

Disclosure of the hydrogen evolution mechanism on [FeFe]-hydrogenases-inspired molecular catalysts – a DFT study
Siyao Qiu A B , Aimin Yu C and Chenghua Sun C *
C *
A
B
C
Abstract
The FeFe bio-inspired molecular catalysts that mimic the [FeFe] hydrogenases have been widely studied. However, the hydrogen evolution mechanism on the molecular catalysts is still not fully understood. In this work, the theoretical calculation was linked with experimental catalytic performance to reveal the possible reaction mechanism of FeFe molecular catalysts. The Density Functional Theory (DFT) calculations on the FeFe molecular catalysts exhibited a good match with the experimental overpotential data, with a R2 of 0.592. The detailed DFT study indicated that the first H+/e− injection was the largest thermodynamic impediment in the whole hydrogen evolution reaction (HER) cycle which follows a proton transfer – electron transfer (PT-ET) mechanism. The injected hydrogen binds to the bridging position between FeFe centre (µ-H) and then transfers to a terminal hydrogen on Fe (t-H). Later, the t-H combines with the second injected hydrogen to form a H2 molecule which is then released from the catalyst. The effect of different ligands on HER was also studied. It was found that different ligands around the FeFe centre could significantly change the PT and ET energy, and some could provide additional binding sites for protons.
Keywords: bioinspired catalysts, catalytic mechanism, DFT, electron transfer, FeFe hydrogenases, H2 production, molecular catalysts, proton transfer.
Introduction
H2, as an environmentally friendly energy, is one of the most promising alternatives to traditional fossil fuels. The existing high-performance inorganic catalysts for HER are often made of expensive metals, like Pt.1,2 In nature, a class of enzyme, hydrogenase, has been found to possess impressive catalytic activity but only consists of earth-abundant metals.3 Based on the metal ions at the active site, the hydrogenases could be further classified into [NiFe], [FeFe] and [Fe] hydrogenases.4,5 It has been reported that [FeFe] hydrogenases present the highest catalytic efficiency for the hydrogen cleavage into proton plus electron.5–8 Hence, a number of [FeFe]-hydrogenases-inspired molecular catalysts were designed and synthesised to mimic the [FeFe] hydrogenases to promote HER.
In the [FeFe] hydrogenases, the bimetallic Fe centre is bridge linked by one SCH2NHCH2S ligand and one carbonyl group. Moreover, one Fe metal (Fep) is terminally linked with a [Fe4S4] cluster, one carbonyl and one cyanide. By contrast, the other Fe (Fed) merely bonds to one carbonyl and one cyanide, leaving a vacant position for hydrogen binding. Molecular catalysts with different Fe2S2 butterfly structures have been synthesised to resemble the active site of the [FeFe] hydrogenases.6 The bridging linkers in these Fe2S2 butterfly molecular catalysts are also dithiolate ones, which have different moieties replacing the NH in SCH2NHCH2S ligand of hydrogenases. There are different kinds of bridging linkers for FeFe molecular catalysts, and we focused on dithiolate ones in this work.7–9 Also, a variety of terminal ligands that bound to Fe in molecular catalysts were synthesised replacing the CN and CO terminal ligands in hydrogenases. The catalytic performance of FeFe molecular catalysts with different ligands have been summarised and compared in previous reviews.10,11
The HER cycle on the active site of [FeFe] hydrogenases has been proposed previously.12–17 The first added proton would bind to the bridging azadithiolate (adt) ligand by N.5,18 Afterwards, accompanied by the second proton injection, the first added proton transfers to Fed and the second injected proton binds to the adt ligand again.19 Subsequently, the second proton will be delivered to Fed, generating H2 and will then be released.
