential defence-related unigenes were identified that were used in further studies (Coram and Pang 2005a, 2006, 2007). Also in 2005, an EST library of chickpea root tissue was made available (Jayashree et al. 2005). The library (>2800 ESTs) was constructed after SSH of root tissue from two closely related chickpea genotypes contrasting for drought avoidance and tolerance. Although these ESTs are yet to be used in functional studies, many potential drought responsive transcripts have been identified and developed into molecular markers. Sequence and annotation information for this EST library is maintained on an independent public database; thus, there are currently only 1311 chickpea ESTs listed in the National Center for Biotechnology Information (NCBI) EST database (GenBank dbEST), which is quite limited when compared with the number available for model legumes (Table 1).
The availability of chickpea ESTs has enabled a series of expression studies aimed at identifying suites of genes responding to particular stresses, thus, acting as a filter to narrow down small subsets of candidate genes that can be studied further. First, Coram and Pang (2005a) constructed a small-scale microarray using only putative defence-related ESTs. This array was used to profile the transcriptional changes occurring in two chickpea genotypes over a time-course after infection with Ascochyta blight (Coram and Pang 2005a). The study provided very limited results, but suggested that several defence-related transcripts were differentially induced in the resistant genotype compared with the susceptible genotype. Furthermore, the authors constructed a larger-scale microarray of >750 features encompassing a broad cross section of ESTs from various functional categories, as well as ESTs from a related temperate legume species (Lathyrus sativus) (Coram and Pang 2006). Differential gene transcription was assessed among four genotypes (including a wild relative) over a time-course after Ascochyta blight inoculation. By comparing the transcriptional profile of resistant and susceptible genotypes, the potential gene ‘signatures’ predictive of effective resistance were identified. These genes were involved in the regulation of known pathogen defence pathways, such as oxidative burst, hypersensitive response (HR), antimicrobial protein accumulation and phenylpropanoid production. Other reported functional studies on chickpea resistance to Ascochyta blight include the work of Cho and Muehlbauer (2004), who studied a small set of potential defence-related ESTs among chickpea genotypes differential in their reaction to both Ascochyta blight and Fusarium wilt. Following transcription quantification using RNA gel blots and reverse transcription–PCR, the expression of fungal response genes was not correlated with either pathotype-dependent resistance to Ascochyta blight, race-specific resistance to Fusarium wilt or non-host resistance to the Fusarium wilt pathogen of field pea.
Recent studies of signalling events inducing local and systemic defence responses in plants have led to the identification of salicylic acid (SA), jasmonic acid (JA) and ethylene (ET) as key regulators of these pathways (Schenk et al. 2000; Salzman et al. 2005; Jalali et al. 2006). Subsequently, Cho and Muehlbauer (2004) studied the response of their selected genes to treatment with SA and methyl jasmonate (MeJA), but found no correlation to the fungal responses. In addition, the authors found that Ascochyta blight resistance in RILs generated from the cross of a resistant and susceptible line did not cosegregate with the expression of the genes induced either by Ascochyta blight inoculation or the signal chemicals. As a result, the authors proposed that fungal resistance in chickpea may be controlled by constitutive or unknown resistance mechanisms independent of SA- or JA-mediated signalling. Coram and Pang (2007) used the same microarray previously used for the Ascochyta blight study to profile potential changes after treatment with SA, MeJA and an ethylene precursor, aminocyclopropane carboxylic acid (ACC). They determined that genotypes resistant to Ascochyta blight displayed a far greater range of defence-related gene inductions with all treatments compared with controls and the susceptible genotype. This indicated that genes within the conserved SA, MeJA and ethylene-type pathways were also likely to be involved in the defence response against Ascochyta blight. Furthermore, there was evidence for the involvement of resistance mechanisms other than SA, MeJA and ACC.
Overall, much functional work has been carried out to determine the action of genes involved in the defence against Ascochyta blight in chickpea. A summary model that represents the hypothetical defence pathways is proposed (Fig. 1). The hypothetical model was synthesised based on evidence gathered from biochemical and gene expression studies and follows the classical mechanisms of an oxidative burst, hypersensitive response, PR proteins and the involvement of the phenylpropanoid pathway. Although not definitive, the model forms a basis for further validation studies.
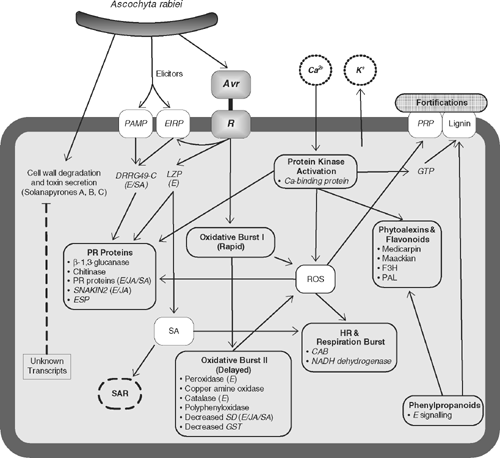
|
Nimbalkar et al. (2006) used a transcriptomics approach for characterisation of the molecular interactions between chickpea and race 1 of Fusarium oxysporum f. sp. ciceris. Transcription changes after root infection of a resistant and susceptible genotype using a cDNA-amplified fragment length polymorphism approach uncovered 19 differentially expressed sequences, potentially involved in a defence response. Notably, many were similar to previously characterised defence-related proteins, including two transcription factors and three nucleotide binding site–leucine rich repeat-type gene sequences (Nimbalkar et al. 2006). The use of existing and further developed EST libraries and microarrays will aid in a fuller characterisation of the mechanisms involved in the Fusarium wilt defence response.
