

Structural diversity in nudibranch chemistry: elucidation of norditerpenes with a dendrillane scaffold from the Australian nudibranch Goniobranchus coi
Louise C. Forster A B , Gregory K. Pierens C , James J. De Voss A and Mary J. Garson A *
A B , Gregory K. Pierens C , James J. De Voss A and Mary J. Garson A *
A
B
C
Abstract
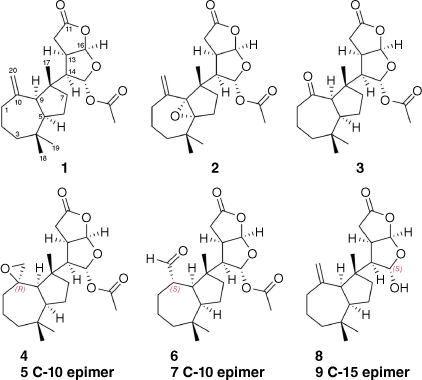
In addition to three known compounds (1–3), two new rearranged spongian diterpenes (4 and 5) with perhydroazulene and dioxabicyclooctane ring systems were isolated from the mantle and viscera of the Australian nudibranch Goniobranchus coi. The relative configuration of the major spiroepoxide 4 was explored by comparison with those of m-chloroperbenzoic acid oxidation products derived from dendrillolide A (1). Aldehydes 6 and 7 were identified as the ring-opened artefacts of the spiroepoxides 5 and 4 respectively, and yielded ketone 3 on storage. The relative configurations of lactols 8 and 9, isolated as an inseparable mixture of diastereomers, were deduced by molecular modelling and computational studies. Acetylation of the lactol mixture provided dendrillolide A (1), further confirming the structural assignments of 8 and 9. Dissection of animal tissue established that the norditerpenoid metabolites were present in both mantle and viscera tissues.
Keywords: dendrillolide, density functional calculations, epoxide, lactol, NMR spectroscopy, norditerpene, nudibranch, perhydroazulene, relative configuration.
Introduction
Goniobranchus coi (Risbec, 1956) is a nudibranch species with distinctive purple markings that is found in tropical Western Pacific locations. In common with other Goniobranchus nudibranchs, these colourful marine animals have a distinctive crawling motion in which the mantle edge flaps rhythmically up and down. According to Rudman and Bergquist,1 ‘Goniobranchus coi is a sponge predator known to feed upon Chelonaplysilla violacea of the Darwinellidae family. It separates and concentrates the noxious distasteful antifeedant chemicals produced by the sponge and contained in its tissues, into the previously mentioned mantle glands for its own defensive purposes.’2 This statement is backed up by photographs on the website SeaSlug Forum that show the mollusc feeding on Chelonaplysilla violacea.3 Although the chemistry of C. violacea has been well documented by Bobzin and Faulkner,4,5 there is limited published data on the secondary metabolite composition of G. coi. Dendrillolide A (1) is the major metabolite of C. violacea from Palau,5 and a minor component in New Zealand collections.6 Dendrillolide A (1) was not reported from C. violacea specimens collected from New South Wales, Australia.7,8
Our research group previously analysed the chemistry of a single specimen of G. coi collected from Mackay, north-east Queensland, and identified the known norditerpenes dendrillolide A (1), norrisolide, macfarlandin E, cheloviolene C and chromolactol.9 We then examined the chemistry of two specimens of G. coi collected from the Percy Isles and Coolum in Queensland and identified 11 known oxygenated terpenes. These two specimens contained a suite of oxygenated metabolites, including a 5,9-epoxy derivative 2 and a (nor)ketone 3, both of which exhibited extensive line broadening in their 1H NMR spectra, and whose structures and stereochemistry were therefore solved by application of variable temperature NMR together with density functional theory (DFT) calculations.10 The current report extends the chemical description of these two specimens of G. coi through structural and stereochemical investigation of additional oxygenated metabolites (Fig. 1). In addition to 10,20-epoxide-functionalised structures 4 and 5, we identified aldehyde rearrangement products 6 and 7 that could be linked to both the epoxide metabolite(s) and the previously reported (nor)ketone 3.11 Both specimens were also found to contain lactol analogues 8 and 9 related to dendrillolide A. Our findings provide an insight into the chemical diversity of G. coi and its dietary relationship with C. violacea.