Though a similar ‘butterfly’ structure could be seen in the molecular catalysts, the HER cycle in the molecular catalysts is still not quite clear.20,21 The previous computational-aided designs of FeFe molecular catalysts have been focused on mimicking the geometrical characteristic of hydrogenase. In the previous works, it was found that bridging binding (µ-H) is more stable compared with terminal H binding (t-H) in FeFe catalysts.22,23 It was found that the µ-H binding is much more stable than t-H binding, whereas the second hydrogen injection on µ-H binding of the FeFe core is more difficult than the t-H binding moiety. In FeFe hydrogenases, a hydrogen atom terminally binds to the FeFe centre. Therefore, previous work by DFT calculation suggested the design of a stable rotated FeFe catalyst, on which the hydrogen could directly bind to the terminal vacant position of Fe, could be possible.24 Moreover, considering that the injected proton initially binds to the bridging dithiolate ligand in FeFe hydrogenase's active site, there is work on FeFe molecular catalysts proposing a reaction path that dithiolate protonates before the formation of µ-hydrides.25
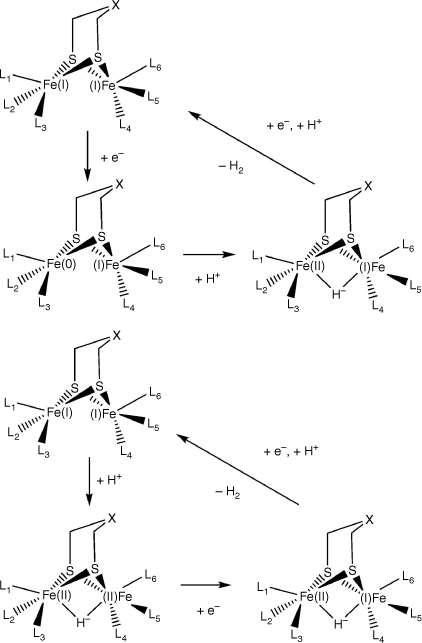
For theoretical work on HER cycle of molecular catalysts, some works proposed that the catalyst is reduced first and then the proton binds to the catalyst to form a H2 molecule. This is named as ET-PT mechanism as shown in Scheme 1.26–29 Scheme 1 also presents the PT-ET mechanism which has also been proposed in previous research.30,31 Moreover, NMR spectroscopy has also suggested the existence of a binding hydride on FeFe molecular catalysts upon the addition of an acid.25,32 A previous study indicated that the injected proton is inclined to form a bridging hydride between Fe ions during the HER process.33 To illustrate the hydrogen evolution mechanism on FeFe molecular catalysts, the energy change during the cycle was calculated. However, the H2 and proton and electron energy were not considered and there is no comparison between theoretical and experimental data.29 In this work, we aimed to link the theoretical calculations with experimental catalytic performance to reveal the possible reaction mechanism. The HER process on the FeFe molecular catalysts was studied to understand HER path on the molecular catalysts. All possible reaction routes for eight different catalysts were considered in our calculation. The largest theoretical energy difference obtained from the calculations were compared with the experimental values.10
Computational methods
Density functional theory (DFT) calculations were carried out for all catalyst models through the Gaussian 09 calculation program.34–36 PBE functional with Def2TZVPP basis set was applied for all models.37 The choice of PBE functional was based on our previous work, which suggested PBE showed the best performance in predicting the redox potential of [FeFe] molecular catalysts compared with experimental data (see Supplementary Table S1 and Fig. S1).38 In all cases, the dispersion of the models was considered by including the empirical dispersion (GD3) compensation.39 Also, the Synchronous Transit-Guided Quasi-Newton (STQN) method (QST3) was applied to determine the transition state (TS). To derive the free energy changes, frequency calculations were conducted for all models. All models were built based on the