To unravel the genes involved in abiotic stress responses in chickpea, a recent microarray study profiled the transcriptional response of tolerant and susceptible genotypes to drought, cold and high salinity (Mantri et al. 2007). Again, the ‘PulseChip’ microarray developed by Coram and Pang (2006) was used. Although developed specifically to hybridise with genes responsive to fungal inoculation the ‘PulseChip’ was considered to be appropriate for profiling gene expression in response to drought, cold and high salinity for the following three reasons: (1) the annotations of many ‘PulseChip’ ESTs were similar to those associated with abiotic stresses (Kreps et al. 2002; Seki et al. 2002; Rabbani et al. 2003), (2) a significant amount of crosstalk was reported between biotic and abiotic stress responses (Chen et al. 2002; Fujita et al. 2006), and (3) the unavailability of targeted abiotic stress-related cDNA libraries or ESTs in chickpea. In the study by Mantri et al. (2007), the abiotic stress treatments were administered carefully to simulate field conditions, and numerous transcripts potentially involved in each abiotic stress response were identified. The majority of differentially expressed transcripts were detected under high-salinity conditions (386), followed by cold (210) and drought (109) stress. The transcripts were categorised on function and a suite of putative candidate stress-related genes were identified that were consistently expressed among the tolerant or susceptible genotypes. Boominathan et al. (2004) carried out a gene expression study of drought adaptation in chickpea using SSH in combination with differential DNA-array hybridisation and northern blot analysis and identified 101 drought-inducible transcripts. These included several transcripts involved in the degradation of starch, which may indicate the importance of small sugar molecules for maintaining osmotic balance in stressed cells. The authors also found that the drought-inducible transcripts were induced by exogenous abscisic acid (ABA) treatment, suggesting a role for ABA-mediated signalling in dehydration tolerance (Boominathan et al. 2004). However, ABA was not involved in the regulation of dehydration-inducible transcripts in an earlier study in chickpea by Romo et al. (2001), who used differential cDNA library screening to identify a lipid transfer protein and late embryo abundant proteins as important for dehydration tolerance. The transcripts were also induced by high-salinity stress, which suggested that they play a role in the protection of cellular functions from damage by high ion concentrations (Romo et al. 2001). In general, the limited functional genomics studies of abiotic stresses in chickpea have suggested that complex mechanisms for tolerance exist, which may prove more difficult to dissect than those conditioning biotic stress tolerances. However, microarray technology has been effective in the identification and study of genes linked to drought, heat, cold and salinity stresses in other major plants (Seki et al. 2002; Rabbani et al. 2003; Buchanan et al. 2005; Rensink et al. 2005).
An alternative approach to improving stress tolerance involves genetically modifying a genome with a gene(s) thought to confer the desired tolerance. Recently, considerable progress was made in developing a stable genetic transformation system for chickpea (for a review see McPhee et al. (2007). Important achievements at the International Crops Research Institute for the Semi-Arid Tropics (ICRISAT) include the development of transgenic plants carrying the cry1Ac and P5CSF genes (Jandhyala 2005; Sharma 2006). The P5CSF129A gene for proline accumulation (Mitra 2001) stabilises degrading proteins (Munns 2005) under osmotic stress. The cry1Ac gene, derived from the bacterium Bacillus thuringiensis, produces a toxin that kills the economically important chickpea pod borer insect pest (Helicoverpa armigera) (Jandhyala 2005). In addition, the dehydration responsive element binding (DREB) gene, DREB1A, has been transferred into chickpea under the control of a stress-inducible promoter via Agrobacterium-mediated genetic transformation (Sharma 2006). The DREB1A construct enhances tolerance to drought, cold and salinity because it encodes a transcription factor that can regulate a suite of genes involved in stress tolerance (Yamaguchi-Shinozaki and Shinozaki 2006). Both DREB1A and P5CSF129A transgenic plants have shown increased drought tolerance and are currently under further study at ICRISAT.
Many of the functional genomics studies carried out in chickpea lack the comprehensiveness required to fully elucidate the genetic mechanisms of stress resistance/tolerance. A weakness of the cDNA microarray approach is that only the genes that encode cDNAs included on the array are assessed. The rather small number of cDNA on the chickpea arrays has limited the power for detecting many potentially important and rare transcripts. To overcome this, without needing to construct very large microarrays, Matsumura et al. (2003) developed the Super Serial Analysis of Gene Expression (SuperSAGE) technique. In brief, this technique is an improvement on SAGE by generating longer 26-bp gene tags that can be more accurately annotated. Very recently, SuperSAGE was used in chickpea to investigate salinity, drought and cold stress (Kahl et al. 2007). The authors exploited the high-power approach to analyse 40 000 unique mRNAs and identified >3000 genes responding to the stresses applied. A disadvantage of this method is that the short sequence tags (26-bp) used in SuperSAGE may be homologous to regions of numerous known sequences with different protein products; thus, there is a risk of mis-annotating the tags. In addition, for studying multiple time points, the SuperSAGE process has to be repeated for each time point, making it laborious and expensive. However, using SuperSAGE to identify large sets of candidate genes responding to a certain stress enables the construction of specialised microarrays that could be used to confirm gene functions by co-expression with other known genes. A combination of SuperSAGE and microarrays would enable the development of a more efficient and effective functional genomics tool to identify genes involved in stress resistance/tolerance. In addition, SuperSAGE appears to be more efficient for capturing rare and low-abundance transcripts than random EST sequencing.
Emerging and future trends for genomics-assisted breeding
Chickpea and other grain legumes have been ‘orphaned’ with regard to investment in molecular research compared with cereals and horticultural crops of high economic value. This scenario is slowly changing with efforts from organisations such as the European Union (EU), who have implemented a Grain Legumes Integrated Project (GLIP) to facilitate coordinated research in grain legumes. Recently, a GLIP dissemination event held in Madrid (Spain) unveiled current and future research interests (The Grain Legumes Portal: www.grainlegumes.com), which are focused on the importance of chickpea alongside major grain crops like field pea and the model legume Medicago truncatula. The chief aim of the GLIP is to understand the interrelationships of the multiple signalling systems that control stress-adaptive responses in legumes. To dissect the mechanisms of abiotic stress tolerance in legumes, gene expression patterns and metabolomic changes induced by various abiotic stresses in field pea, chickpea and M. truncatula will be analysed using various genomic approaches. This is coupled with detailed genetic mapping of crosses between salinity tolerant and sensitive varieties in chickpea and M. truncatula. The approach was implemented to help evaluate control mechanisms exerted by QTL on gene expression patterns and to identify regulators of gene expression and metabolic adaptation. The proposed outcomes of this project are: (1) identification of candidate genes induced by salinity, drought or cold stress in M. truncatula, field pea and chickpea, (2) generation of SSH cDNA libraries of field pea, chickpea and M. truncatula exposed to drought and salinity stress conditions, (3) identification of molecular markers associated with QTL linked to abiotic stress tolerances in M. truncatula and chickpea, (4) fine mapping of M. truncatula and chickpea QTL for salinity tolerance, and (5) generation of a ‘LeguStressChip’ to serve as a diagnostic tool to screen legume germplasm for stress tolerance (The Grain Legumes Portal: www.grainlegumes.com). The GLIP is also using a genomics approach to develop tools for transferring the information gained from model plants (including M. truncatula, Lotus japonicus and Arabidopsis thaliana) to grain legume crops, such as chickpea, field pea, faba bean, alfalfa and clover. Such a large-scale coordinated research project will greatly accelerate our understanding of stress tolerance in chickpea and other legumes and will boost the technology transfer from model crops to cultivated species.