The nudibranch specimens were collected under permit, with specimen number 1426 collected by Dr A Roberts-Thomson (Salty Pets Pty Ltd, Qld) and specimen number 1647 collected by Gary Cobb (permit number 183990, General Fisheries, Qld).
Results and discussion
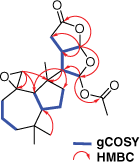
Two specimens of G. coi were collected from Percy Isles (2015) and Coolum (2017), Australia, and chemical extracts of the mantle and gut tissue prepared and analysed as described previously.11 Metabolite 4 was isolated from both the mantle and viscera as a colourless oil and displayed a sodiated molecular ion peak in the high-resolution electrospray ionisation mass spectrum (HRESIMS) at m/z 415.2083 [M + Na]+, corresponding to a molecular formula of C22H32O6. Comparison of the 1H NMR and 13C NMR spectroscopic data (Table 1) of 4 with those of dendrillolide A (1)12,13 supported a dendrillane-derived skeleton. Gradient correlation spectroscopy (gCOSY) correlations confirmed the H2-1/H2-2/H2-3 and H-9/H-5/H2-6/H2-7 spin systems of a perhydroazulene fragment shown to be linked to the 2,8-dioxabicyclo[3.3.0]octane ring system through heteronuclear multiple bond correlations (HMBC) from H-14 to C-7 (δC 38.4), C-8 (δC 47.7) and C-9 (δC 53.4) (Fig. 2). In the 2,8-dioxabicyclo[3.3.0]octane ring system, gCOSY connectivities linked H-14 (δH 2.70) to both H-13 (δH 3.05) and H-15 (δH 6.38), as well as H-13 to H-16 (δH 5.98). There were HMBC correlations from H-15 to C-14 at δC 56.2 and the acetate carbonyl at δC 169.5, as well as correlations from H-16 to C-15 (δC 96.9), C-13 (δC 42.4), C-12 (δC 29.1) and the lactone at δC 175.7 (C-11). The characteristic exomethylene signals of dendrillolide A at δH 4.83 (d) and 4.57 (d) were replaced with signals typical of an epoxide at δH 2.64 (d, J = 5.2 Hz) and 2.40 (dd, J = 5.2, 1.5 Hz). HMBC correlations from the epoxide protons (H2-20) to signals at δC 37.2 (C-1), 53.4 (C-9) and 61.2 (C-10) established the spiroepoxide at the C-10 position. Accordingly, 4 was named 10,20-epoxydendrillolide A.