X-ray crystallographic data of different models. Experimental data of overpotentials of molecular catalysts including the potential of HER in nonaqueous solvents and pKa correction were reported by Felton et al.40 Since the experimental values were corrected for all catalysts, the energy difference for the H+/e− added step in this work could be approximated as half of the free energy of the H2 molecule, that is, G(H+ + e−) = ½ G(H2) v. SHE. Proton energy was estimated according to the previous work by high level first principles calculation, being −262.4 kcal mol–1.41
Results and discussion
Eight different catalysts were studied in this work (see Fig. 1), including (µ-SCH2CH2)2OFe2(CO)6,42 (µ-SCH2CH2CH2S)-Fe2(CO)5SO(CH3)2,43 (µ-pdt)Fe2(CO)5P(NC4H8)3,44 Fe2[µ-S2(CH2)3](CO)5P(CH2)6N3,26 Fe2[µ-S2(CH2)3] (CO)5IMes,45 [µ-S-2-(4-FC6H4)CONHC6H4]2Fe2(CO)6,46 (µ-bdt)Fe2(CO)647 and CH3CH2CH2N(µ-SCH2)2[Fe2(CO)5DAPTA](2-DAPTA).48
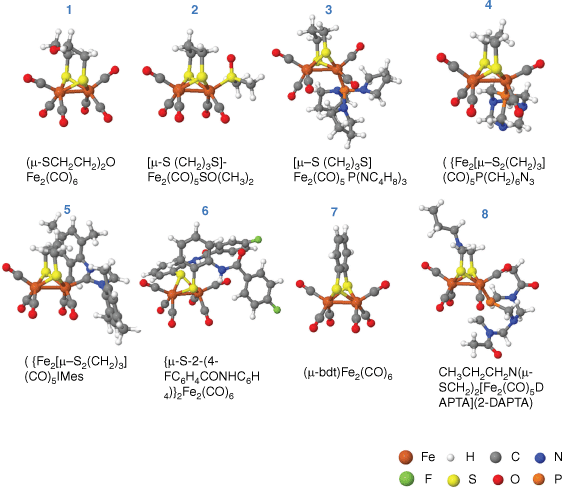
The (µ-SCH2CH2)2OFe2(CO)6 is one of the simplest structures among the eight catalysts. Possible hydrogen binding sites of this catalyst were calculated and are shown in Fig. 2. Five different possible routes werecalculated. Route 1, 2 and 3 are three different mechanisms for H+/e− injection to FeFe µ-H binding sites, which are the proton transfer – electron transfer (PT-ET), the electron transfer – proton transfer (ET-PT) and the concerted proton electron transfer (CPET) respectively. It was found that it is impossible for two H atoms that both bind to the bridging site to generate a H2 molecule. The two H atoms preferred to bind separately on two Fe metals. Therefore, to generate and release H2, the bridge binding H transferred to the terminal binding H on one Fe, and then combined with the second injected H. For direct t-H binding in Route 4, the FeFe catalyst first rotated to form an unsymmetrical conformation, and then the H+/e− was injected onto the terminal Fe forming the t-H. In Route 5, a H bound to the bridging ligand and then transferred to the terminal Fe to form the t-H. An additional conformation with one hydrogen on terminal Fe and the other on a bridging ligand, which is similar to the HhydH+ state in FeFe hydrogenase,18 was also calculated. However, such a conformation was not stable, as the proton on the ligand would spontaneously move to the hydride that's terminally binding on Fe and generate H2. The calculation results showed that the energy through Route 1 with the PT-ET mechanism was the lowest among the five routes. The largest thermodynamic step was merely +0.61 eV (+14.0 kcal mol–1) for the PT-ET mechanism, whereas the CPET required +0.72 eV (+16.5 kcal mol–1) and the ET-PT needed even more energy than the CPET route, being +1.29 eV (+29.9 kcal mol–1). The lowest reaction route for HER is highlighted in green in Fig. 2. To generate the H2 molecule, the bridging hydride rotated to the terminal binding position. The energy barrier for the rotation step was +1.10 eV (+25.4 kcal mol–1). After the first H+/e− injection, the second hydrogen atom was directly added to the Fe, with an energy demand of +0.11 eV (+2.6 kcal mol–1). Then, a H2 molecule was spontaneously released from the catalyst.
HER pathway followed by the (µ-SCH2CH2)2OFe2(CO)6 molecular catalysts. Gibbs free energy differences, in electronvolts (kcal mol–1), are shown. Binding H atoms are highlighted in pink.