Chickpea gene expression studies carried out to date using microarrays and SuperSAGE have identified candidate chickpea genes for resistance/tolerance to major biotic and abiotic stresses. A major criticism of these gene expression studies is that contrasting genotypes were used as resistant/tolerant and susceptible sources. A more strategic approach would be to use near-isogenic lines (NILs) that differ only for the trait of interest. The use of NILs or RILs could theoretically eliminate the gene expression responses detected as a result of background genetic differences that are not necessarily attributable to conferring resistance/susceptibility to a given stress. However, such germplasm has not been available for most previous studies, and may be difficult to generate for quantitative/polygenic traits. In addition, although ideal to study the expression profiles of all genes in the chickpea genome in response to particular stresses, the microarrays constructed so far are limited by the ESTs available and are likely to be under representative. Combining larger-scale gene expression profiling (e.g. SuperSAGE and/or increased EST generation) with the use of near-isogenic germplasm contrasting only for the trait of interest will greatly enhance the identification of genes directly involved in resistance/tolerance to key biotic and abiotic stresses.
Recently there have emerged a series of powerful functional genomics tools for model legumes and chickpea that will shape the future of research in this field (Table 1). For chickpea, a relatively dense integrated genetic map with most linkage groups related to chromosomes was developed (Vlacilova et al. 2002). Together with the existence of several bacterial artificial chromosome (BAC) libraries, this will greatly facilitate map-based gene/QTL cloning, genome sequencing and physical map construction. In fact, positional cloning of Ascochyta blight resistance genes from QTL1 is currently in progress (FJ Muehlbauer, pers. comm.; X Bian, pers. comm.). In addition, colinear mapping, making use of cross-species synteny, has enabled the recent placement of the same QTL from chickpea from different genetic backgrounds on the M. truncatula genome (Bian et al. 2007). With the advent of efficient chickpea transformation protocols (Senthil et al. 2004), important clones from the binary BAC library (Lichtenzveig et al. 2005) may be readily used in high-throughput transgenic studies. Also of great importance is the development of powerful and high-throughput array-based genotyping tools, such as Diversity Array Technology (DArT) and Tagged-Array Marker (TAM) (Table 1), which are beginning to be applied to legumes and have the capacity to enhance chickpea genomics.
A logical first step for many of the candidate genes identified by expression studies is to determine if they localise to mapped trait QTL regions. Marker development is greatly aided by the existence of the BAC libraries, from which full-length genomic sequences may be obtained. A candidate gene that can be placed under an existing QTL provides stronger evidence of associated function. The progression to map-based cloning of such genes may then be carried out through probing BAC libraries and/or comparative genomics. Sequencing of BAC clones under a QTL may also generate focused oligonucleotide microarrays that could be used to dissect a QTL region and identify the exact genes involved in the trait of interest. The major limitation of this approach is the location of the mapped probe(s) used to search for the associated BACs. If the marker is a large physical distance (~ >1 cM) from the candidate gene, then BAC contig walking becomes a lengthy and sometimes dead-ended process. This is because the genome coverage of the BAC library may be incomplete, particularly when BAC libraries are generated for large genomes, such as chickpea.
Much work is needed to take the step from candidate gene identification to proving gene function in traits such as resistance/tolerance, and then applying this information to improve chickpea breeding. Reverse/forward genetics approaches to fully determine gene function, such as Targeted Induced Local Lesions in Genomes (TILLING), insertional mutagenesis and activation tagging have been applied in the model legumes (Table 1). A recent development for chickpea was the generation of TILLING mutant populations that may be screened for change in function associated with previously identified candidate genes (Rajesh et al. 2007a; C. Pittock, pers. comm.). As a result, individuals carrying point mutations in the genes of interest can be phenotypically assessed to determine the association to function(s) of each gene. In addition to this valuable tool, the function of candidate genes may also be determined by transgenics and gene knockdown methods, such as RNA interference (RNAi) and Viral Induced Gene Silencing (VIGS), although these are yet to be applied in chickpea. Finally, it is important to note the emerging proteomics studies in chickpea, which have profiled the proteome of the chickpea cell wall (Bhushan et al. 2006) and nucleus (Pandey et al. 2006). These studies have functionally classified many chickpea proteins and are vitally important for linking the transcriptome to the proteome.
In an interesting recent development, Winter et al. (2006) built on SuperSAGE results by using 3′-Rapid Amplification of cDNA Ends (RACE) to generate expression markers in response to abiotic stresses. By searching the 3′-RACE products for single nucleotide polymorphisms (SNPs) via cleaved amplified polymorphic sequences (CAPS) and EcoTILLING (Comai et al. 2004), polymorphic markers were used to generate a chickpea expression map that was compared with the M. truncatula physical map to determine gene localisation. Expanding on this, a more optimistic future direction for chickpea functional genomics would include the use of microarrays to discover expression level polymorphisms that can be mapped with molecular markers as expression QTLs (eQTLs). Such gene expression markers would represent significant differences in transcript level between the parents of a segregating population. West et al. (2006) recently used this technique to successfully identify gene expression markers in an A. thaliana RIL population responding to treatment with a surfactant. In addition, single-feature polymorphisms (SFPs) may be identified between individual probes, which may represent SNPs that could be used as gene-specific markers. However, this type of study is more suited to large oligonucleotide microarrays, such as whole genome chips; thus, it is unlikely to be applied in chickpea research for some time. Finally, Massively Parallel Signature Sequencing (MPSS) (Brenner et al. 2000) is a similar, but more powerful, method to SAGE that can provide a representation of the mRNA population of a sample on a much larger scale. However, MPSS is very costly and has only been used in A. thaliana, rice and grape (http://mpss.udel.edu).