| Position | 4 | 5 | 6 | 7 | |||||
|---|---|---|---|---|---|---|---|---|---|
| 13C | 1H, mult. (J, Hz) | 13C | 1H, mult. (J, Hz) | 13C | 1H, mult. (J, Hz) | 13C | 1H, mult. (J, Hz) | ||
| 1 | 37.2, CH2 | a2.06, m | 35.5, CH2 | a1.82, m | 30.3, CH2 | a1.72, m | 32.7, CH2 | a2.19, dt (14.8, 4.0) | |
| b1.21, br dd (13.7, 4.9) | b1.27, m | b1.53, m | b1.46, m | ||||||
| 2 | 24.1, CH2 | a1.66, m | 22.1, CH2 | a1.77, m | 24.5, CH2 | a1.66, m | 20.3, CH2 | a1.84, m | |
| b1.45, m | b1.50, m | b1.66, m | b1.71, m | ||||||
| 3 | 37.2, CH2 | a1.58, m | 37.7, CH2 | a1.61, m | 44.1, CH2 | a1.62, m | 39.4, CH2 | a1.79, m | |
| b1.32, m | b1.41, m | b1.35, m | b1.30, m | ||||||
| 4 | 36.3, C | – | 35.8, C | – | 36.4, C | – | 35.1, C | – | |
| 5 | 54.1, CH | 1.69, m | 55.7, CH | 1.72, m | 52.6, CH | 2.10, m | 55.0, CH | 1.95, m | |
| 6 | 27.2, CH2 | a1.69, m | 26.9, CH2 | a1.73, m | 26.9, CH2 | a1.81, m | 27.0, CH2 | a1.70, m | |
| b1.69, m | b1.70, m | b1.62, m | b1.61, m | ||||||
| 7 | 38.4, CH2 | a1.62, m | 37.8, CH2 | a1.73, m | 36.5, CH2 | a1.72, m | 38.0, CH2 | a1.68, m | |
| b1.41, m | b1.46, m | b1.44, m | b1.39, m | ||||||
| 8 | 47.7, C | – | 46.4, C | – | 46.3, C | – | 46.4, C | – | |
| 9 | 53.4, CH | 1.73, m | 54.4, CH | 1.71, m | 49.5, CH | 2.39, t (9.9) | 54.7, CH | 2.58, m | |
| 10 | 61.2, C | – | 60.1, C | – | 52.8, C | 2.55, m | 54.7, C | 2.58, m | |
| 11 | 175.7, C | – | 175.1, C | – | 175.1, C | – | 174.9, C | – | |
| 12 | 29.1, CH2 | a2.75, dd (18.0, 9.2) | 28.4, CH2 | a2.72, mA | 29.5, CH2 | a2.64, dd (17.1, 9.9) | 28.7, CH2 | a2.61, m | |
| b2.67, dd (18.0, 10.1) | b2.52, dd (18.0, 9.8) | b2.52, m | b2.57, m | ||||||
| 13 | 42.4, CH | 3.05, tdd (9.6, 6.4, 4.4) | 42.0, CH | 3.13, mA | 41.8, CH | 3.05, tdd (9.7, 6.6, 4.4) | 41.6, CH | 3.05, tdd (9.7, 6.6, 4.6) | |
| 14 | 56.2, CH | 2.70, t (6.6) | 55.9, CH | 2.65, mA | 52.1, CH | 2.54, m | 53.3, CH | 2.63, m | |
| 15 | 96.9, CH | 6.38, d (6.8) | 97.0, CH | 6.42, d (6.6) | 96.7, CH | 6.41, d (6.7) | 96.7, CH | 6.39, d (6.8) | |
| 16 | 104.8, CH | 5.98, d (4.3) | 105.0, CH | 6.04, d (4.5) | 104.7, CH | 6.00, d (4.5) | 104.3, CH | 6.01, d (4.2) | |
| 17 | 24.1, CH3 | 1.34, s | 25.8, CH3 | 1.19, s | 22.1, CH3 | 1.00, s | 26.6, CH3 | 1.14, s | |
| 18 | 34.8, CH3 | 0.97, s | 34.7, CH3 | 1.97, s | 25.1, CH3 | 0.86, s | 26.0, CH3 | 1.05, s | |
| 19 | 26.5, CH3 | 0.99, s | 25.8, CH3 | 1.11, s | 32.8, CH3 | 0.99, s | 32.7, CH3 | 1.03, s | |
| 20 | 56.7, CH3 | 2.64, d (5.2) | 56.4, CH3 | 2.84, d (4.9) | 204.8, CH | 9.57, d (4.4) | 204.4, CH | 9.95, s | |
| 2.40, dd (5.2, 1.5) | 2.66, mA | ||||||||
| 15-OAc | 169.5, C | – | 169.5, C | – | 169.6, C | – | 169.2, C | – | |
| 15-OAc | 21.1, CH3 | 2.09, s | 21.1, CH3 | 2.09, s | 21.4, CH3 | 2.14, s | 21.2, CH3 | 2.15, s | |
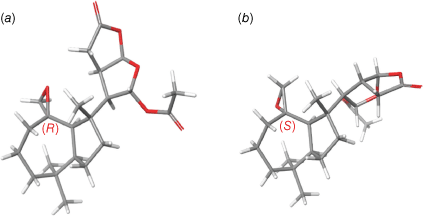