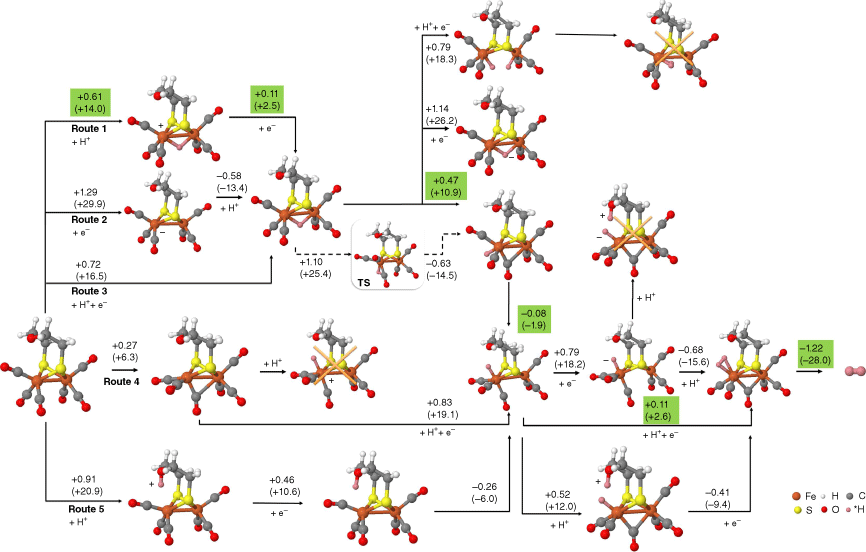
The reaction path for the CH3CH2CH2N(µ-SCH2)2[Fe2(CO)5DAPTA](2-DAPTA), (µ-bdt)Fe2(CO)6, Fe2[µ-S2(CH2)3](CO)5P(CH2)6N3, Fe2[µ-S2(CH2)3](CO)5Imes, Fe2[µ-S2(CH2)2N(2-C4H3O](CO)6 and (µ-SCH2CH2CH2S)-Fe2(CO)5SO(CH3)2 are similar with the (µ-SCH2CH2)2OFe2(CO)6 catalyst, in which the hydrogen production also followed the Route 1 PT-ET mechanism. The proton initially bound to the bridging position between FeFe, and then electron transferred to the FeFe centre. Later, the bridging hydride rotated to the terminal site on a single Fe ion, and combined with the second injected hydrogen forming H2. Moreover, we found the second proton and electron injection only required a small amount of energy, whereas the first hydrogen atom injection or its rotation was usually the largest thermodynamic impediment. For example, the largest thermodynamic step for the (µ-bdt)Fe2(CO)6 molecule was the addition of the proton to the bridging position, being +0.67 eV (+15.5 kcal mol–1), as shown in Supplementary Fig. S2. The energy differences for hydrogen production in (µ-bdt)Fe2(CO)6 are quite similar with (µ-SCH2CH2)2OFe2(CO)6, suggesting the change of bridging dithiolate ligand from CH2OCH2 to bdt had limited influence on the electronic property of Fe.
However, the largest impeding step for the CH3CH2CH2N(µ-SCH2)2[Fe2(CO)5DAPTA] (2-DAPTA) molecule was the electron injection step after the first proton was injected to the bridging position. As seen from Supplementary Fig. S3, it required +0.19 eV (+4.4 kcal mol–1) for the PT step and +0.49 eV (+11.2 kcal mol–1) for the ET step. This indicates that the ligands could change the PT and ET energy required for HER. A similar energy profile could be seen in Supplementary Fig. S4 for the (µ-pdt)Fe2(CO)5P(NC4H8)3. Likewise, for Fe2[µ-S2(CH2)3](CO)5P(CH2)6N3, the largest energy difference step was also the ET step after the first proton injection, which was +0.52 eV (+12.1 kcal mol–1). Similarly, Fe2[µ-S2(CH2)3](CO)5IMes showed a larger ET step energy of +0.62 eV (+14.3 kcal mol–1) than the PT step of +0.16 eV (+3.7 kcal mol–1). The results indicated that energy for PT was reduced when a more electron-abundant ligand was introduced. In previous work, electron-abundant ligands were widely introduced to mimic the [Fe4S4] cluster in hydrogenase.49 However, our calculation suggested the more nucleophilic the ligand, the larger the energy required for the first electron injection. Therefore, to obtain a smaller overpotential, we should find a ligand that could balance the energy difference between PT and ET. For instance, (µ-SCH2CH2CH2S)-Fe2(CO)5SO(CH3)2 molecule, which has a −SO(CH3)2 ligand terminally linked to the Fe ion, the first proton transferred to the bridging position required +0.33 eV (+7.6 kcal mol–1), and the following ET required +0.23 eV (+5.2 kcal mol–1), as shown in Supplementary Fig. S5. The largest energy requirement was the rotation of the bridging hydride to the terminally bounded proton, which was +0.4 eV (+9.1 kcal mol–1).
The ligands of molecular catalysts could affect the reaction energy not only by influencing the proton or electron injection to the FeFe centre but also by providing available binding sites for a proton before proton transfer to the FeFe centre. For example, calculation on [µ-S-2-(4-FC6H4) CONHC6H4]2Fe2(CO)6 found, without considering the bridging sulfide ligand, the first H+/e− injection required 0.73 eV (+16.9 kcal mol–1). However, the first proton injection energy was reduced to 0.48 eV (+11.1 kcal mol–1), when the proton was injected onto the ketone CO on the sulfide ligand. Later, with the 0.26 eV (+6.0 kcal mol–1) energy injection, the proton was transferred to the bridging position between FeFe accompanied by the first electron injection (see Supplementary Fig. S6).