Comparative genomics
The model legumes M. truncatula and L. japonicus were chosen as the representative model legume species; thus, the tools available for these species are far broader and more developed than for chickpea (Table 1). As a result, comparative genomics using these models may accelerate aspects of chickpea functional genomics studies. Extensive genome sequence data and ESTs, saturated genetic/physical maps, large-scale microarrays, reverse/forward genetics tools, and bioinformatics resources are available for the model legumes. An important requirement for comparative genomics is the presence of sufficient similarity between the model genome/s and the genome of interest, which is referred to as macro- and micro-synteny. Macro-synteny refers to the presence of genes on the same chromosome region, while micro-synteny (or colinearity) refers to the conservation of gene order. Many recent studies demonstrate macro- and micro-synteny between the model and crop legumes (see the review by Cronk et al. 2006), which enabled positional gene cloning of, for example, a symbiosis gene in field pea (Stracke et al. 2004). An important resource for plant comparative genomics is the Phytome project (www.phytome.org) (Hartmann et al. 2006), which contains genomic data for the model legumes and can be used to identify orthologous and paralogous sequences, and for creating sequence-anchored maps from different species.
Chickpea is more closely related to M. truncatula than L. japonicus; thus, comparative gene mapping between chickpea and M. truncatula may be more successful. However, synteny between soybean and M. truncatula has been shown despite their evolutionary divergence (Mudge et al. 2005). Only one very recent study has assessed the extent of synteny between chickpea and M. truncatula based on 500 Kb of sequence from 11 BAC clones (Rajesh et al. 2007b). This study found evidence for macro-synteny, but relatively little micro-synteny. Furthermore, Coram and Pang (2005b) showed that levels of similarity between chickpea EST sequences and the model legumes were only marginally superior than those observed for A. thaliana; thus, the use of the models for the study of chickpea on a gene sequence level may be limited. Overall, the model legumes will be valuable for comparative mapping and positional cloning of chickpea genes, accelerating the process towards identifying the genes involved in stress resistance/tolerance. To a lesser extent because of a limit in the developed tools, other crop legumes, such as field pea and lentil, will also be valuable. Localisation of important markers/QTL on the physical maps of the model legumes, as performed by Winter et al. (2006) after SuperSAGE, will greatly aid the cloning of important genes.
Conclusions and perspectives
A major aim of chickpea breeding is the development of cultivars with adequate resistance/tolerance to yield-reducing stresses. However, breeding for abiotic stress tolerance is very complicated because of the combination of factors affecting the trait, and molecular breeding has had only limited success in improving tolerance. Traditional and molecular breeding targeting resistance to biotic stresses has been the focus of much research in the past 30 years. Most work has focused on improving either Ascochyta blight or Fusarium wilt resistance and, although some moderately resistant varieties have been released, the genes and pathways of gene regulation controlling these traits remain largely unknown. To overcome this gap, recent studies have focused on the use of functional genomics tools in an attempt to uncover important genes involved in resistance/tolerance to both biotic and abiotic stresses. Subsequently, several significant achievements have been made through functional genomics, including the generation of valuable resources, such as an integrated genetic map, EST libraries, BAC libraries, microarrays and SuperSAGE. The use of model legumes for comparative genomics also promises to enhance breeding efforts. However, the application of these resources to identifying the genes involved in mechanisms of biotic and abiotic stress resistance/tolerance is in its infancy. Candidate genes involved in biotic and abiotic stress tolerances have been identified through the use of both microarray and SuperSAGE techniques. Despite these achievements, there have been no reports of using candidate genes from functional genomics to improve chickpea cultivars in the field. However, this may change in the near future because the results of most functional genomics studies are relatively recent. The challenge for researchers now is to use the functional genomics resources available to develop definitive lists of candidate genes that may be important for each important stress. Generating narrow lists of candidates will enable the use of reverse/forward genetics approaches to validate the function of particular genes. This information can then be applied to breeding programs. To achieve such goals, the chickpea research community needs to bring together the strengths of various research groups around the world. Major groups working on functional genomics include a group in Australia (RMIT University, Bundoora and University of Melbourne) who possess a stem and leaf EST library and expertise in microarrays, a group at ICRISAT (India) who possess a root EST library valuable for drought studies and a well-developed transformation methodology, a group at The University of Frankfurt (Germany) who have pioneered the high-powered SuperSAGE technique in chickpea, and a group in the USA (USDA-ARS, Pullman) who possess valuable resources, including a TILLING population, BAC library and desirable germplasm (RILs and NILs). Continued exchange and collaboration of these resources is required to make a more rapid and significant progress towards function-associated chickpea breeding. Respective research groups should take more advantage of the International Chickpea Genomics Consortium (www.icgc.wsu.edu) to establish collaborative efforts that could allow for high-powered studies on ideal germplasm lines; thus, fast-tracking the overall development of resistant/tolerant cultivars. Finally, a database and bioinformatics resource capable of integrating and mining chickpea functional genomics data is lacking. Such resources are available for the model legumes and have greatly enhanced the coordination of research efforts and have allowed in-depth analysis of research results. The near future of functional genomics will see the development of many new and powerful techniques, such as expression QTL mapping and whole-genome sequencing. The current focus in chickpea functional genomics should be to coordinate valuable resources around the world and take full advantage of the power of functional genomics for improving chickpea crops.
Acknowledgements
Research at RMIT University was supported by a Grains Research Scholarship (Grains Research and Development Corporation; GRS52), a Vavilov–Frankel Fellowship (Biodiversity International), an Australian Postgraduate Award (Australian Research Council) and the Department of Primary Industry (Horsham).
Abbo S,
Berger J, Turner N
(2003) Evolution of cultivated chickpea: four bottlenecks limit diversity and constrain adaptation. Functional Plant Biology 30, 1081–1087.
| Crossref | GoogleScholarGoogle Scholar |

Arumuganathan K, Earle E
(1991) Nuclear DNA content of some important plant species. Plant Molecular Biology Reporter 9, 208–218.