Nuclear Overhauser Enhancement spectroscopy correlations (NOESY) between H-5/H-9, and between H-13/H-16, H-14/H-16, and H-13/14 were fully consistent with the relative configuration of dendrillolide A (1). Although NOESY correlations were observed from the epoxide proton at δH 2.40 (H-20b) to Me-19 (δH 0.99) and H-9 (δH 1.73), inspection of molecular models revealed that the configuration at C-10 could not be conclusively assigned from the NOESY data. Two candidate stereostructures, 4 or its C-10 epimer 5, were considered. A Monte Carlo Multiple Minimum (MCMM) conformational search was undertaken using MacroModel (ver. 2018-1, Schrödinger Inc., New York, NY, USA) providing eight conformers for 4 and 12 conformers for 5. The lowest energy conformers (<3 kcal mol−1 of the global minimum) were next optimised by DFT calculations using the B3LYP/6-31G(d,p) method with the integral equation formalism polarisable continuum model (IEF-PCM) for chloroform solvent (Gaussian, ver. 16W, revision B.01, Gaussian, Inc., Wallingford, CT, USA), converging to six and five conformers for 4 and 5 respectively. The two lowest energy conformers of 4 were within 0.07 kcal mol−1 of the global minimum and thus represented >75.6% of the conformational population. These two conformers both had a chair conformation for the cycloheptane ring and differed only in the orientation of the 2,8-dioxabicyclo[3.3.0]octane moiety relative to the perhydroazulene fragment. The two conformers of 4 showed a C(13)–C(14)–C(8)–C(9) dihedral angle of 68.5 and 173.2° respectively. The lowest energy conformer for 4 represented 40.3% of the conformational population. The lowest energy conformer of 5, which represented >84.6% of the conformational population, had a chair conformation for the cycloheptane ring and a C(13)–C(14)–C(8)–C(9) dihedral angle of 175.7° (Fig. 3). The free energies of 4 and 5 were used to calculate the Boltzmann-weighted 1H and 13C NMR chemical shifts in chloroform solvent (IEF-PCM) (see Tables S1–S4 in the Supplementary material). The calculated chemical shifts were then examined using the modified DP4 computational approach developed by Grimblat et al. to assign the most probable diastereomer.14 Using both the 1H and 13C NMR data, the probability was 99.99% that the C-10 configuration of 4 was R.
An aldehyde product 6 was purified as a colourless oil from both the mantle and the viscera and displayed a sodiated molecular ion peak in the HRESIMS at m/z 415.2092 [M + Na]+. The corresponding molecular formula of C22H32O6 was identical to that of spiroepoxide 4. Comparison of the 1H NMR spectroscopic data (Table 1) of 6 with those of dendrillolide A (1)12,13 again supported a dendrillane-derived skeleton, except that the exomethylene signals of dendrillolide A (1) were replaced by a methine signal at δH 2.55 (m) and an aldehyde methine at δH 9.57 (d, J = 4.4 Hz, H-20). HMBC correlations from the aldehyde proton (H-20) to the signals at δC 30.3 (C-1), 49.5 (C-9) and 52.8 (C-10) established the position of an aldehyde at the C-10 position. By inspection of NOESY and coupling constant data, the relative configurations of the perhydroazulene and 2,8-dioxabicyclo[3.3.0]octane moieties were found to be consistent with a dendrillolide A-like scaffold. Signals for aldehyde 6 were not observed in the 1H NMR spectrum of the crude extract and were only observed after the sample was purified by silica chromatography, implying that 6 was a rearrangement product formed during the purification process.