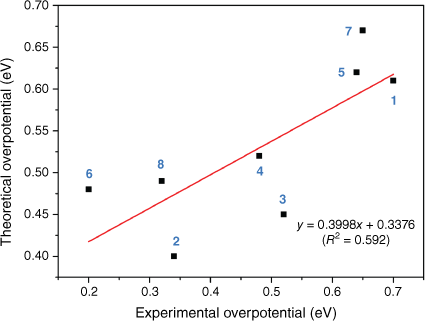
The experimental overpotential data are shown compared with the largest energy difference along the reaction for each complex by calculation in Fig. 3. Our calculation exhibited a good match with the experimental overpotential, with a R2 of 0.592. The calculated result was sensitive to the free energy of the proton, and the energy value used here was also recommended by a recent paper.32 The deviation may also be attributed to the different experimental environments and the error that exists between the theoretical prediction and experimental result.
Summary and conclusions
In summary, our DFT calculation matched the experimental overpotential with a R2 value of 0.592. Based on the calculation, the mechanism of the hydrogen evolution process on FeFe molecular catalysts has been proposed; the first H+/e− injection of the HER on the FeFe molecular catalysts follows the PT-ET mechanism. The proton is added to the bridging position between Fe ions and then rotates to the terminal position of a single Fe. Then the second hydrogen binds terminally to the Fe resulting in the formation of dihydrogen homolytically. The largest thermodynamic impediment was found to be the first proton transfer step. The ligand of molecular catalysts could affect the required HER energy in two ways; one is influencing the PT and ET energy, and the other is introducing a new intermediate step by providing a proton binding position by the modified ligands. Our findings on the FeFe molecular catalysts enables the screening of molecular catalysts with low overpotentials by theoretical calculations, contributing to the future development of bio-inspired molecular catalysts.
Supplementary material
HER pathways of FeFe molecular catalysts. Supplementary material is available online.
Data availability
The data that support this study will be shared upon reasonable request to the corresponding author.
Declaration of funding
This research was sponsored by a grant to Siyao Qiu from the Guangdong Basic and Applied Basic Research Foundation (2020A1515110978).
References
1 Conway BE, Tilak BV. Interfacial processes involving electrocatalytic evolution and oxidation of H2, and the role of chemisorbed H. Electrochimica Acta 2002; 47(22): 3571-3594.
| Crossref | Google Scholar |
2 Nørskov JK, Bligaard T, Logadottir A, Kitchin JR, Chen JG, Pandelov S, Stimming U. Trends in the exchange current for hydrogen evolution. J Electrochem Soc 2005; 152(3): J23.
| Crossref | Google Scholar |
3 Madden C, Vaughn MD, Díez-Pérez I, Brown KA, King PW, Gust D, Moore AL, Moore TA. Catalytic turnover of [FeFe]-hydrogenase based on single-molecule imaging. J Am Chem Soc 2012; 134(3): 1577-1582.
| Crossref | Google Scholar | PubMed |
4 Vignais PM, Billoud B. Occurrence, classification, and biological function of hydrogenases: an overview. Chem Rev 2007; 107(10): 4206-4272.
| Crossref | Google Scholar | PubMed |
5 Lubitz W, Ogata H, Rüdiger O, Reijerse E. Hydrogenases. Chem Rev 2014; 114(8): 4081-4148.
| Crossref | Google Scholar | PubMed |
6 Li Y, Rauchfuss TB. Synthesis of diiron(I) dithiolato carbonyl complexes. Chem Rev 2016; 116(12): 7043-7077.
| Crossref | Google Scholar | PubMed |
7 Gimbert-Suriñach C, Bhadbhade M, Colbran SB. Bridgehead hydrogen atoms are important: unusual electrochemistry and proton reduction at iron dimers with ferrocenyl-substituted phosphido bridges. Organometallics 2012; 31(9): 3480-3491.
| Crossref | Google Scholar |
8 Korb M, Liu X, Walz S, Mahrholdt J, Popov AA, Lang H. Structural variety of iron carbonyl clusters featuring ferrocenylphosphines. Euro J Inorg Chem 2021; 2021(21): 2017-2033.