Asamizu E,
Nakamura Y,
Sato S, Tabata S
(2005) Comparison of the transcript profiles from the root and the nodulating root of the model legume Lotus japonicus by serial analysis of gene expression. Molecular Plant—Microbe Interactions 18, 487–498.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Baldwin I
(1998) Jasmonate-induced responses are costly but benefit plants under attack in native populations. Proceedings of the National Academy of Sciences of the United States of America 95, 8113–8118.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Barz W, Mackenbrock U
(1994) Constitutive and elicitation induced metabolism of isoflavones and pterocarpans in chickpea (Cicer arietinum) cell-suspension cultures. Plant Cell, Tissue and Organ Culture 38, 199–211.
| Crossref | GoogleScholarGoogle Scholar |
Bhushan D,
Pandey A,
Chattopadhyay A,
Choudhary M,
Chakraborty S,
Datta A, Chakraborty N
(2006) Extracellular matrix proteome of chickpea (Cicer arietinum L.) illustrates pathway abundance, novel protein functions and evolutionary perspect. Journal of Proteome Research 5, 1711–1720.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Bian X,
Ford R,
Han T,
Coram T,
Pang E, Taylor P
(2007) Approaching chickpea quantitative trait loci conditioning resistance to Ascochyta rabiei via comparative genomics. Australasian Plant Pathology ,
Boominathan P,
Shukla R,
Kumar A,
Manna D,
Negi D,
Verma P, Chattopadhyay D
(2004) Long term transcript accumulation during the development of dehydration adaptation in Cicer arietinum. Plant Physiology 135, 1608–1620.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Brenner S,
Johnson M,
Bridgham J,
Golda J, Lloyd D
(2000) Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays. Nature Biotechnology 18, 630–634.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Buchanan C,
Lim S,
Salzman RA,
Kagiampakis I, Morishige DT , et al.
(2005) Sorghum bicolor’s transcriptome response to dehydration, high salinity and ABA. Plant Molecular Biology 58, 699–720.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Cannon S,
Crow J,
Heuer ML,
Wang X, Cannon EK , et al.
(2005) Databases and information integration for the Medicago truncatula genome and transcriptome. Plant Physiology 138, 38–46.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Cato S,
Gardner R,
Kent J, Richardson T
(2001) A rapid PCR-based method for genetically mapping ESTs. Theoretical and Applied Genetics 102, 296–306.
| Crossref | GoogleScholarGoogle Scholar |
Chen W,
Provart N,
Glazebrook J,
Katagiri F, Chang HS , et al.
(2002) Expression profile matrix of Arabidopsis transcription factor genes suggests their putative functions in response to environmental stresses. The Plant Cell 14, 559–574.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Cho R,
Mindrinos M,
Richards DR,
Sapolsky RJ, Anderson M , et al.
(1999) Genome-wide mapping with biallelic markers in Arabidopsis thaliana. Nature Genetics 23, 203–207.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Cho S, Muehlbauer F
(2004) Genetic effect of differentially regulated fungal response genes on resistance to necrotrophic fungal pathogens in chickpea (Cicer arietinum L.). Physiological and Molecular Plant Pathology 64, 57–66.
| Crossref | GoogleScholarGoogle Scholar |
Cobos M,
Rubio J,
Strange R,
Moreno M,
Gil J, Millan T
(2006) A new QTL for Ascochyta blight resistance in a RIL population derived from an interspecific cross in chickpea. Euphytica 149, 105–111.
| Crossref | GoogleScholarGoogle Scholar |
Collard B,
Ades P,
Pang E,
Brouwer J, Taylor P
(2001) Prospecting for sources of resistance to ascochyta blight in wild Cicer species. Australasian Plant Pathology 30, 271–276.
| Crossref | GoogleScholarGoogle Scholar |
Collard B,
Pang E,
Ades P, Taylor P
(2003) Preliminary investigation of QTLs associated with seedling resistance to ascochyta blight from Cicer echinospermum, a wild relative of chickpea. Theoretical and Applied Genetics 107, 719–729.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Collard B,
Jahufer M,
Brouwer J, Pang E
(2005) An introduction to markers, quantitative trait loci (QTL) mapping and marker-assisted selection for crop improvement: the basic concepts. Euphytica 142, 169–196.
| Crossref | GoogleScholarGoogle Scholar |
Comai L,
Young K,
Till BJ,
Reynolds SH, Greene EA , et al.
(2004) Efficient discovery of DNA polymorphisms in natural populations by Ecotilling. The Plant Journal 37, 778–786.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Constantin GD,
Krath1 BN,
MacFarlane SA,
Nicolaisen M,
Johansen IE, Lund OS
(2004) Virus-induced gene silencing as a tool for functional genomics in a legume species. The Plant Journal 40, 622–631.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Coram T, Pang E
(2005a) Isolation and analysis of candidate ascochyta blight defence genes in chickpea. Part II. Microarray expression analysis of putative defence-related ESTs. Physiological and Molecular Plant Pathology 66, 201–210.
| Crossref | GoogleScholarGoogle Scholar |
Coram T, Pang E
(2005b) Isolation and analysis of candidate ascochyta blight defence genes in chickpea. Part I. Generation and analysis of an expressed sequence tag (EST) library. Physiological and Molecular Plant Pathology 66, 192–200.
| Crossref | GoogleScholarGoogle Scholar |
Coram T, Pang E
(2006) Expression profiling of chickpea genes differentially regulated during a resistance response to Ascochyta rabiei. Plant Biotechnology Journal 4, 647–666.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Coram T, Pang E
(2007) Transcriptional profiling of chickpea genes differentially regulated by salicylic acid, methyl jasmonate, and aminocyclopropane carboxylic acid to reveal pathways of defence-related gene regulation. Functional Plant Biology 34, 52–64.
| Crossref | GoogleScholarGoogle Scholar |
Cornels H,
Ichinose Y, Barz W
(2000) Characterization of cDNAs encoding two glycine-rich proteins in chickpea (Cicer arietinum L.): accumulation in response to fungal infection and other stress factors. Plant Science 154, 83–88.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Cronk Q,
Ojeda I, Pennington R
(2006) Legume comparative genomics: progress in phylogenetics and phylogenomics. Current Opinion in Plant Biology 9, 99–103.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Croser J,
Clarke H,
Siddique K, Khan T
(2003) Low temperature stress: implications for chickpea (Cicer arietinum L.) improvement. Critical Reviews in Plant Sciences 22, 185–219.