| Crossref | Google Scholar |
9 Arrigoni F, Rizza F, Vertemara J, Breglia R, Greco C, Bertini L, Zampella G, De Gioia L. Rational design of Fe2(µ-PR2)2(L)6 coordination compounds featuring tailored potential inversion. ChemPhysChem 2020; 21(20): 2279-2292.
| Crossref | Google Scholar | PubMed |
10 Felton GAN, Mebi CA, Petro BJ, Vannucci AK, Evans DH, Glass RS, Lichtenberger DL. Review of electrochemical studies of complexes containing the Fe2S2 core characteristic of [FeFe]-hydrogenases including catalysis by these complexes of the reduction of acids to form dihydrogen. J Organometallic Chem 2009; 694(17): 2681-2699.
| Crossref | Google Scholar |
11 Amaro-Gahete J, Pavliuk MV, Tian H, Esquivel D, Romero-Salguero FJ, Ott S. Catalytic systems mimicking the [FeFe]-hydrogenase active site for visible-light-driven hydrogen production. Coord Chem Rev 2021; 448: 214172.
| Crossref | Google Scholar |
12 Wittkamp F, Senger M, Stripp ST, Apfel UP. [FeFe]-Hydrogenases: recent developments and future perspectives. Chem Commun 2018; 54(47): 5934-5942.
| Crossref | Google Scholar | PubMed |
13 Mulder DW, Ratzloff MW, Shepard EM, Byer AS, Noone SM, Peters JW, Broderick JB, King PW. EPR and FTIR analysis of the mechanism of H2 activation by [FeFe]-hydrogenase HydA1 from Chlamydomonas reinhardtii. J Am Chem Soc 2013; 135(18): 6921-6929.
| Crossref | Google Scholar | PubMed |
14 Silakov A, Kamp C, Reijerse E, Happe T, Lubitz W. Spectroelectrochemical characterization of the active site of the [FeFe] hydrogenase HydA1 from Chlamydomonas reinhardtii. Biochemistry 2009; 48(33): 7780-7786.
| Crossref | Google Scholar | PubMed |
15 Adamska A, Silakov A, Lambertz C, Rüdiger O, Happe T, Reijerse E, Lubitz W. Identification and characterization of the “super-reduced” state of the H-cluster in [FeFe] hydrogenase: a new building block for the catalytic cycle? Angew Chem Int Ed 2012; 51(46): 11458-11462.
| Crossref | Google Scholar | PubMed |
16 Sbraccia C, Zipoli F, Car R, Cohen MH, Dismukes GC, Selloni A. Mechanism of H2 production by the [FeFe]H subcluster of di-iron hydrogenases: implications for abiotic catalysts. J Phys Chem B 2008; 112(42): 13381-13390.
| Crossref | Google Scholar | PubMed |
17 Bruschi M, Zampella G, Fantucci P, De Gioia L. DFT investigations of models related to the active site of [NiFe] and [Fe] hydrogenases. Coord Chem Rev 2005; 249(15): 1620-1640.
| Crossref | Google Scholar |
18 Birrell JA, Pelmenschikov V, Mishra N, Wang H, Yoda Y, Tamasaku K, Rauchfuss TB, Cramer SP, Lubitz W, DeBeer S. Spectroscopic and computational evidence that [FeFe] hydrogenases operate exclusively with CO-bridged intermediates. J Am Chem Soc 2020; 142(1): 222-232.
| Crossref | Google Scholar | PubMed |
19 Bruschi M, Greco C, Kaukonen M, Fantucci P, Ryde U, De Gioia L. Influence of the [2Fe]H subcluster environment on the properties of key intermediates in the catalytic cycle of [FeFe] hydrogenases: hints for the rational design of synthetic catalysts. Angew Chem Int Ed 2009; 48(19): 3503-3506.
| Crossref | Google Scholar | PubMed |
20 Schilter D, Camara JM, Huynh MT, Hammes-Schiffer S, Rauchfuss TB. Hydrogenase enzymes and their synthetic models: the role of metal hydrides. Chem Rev 2016; 116(15): 8693-8749.
| Crossref | Google Scholar | PubMed |
21 Gloaguen F, Rauchfuss TB. Small molecule mimics of hydrogenases: hydrides and redox. Chem Soc Rev 2009; 38(1): 100-108.
| Crossref | Google Scholar | PubMed |
22 Barton BE, Rauchfuss TB. Terminal hydride in [FeFe]-hydrogenase model has lower potential for H2 production than the isomeric bridging hydride. Inorg Chem 2008; 47(7): 2261-2263.
| Crossref | Google Scholar | PubMed |
23 Filippi G, Arrigoni F, Bertini L, De Gioia L, Zampella G. DFT dissection of the reduction step in H2 catalytic production by [FeFe]-hydrogenase-inspired models: can the bridging hydride become more reactive than the terminal isomer? Inorg Chem 2015; 54(19): 9529-9542.
| Crossref | Google Scholar | PubMed |
24 Arrigoni F, Rizza F, Bertini L, De Gioia L, Zampella G. Toward diiron dithiolato biomimetics with rotated conformation of the [FeFe]-hydrogenase active site: a dft case study on electron-rich, isocyanide-based scaffolds. Euro J Inorg Chem 2022; 2022(17): e202200153.
| Crossref | Google Scholar |
25 Zhou X, Barton BE, Chambers GM, Rauchfuss TB, Arrigoni F, Zampella G. Preparation and protonation of Fe2(pdt)(CNR)6, electron-rich analogues of Fe2(pdt)(CO)6. Inorg Chem 2016; 55(7): 3401-3412.
| Crossref | Google Scholar | PubMed |
26 Mejia-Rodriguez R, Chong D, Reibenspies JH, Soriaga MP, Darensbourg MY. The hydrophilic phosphatriazaadamantane ligand in the development of H2 production electrocatalysts: iron hydrogenase model complexes. J Am Chem Soc 2004; 126(38): 12004-12014.
| Crossref | Google Scholar | PubMed |
27 Ghosh S, Hollingsworth N, Warren M, Hrovat DA, Richmond MG, Hogarth G. Hydrogenase biomimics containing redox-active ligands: Fe2(CO)4(µ-edt)(κ2-bpcd) with electron-acceptor 4,5-bis(diphenylphosphino)-4-cyclopenten-1,3-dione (bpcd) as a potential [Fe4–S4]H surrogate. Dalton Trans 2019; 48(18): 6051-6060.
| Crossref | Google Scholar | PubMed |
28 Orton GRF, Ringenberg MR, Hogarth G. Biomimics of [FeFe]-hydrogenases incorporating redox-active ligands: ferrocene-bridged dithiolate complexes [Fe2(CO)6(µ-EC5H4FeC5H4E)] (E = S, Se). J Organometallic Chem 2022; 978: 122472.
| Crossref | Google Scholar |
29 Natarajan M, Kumar N, Joshi M, Stein M, Kaur-Ghumaan S. Mechanism of diiron hydrogenase complexes controlled by nature of bridging dithiolate ligand. ChemistryOpen 2022; 11(1): e202100238.
| Crossref | Google Scholar | PubMed |
30 Rauchfuss TB. Diiron azadithiolates as models for the [FeFe]-hydrogenase active site and paradigm for the role of the second coordination sphere. Acc Chem Res 2015; 48(7): 2107-2116.
| Crossref | Google Scholar | PubMed |
31 Ghosh S, Sanchez BE, Richards I, Haque MN, Holt KB, Richmond MG, Hogarth G. Biomimetics of the [FeFe]-hydrogenase enzyme: Identification of kinetically favoured apical-basal [Fe2(CO)4(µ-H){κ2-Ph2PC(Me2)PPh2}(µ-pdt)] + as a proton-reduction catalyst. J Organometallic Chem 2016; 812: 247-258.
| Crossref | Google Scholar |
32 Orton GRF, Ghosh S, Alker L, Sarker JC, Pugh D, Richmond MG, Hartl F, Hogarth G. Biomimics of [FeFe]-hydrogenases incorporating redox-active ligands: synthesis, redox properties and spectroelectrochemistry of diiron–dithiolate complexes with ferrocenyl-diphosphines as Fe4S4 surrogates. Dalton Trans 2022; 51(25): 9748-9769.
| Crossref | Google Scholar | PubMed |
33 Tschierlei S, Ott S, Lomoth R. Spectroscopically characterized intermediates of catalytic H2 formation by [FeFe] hydrogenase models. Energy Environ Sci 2011; 4(7): 2340-2352.
| Google Scholar |
34 Perdew JP. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys Rev B 1986; 33(12): 8822-8824.
| Crossref | Google Scholar | PubMed |
35 Becke AD. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev A 1988; 38(6): 3098-3100.
| Crossref | Google Scholar | PubMed |
36 Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery Jr JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam NJ, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. GAUSSIAN09, Revision D.01. Wallingford, CT, USA: Gaussian, Inc.; 2009.