| Crossref | GoogleScholarGoogle Scholar |
Dalal R,
Strong W,
Weston E,
Cooper J,
Wildermuth G,
Lehane K,
King A, Holmes C
(1998) Sustaining productivity of a Vertisol at Warra, Queensland, with fertilisers, no-tillage, or legumes 5. Wheat yields, nitrogen benefits and water-use efficiency of chickpea–wheat rotation. Australian Journal of Experimental Agriculture 38, 489–501.
| Crossref | GoogleScholarGoogle Scholar |
Dita M,
Rispail N,
Prats E,
Rubiales D, Singh K
(2006) Biotechnology approaches to overcome biotic and abiotic stress constraints in legumes. Euphytica 147, 1–24.
| Crossref | GoogleScholarGoogle Scholar |
Epple P,
Apel K, Bohlmann H
(1997) ESTs reveal a multigene family for plant defensins in Arabidopsis thaliana. FEBS Letters 400, 168–172.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Firnhaber C,
Puhler A, Kuster H
(2005) EST sequencing and time course microarray hybridizations identify more than 700 Medicago truncatula genes with developmental expression regulation in flowers and pods. Planta 222, 269–283.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Flavell A,
Bolshakov V,
Booth A,
Jing R,
Russell J,
Ellis T, Isaac P
(2003) A microarray-based high throughput molecular marker genotyping method: the tagged microarray marker (TAM) approach. Nucleic Acids Research 31, e115.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Fujita M,
Fujita Y,
Noutoshi Y,
Takahashi F,
Narusaka Y,
Yamaguchi-Shinozaki K, Shinozaki K
(2006) Crosstalk between abiotic and biotic stress responses: a current view from the points of convergence in the stress signaling networks. Current Opinion in Plant Biology 9, 436–442.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Fujiwara S,
Tanaka N,
Kaneda T,
Takayama S,
Isogai A, Che F-S
(2004) Rice cDNA microarray-based gene expression profiling of the response to flagellin perception in cultured rice cells. Molecular Plant—Microbe Interactions 17, 986–998.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Gibly A,
Bonshtien A,
Balaji V,
Debbie P,
Martin G, Sessa G
(2004) Identification and expression profiling of tomato genes differentially regulated during a resistance response to Xanthomonas campestris pv. vesicatoria. Molecular Plant—Microbe Interactions 17, 1212–1222.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Gil J,
Nadal S,
Luna D,
Moreno M, de Haro A
(1996) Variability of some physico-chemical characters in Desi and Kabuli chickpea types. Journal of the Science of Food and Agriculture 71, 179–184.
| Crossref | GoogleScholarGoogle Scholar |
Giri A,
Harsulkar A,
Patankar A,
Gupta V,
Sainani M,
Deshpande V, Ranjekar P
(1998) Association of induction of protease and chitinase in chickpea roots with resistance to Fusarium oxysporum f. sp. ciceri. Plant Pathology 47, 693–699.
| Crossref | GoogleScholarGoogle Scholar |
Hanselle T, Barz W
(2001) Purification and characterisation of the extracellular PR-2b (beta)-1,3-glucanase accumulating in different Ascochyta rabiei-infected chickpea (Cicer arietinum L.) cultivars. Plant Science 161, 773–781.
| Crossref | GoogleScholarGoogle Scholar |
Hartmann S,
Lu D,
Phillips J, Vision T
(2006) Phytome: a platform for plant comparative genomics. Nucleic Acids Research 34, D724–D730.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Huitema E,
Vleeshouwers V,
Francis D, Komoun S
(2003) Active defence responses associated with non-host resistance of Arabidopsis thaliana to the oomycete pathogen Phytophthora infestans. Molecular Plant Pathology 4, 487–500.
| Crossref | GoogleScholarGoogle Scholar |
Ichinose Y,
Tiemann K,
Schwenger-Erger C,
Toyoda K,
Hein F,
Hanselle T,
Cornels H, Barz W
(2000) Genes expressed in Ascochyta rabiei-inoculated chickpea plants and elicited cell cultures as detected by differential cDNA-hybridization. Zeitschrift für Naturforschung. C, Journal of Biosciences 55, 44–54.
| PubMed |
Imaizumi R,
Sato S,
Kameya N,
Nakamura I,
Nakamura Y,
Tabata S,
Ayabe S, Aoki T
(2005) Activation tagging approach in a model legume, Lotus japonicus. Journal of Plant Research 118, 391–399.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Iruela M,
Castro P,
Rubio J,
Cubero J,
Jacinto C,
Millan T, Gil J
(2007) Validation of a QTL for resistance to ascochyta blight linked to resistance to fusarium wilt race 5 in chickpea (Cicer arietinum L.). European Journal of Plant Pathology ,
| Crossref | GoogleScholarGoogle Scholar |
Jalali B,
Bhargava S, Kamble A
(2006) Signal transduction and transcriptional regulation of plant defence responses. Journal of Phytopathology 154, 65–74.
| Crossref | GoogleScholarGoogle Scholar |
Jayashree B,
Buhariwalla H,
Shinde S, Crouch J
(2005) A legume genomics resource: the chickpea root expressed sequence tag database. Electronic Journal of Biotechnology 8, 128–133.
| Crossref | GoogleScholarGoogle Scholar |
Kouchi H,
Shimomura K,
Hata S,
Hirota A, Wu G-J , et al.
(2004) Large-scale analysis of gene expression profiles during early stages of root nodule formation in a model legume, Lotus japonicus. DNA Research 11, 263–274.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Kreps J,
Wu Y,
Chang H,
Zhu T,
Wangand X, Harper J
(2002) Transcriptome changes for Arabidopsis in response to salt, osmotic, and cold stress. Plant Physiology 130, 2129–2141.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Kumagai H, Kouchi H
(2003) Gene silencing by expression of hairpin RNA in Lotus japonicus roots and root nodules. Molecular Plant—Microbe Interactions 16, 663–668.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Kumar J, Abbo S
(2001) Genetics of flowering time in chickpea and its bearing on productivity in semiarid environments. Advances in Agronomy 72, 107–138.
Ladizinsky G
(1975) A new Cicer from Turkey. Notes of the Royal Botanic Garden Edinburgh 34, 201–202.
Lan L,
Chen W,
Lai Y,
Suo J, Kong Z , et al.
(2004) Monitoring of gene expression profiles and isolation of candidate genes involved in pollination and fertilization in rice (Oryza sativa L.) with a 10K cDNA microarray. Plant Molecular Biology 54, 471–487.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Lichtenzveig J,
Scheuring C,
Dodge J,
Abbo S, Zhang H
(2005) Construction of BAC and BIBAC libraries and their applications for generation of SSR markers for genome analysis of chickpea, Cicer arietinum L. Theoretical and Applied Genetics 110, 492–510.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Limpens E,
Ramos J,
Franken C,
Raz V,
Compaan B,
Franssen H,
Bisseling T, Geurts R
(2004) RNA interference in Agrobacterium rhizogenes-transformed roots of Arabidopsis and Medicago truncatula. Journal of Experimental Botany 55, 983–992.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Lopez C,
Soto M,
Restrepo S,
Piegu B,
Cooke R,
Delseny M,
Tohme J, Verdier V
(2005) Gene expression profile in response to Xanthomonas axonopodis pv. manihotis in cassava using a cDNA microarray. Plant Molecular Biology 57, 393–410.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Mackenbrock U,
Gunia W, Barz W
(1993) Accumulation and metabolism of medicarpin and maackiain malonylglucosides in elicited chickpea (Cicer arietinum L.) cell-suspension cultures. Journal of Plant Physiology 142, 385–391.
Mantri NL,
Ford R,
Coram TE, Pang ECK
(2007) Transcriptional profiling of chickpea genes differentially regulated in response to high-salinity, cold and drought. BMC Genomics 8, 303.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Marcellos H,
Felton WJ, Herridge DF
(1998) Chickpea in wheat-based cropping systems of northern New South Wales - I. N-2 fixation and influence on soil nitrate and water. Australian Journal of Agricultural Research 49, 391–400.
| Crossref | GoogleScholarGoogle Scholar |
Matsumura H,
Reich S,
Ito A,
Saitoh H,
Kamoun S,
Winter P,
Kahl G,
Reuter M,
Krüger DH, Terauchi R
(2003) Gene expression analysis of plant host–pathogen interactions by SuperSAGE. Proceedings of the National Academy of Sciences of the United States of America 100, 15718–15723.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Millan T,
Clarke H,
Siddique K,
Buhariwalla H,
Gaur P,
Kumar J,
Gil J,
Kahl G, Winter P
(2006) Chickpea molecular breeding: new tools and concepts. Euphytica 147, 81–103.
| Crossref | GoogleScholarGoogle Scholar |
Mitra J
(2001) Genetics and genetic improvement of drought resistance in crop plants. Current Science 80, 758–763.
Moy P,
Qutob D,
Chapman B,
Atkinson I, Gijzen M
(2004) Patterns of gene expression upon infection of soybean plants by Phytophthora sojae. Molecular Plant—Microbe Interactions 17, 1051–1062.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Mudge J,
Cannon S,
Kalo P,
Oldroyd G,
Roe B,
Town C, Young N
(2005) Highly syntenic regions in the genomes of soybean, Medicago truncatula, and Arabidopsis thaliana. BMC Plant Biology 5, 15.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Munns R
(2005) Genes and salt tolerance: bringing them together. The New Phytologist 167, 645–663.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Nimbalkar S,
Harsulkar A,
Giri A,
Sainani M,
Franceschi V, Gupta V
(2006) Differentially expressed gene transcripts in roots of resistant and susceptible chickpea plant (Cicer arietinum L.) upon Fusarium oxysporum infection. Physiological and Molecular Plant Pathology 68, 176–188.
| Crossref | GoogleScholarGoogle Scholar |
Otte O,
Pachten A,
Hein F, Barz W
(2001) Early elicitor-induced events in chickpea cells: functional links between oxidative burst, sequential occurrence of extracellular alkalinisation and acidification, K+/H+ exchange and defence-related gene activation. Zeitschrift für Naturforschung. C, Journal of Biosciences 56, 65–76.
| PubMed |
Pandey A,
Choudhary M,
Bhushan D,
Chattopadhyay A,
Chakraborty S,
Datta A, Chakraborty N
(2006) The nuclear proteome of chickpea (Cicer arietinum L.) reveals predicted and unexpected proteins. Journal of Proteome Research 5, 3301–3311.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Pedrosa A,
Sandal N,
Stougaard J,
Schweizer D, Bachmair A
(2002) Chromosomal map of the model legume Lotus japonicus. Genetics 161, 1661–1672.
| PubMed |
Perry J,
Wang T,
Welham T,
Gardner S,
Pike J,
Yoshida S, Parniske M
(2003) A TILLING reverse genetics tool and a web accessible collection of mutants of the legume Lotus japonicus. Plant Physiology 131, 866–871.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Rabbani MA,
Maruyama K,
Abe H,
Khan MA,
Katsura K,
Ito Y,
Yoshiwara K,
Seki M,
Shinozaki K, Yamaguchi-Shinozaki K
(2003) Monitoring expression profiles of rice genes under cold, drought, and high-salinity stresses and abscisic acid application using cDNA microarray and RNA gel-blot analyses. Plant Physiology 133, 1755–1767.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Rajesh P,
Tekeoglu M,
Gupta V,
Ranjekar P, Muehlbauer F
(2002) Molecular mapping and characterisation of an RGA locus RGAPtokin1–2 171 in chickpea. Euphytica 128, 427–433.
| Crossref | GoogleScholarGoogle Scholar |
Rajesh P,
Coyne C,
Meksem K,
Dev Sharma K,
Gupta V, Muehlbauer F
(2004) Construction of a HindIII bacterial artificial chromosome library and its use in identification of clones associated with disease resistance in chickpea. Theoretical and Applied Genetics 108, 663–669.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Rensink W,
Iobst S,
Hart A,
Stegalkina S,
Liu J, Buell R
(2005) Gene expression profiling of potato responses to cold, heat and salt stress. Functional and Integrative Genomics 5, 201–207.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Romo S,
Labrador E, Dopico B
(2001) Water stress-regulated gene expression in Cicer arietinum seedlings and plants. Plant Physiology and Biochemistry 39, 1017–1026.
| Crossref | GoogleScholarGoogle Scholar |
Saikia R,
Singh B,
Kumar R, Arora D
(2005) Detection of pathogenesis-related proteins-chitinase and beta-1,3-glucanase in induced chickpea. Current Science 89, 659–663.