37 Weigend F, Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys Chem Chem Phys 2005; 7(18): 3297-3305.
| Crossref | Google Scholar | PubMed |
38 Qiu S, Yu A, Sun C. Assessment of density functional theory methods for the [FeFe]-hydrogenases-inspired molecular catalysts. Chem Phys Lett 2025; 860: 141805.
| Crossref | Google Scholar |
39 Grimme S, Antony J, Ehrlich S, Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J Chem Phys 2010; 132(15): 154104.
| Crossref | Google Scholar | PubMed |
40 Felton GAN, Glass RS, Lichtenberger DL, Evans DH. Iron-only hydrogenase mimics. Thermodynamic aspects of the use of electrochemistry to evaluate catalytic efficiency for hydrogen generation. Inorg Chem 2006; 45(23): 9181-9184.
| Crossref | Google Scholar | PubMed |
41 Zhan C-G, Dixon DA. Absolute hydration free energy of the proton from first-principles electronic structure calculations. J Phys Chem A 2001; 105(51): 11534-11540.
| Crossref | Google Scholar |
42 Song L-C, Gao J, Wang H-T, Hua Y-J, Fan H-T, Zhang X-G, Hu Q-M. Synthesis and structural characterization of metallocrown ethers containing butterfly Fe2S2 cluster cores. Biomimetic hydrogen evolution catalyzed by Fe2(µ-SCH2CH2OCH2CH2S-µ)(CO)6. Organometallics 2006; 25(24): 5724-5729.
| Crossref | Google Scholar |
43 Hu M-Q, Ma C-B, Si Y-T, Chen C-N, Liu Q-T. Diiron models for the active site of Fe-only hydrogenase with terminal organosulfur ligation: synthesis, structures and electrochemistry. J Inorg Biochem 2007; 101(10): 1370-1375.
| Crossref | Google Scholar | PubMed |
44 Hou J, Peng X, Zhou Z, Sun S, Zhao X, Gao S. Tris(N-pyrrolidinyl)phosphine substituted diiron dithiolate related to iron-only hydrogenase active site: synthesis, characterization and electrochemical properties. J Organometallic Chem 2006; 691(22): 4633-4640.
| Crossref | Google Scholar |
45 Tye JW, Lee J, Wang H-W, Mejia-Rodriguez R, Reibenspies JH, Hall MB, Darensbourg MY. Dual electron uptake by simultaneous iron and ligand reduction in an N-heterocyclic carbene substituted [FeFe] hydrogenase model compound. Inorg Chem 2005; 44(16): 5550-5552.
| Crossref | Google Scholar | PubMed |
46 Yu Z, Wang M, Li P, Dong W, Wang F, Sun L. Diiron dithiolate complexes containing intra-ligand NH⋯S hydrogen bonds: [FeFe] hydrogenase active site models for the electrochemical proton reduction of HOAc with low overpotential. Dalton Trans 2008; 2008(18): 2400-2406.
| Crossref | Google Scholar | PubMed |
47 Cabeza JA, Martínez-García MA, Riera V, Ardura D, García-Granda S. Binuclear iron(I), ruthenium(I), and osmium(I) hexacarbonyl complexes containing a bridging benzene-1,2-dithiolate ligand. Synthesis, X-ray structures, protonation reactions, and EHMO calculations. Organometallics 1998; 17(8): 1471-1477.
| Crossref | Google Scholar |
48 Wang Z, Liu J, He C, Jiang S, Åkermark B, Sun L. Diiron azadithiolates with hydrophilic phosphatriazaadamantane ligand as iron-only hydrogenase active site models: synthesis, structure, and electrochemical study. Inorg Chim Acta 2007; 360(7): 2411-2419.
| Crossref | Google Scholar |
49 Arrigoni F, Elleouet C, Mele A, Pétillon FY, De Gioia L, Schollhammer P, Zampella G. Insights into the two-electron reductive process of [FeFe]H2ase biomimetics: cyclic voltammetry and DFT investigation on chelate control of redox properties of [Fe2(CO)4(κ2-chelate)(µ-dithiolate)]. Chem. Eur. J. 2020; 26(72): 17536-17545.
| Crossref | Google Scholar | PubMed |