Salzman RA,
Brady JA,
Finlayson SA,
Buchanan CD, Summer EJ , et al.
(2005) Transcriptional profiling of sorghum induced by methyl jasmonate, salicylic acid, and aminocyclopropane carboxylic acid reveals cooperative regulation and novel gene responses. Plant Physiology 138, 352–368.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Sandal N,
Krusell L,
Radutoiua S,
Olbryta M, Pedrosab A , et al.
(2002) A genetic linkage map of the model legume Lotus japonicus and strategies for fast mapping of new loci. Genetics 161, 1673–1683.
| PubMed |
Sato S, Tabata S
(2006) Lotus japonicus as a platform for legume research. Current Opinion in Plant Biology 9, 128–132.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Schauser L,
Handberg K,
Sandal N,
Stiller J,
Thykjaer T,
Pajuelo E,
Nielsen A, Stougarrd J
(1998) Symbiotic mutants deficient in nodule establishment identified after T-DNA transformation of Lotus japonicus. Molecular and General Genetics 259, 414–423.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Schauser L,
Roussis A,
Stiller J, Stougaard J
(1999) A plant regulator controlling development of symbiotic root nodules. Nature 402, 191–195.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Schenk P,
Kazan K,
Wilson I,
Anderson J,
Richmond T,
Somerville S, Manners J
(2000) Coordinated plant defense responses in Arabidopsis revealed by microarray analysis. Proceedings of the National Academy of Sciences of the United States of America 97, 11655–11660.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Scholte M,
d’Erfurth I,
Rippa S,
Mondy S, Cosson V , et al.
(2002) T-DNA tagging in the model legume Medicago truncatula allows efficient gene discovery. Molecular Breeding 10, 203–215.
| Crossref | GoogleScholarGoogle Scholar |
Seki M,
Narusaka M,
Ishida J,
Nanjo T, Fujita M , et al.
(2002) Monitoring the expression profiles of 7000 Arabidopsis genes under drought, cold and high-salinity stresses using a full-length cDNA microarray. The Plant Journal 31, 279–292.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Senthil G,
Williamson B,
Dinkins R, Ramsay G
(2004) An efficient transformation system for chickpea (Cicer arietinum L.). Plant Cell Reports 23, 297–303.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Singh K
(1997) Chickpea (Cicer arietinum L.). Field Crops Research 53, 161–170.
| Crossref | GoogleScholarGoogle Scholar |
Singh K, Ocampo B
(1993) Interspecific hybridisation in annual Cicer species. Journal of Genetics and Breeding 47, 199–204.
Singh K,
Hawtin G,
Nene Y, Reddy M
(1981) Resistance in chickpeas to Ascochyta rabiei. Plant Disease 65, 586–587.
Stevenson P,
Turner H, Haware M
(1997) Phytoalexin accumulation in the roots of chickpea (Cicer arietinum L.) seedlings associated with resistance to fusarium wilt (Fusarium oxysporum f. sp. ciceris). Physiological and Molecular Plant Pathology 50, 167–178.
| Crossref | GoogleScholarGoogle Scholar |
Stiller J,
Martirani L,
Tuppale S,
Chian R,
Chiurazzi M, Gresshoff P
(1997) High frequency transformation and regeneration of transgenic plants in the model legume Lotus japonicus. Journal of Experimental Botany 48, 1357–1365.
| Crossref | GoogleScholarGoogle Scholar |
Stracke S,
Sato S,
Sandal N,
Koyama M,
Kaneko T,
Tabata S, Parniske M
(2004) Exploitation of colinear relationships between the genomes of Lotus japonicus, Pisum sativum and Arabidopsis thaliana, for positional cloning of a legume symbiosis gene. Theoretical and Applied Genetics 108, 442–449.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Tekeoglu M,
Santra D,
Kaiser W, Muehlbauer F
(2000) Ascochyta blight resistance inheritance in three chickpea recombinant inbred line populations. Crop Science 40, 1251–1256.
Trieu AT,
Burleigh SH,
Kardailsky IV,
Maldonado-Mendoza IE, Versaw WK , et al.
(2000) Transformation of Medicago truncatula via infiltration of seedlings or flowering plants with Agrobacterium. The Plant Journal 22, 531–541.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Vlacilova K,
Ohri D,
Vrana J,
Cihalikova J,
Kubalakova M,
Kahl G, Dolezel J
(2002) Development of flow cytogenetics and physical genome mapping in chickpea (Cicer arietinum L.). Chromosome Research 10, 695–706.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
West M,
van Leeuwen H,
Kozik A,
Kliebenstein D,
Doerge R,
St. Clair D, Michelmore R
(2006) High-density haplotyping with microarray-based expression and single feature polymorphism markers in Arabidopsis. Genome Research 16, 787–795.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Winter P,
Benko-Iseppon A-M,
Hüttel B,
Ratnaparkhe M, Tullu A , et al.
(2000) A linkage map of the chickpea (Cicer arietinum L.) genome based on recombinant inbred lines from a C-arietinum × C-reticulatum cross: localization of resistance genes for fusarium wilt races 4 and 5. Theoretical and Applied Genetics 101, 1155–1163.
| Crossref | GoogleScholarGoogle Scholar |
Yamaguchi-Shinozaki K, Shinozaki K
(2006) Transcriptional regulatory networks in cellular responses and tolerance to dehydration and cold stresses. Annual Review of Plant Biology 57, 781–803.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Yang S,
Pang W,
Ash G,
Harper J,
Carling J,
Wenzl P,
Huttner E,
Zong X, Kilian A
(2006) Low level of genetic diversity in cultivated Pigeonpea compared to its wild relatives is revealed by diversity arrays technology. Theoretical and Applied Genetics 113, 585–595.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Yu J,
La Rota M,
Kantety R, Sorrells M
(2004) EST derived SSR markers for comparative mapping in wheat and rice. Molecular Genetics and Genomics 271, 742–751.
| Crossref | GoogleScholarGoogle Scholar | PubMed |