Epoxides isomerise into carbonyl compounds under mild acid conditions (House–Meinwald rearrangement) by hydride migration across the unhindered face of the epoxide, antiperiplanar to the breaking C–O bond.15,16 As a result of this preference, an epoxide with 10R configuration was expected to convert into an aldehyde with 10R configuration, whereas the diastereomeric S-epoxide would convert into the 10S aldehyde.
To establish the stereochemical relationship between epoxide 4 and aldehyde 6, a sample of dendrillolide A (1) was reacted with m-chloroperbenzoic acid (mCPBA) in a biphasic solution with phosphate buffer and DCM for 3 days, yielding a mixture of the 10R and 10S spiroepoxides (Scheme 1). From the 1H NMR data of the crude reaction mixture, the signals for the major component matched those of the 10R-epoxide 4 whereas the signals of the minor epoxide were in agreement with the computationally derived data for 10S-epoxide 5. During acquisition of 2-D NMR data, the signals for the epoxide diastereomers disappeared, and aldehyde signals became apparent. Fortunately, the degradation process was relatively slow, so the initially collected heteronuclear single quantum correlation (HSQC) and HMBC data provided sufficient information for full characterisation of the minor epoxide 5 from the reaction mixture (Table 1). Re-inspection of the 1H NMR data then revealed evidence of traces of epoxide 5 in the G. coi crude extract.
Chemical correlation of spiroepoxides 4 and 5 and aldehyde products 6 and 7 following epoxidation of dendrillolide A (1) with mCPBA.
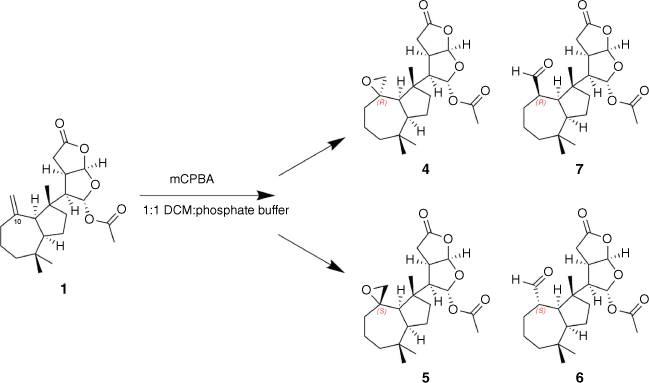
Subsequent normal-phase high-performance liquid chromatography (NP-HPLC) (25% EtOAc in hexanes) of the mCPBA reaction mixture afforded directly two aldehyde products, one of which had 1H NMR data that matched those for the isolated aldehyde from G. coi. Inspection of a model of 4 revealed the epoxide protons are orientated towards the centre of the cycloheptane ring, placing them in proximity (2.65 Å) to one of the geminal dimethyl groups (Me-19). This steric hindrance shields the epoxide from undergoing rapid rearrangement to form the 10R-aldehyde 7. By contrast, in 5 the epoxide protons are orientated outward from the cycloheptane ring, placing an epoxide proton in proximity to Me-17 (2.54 Å), and the epoxide can undergo isomerisation to the 10S-aldehyde product 6. Consequently, it was inferred that both epoxides were originally present in the crude extracts of G. coi, however, when the extract was exposed to silica chromatography, the 10S-epoxide 5 isomerised to form the 10S-aldehyde product 6. On inspection, traces of aldehyde 7 were also detectable in the 1H NMR spectrum of the crude extract. The specific optical rotation measurement of the major aldehyde reaction product gave a value of [α]21D +11 (c 0.042, CHCl3) that was comparable to that of the isolated aldehyde artefact 6 [α]21D +16 (c 0.055, CHCl3). The sample of the 10R-aldehyde reaction product 7 degraded on storage to the (nor)ketone, 10-oxonordendrillolide A (3), which we previously reported from G. coi11; the specific optical rotation of 7 could not therefore be measured.
In a recent study on plant sesquiterpenes,17 Bombarda et al. reported that oxidation of an alloaromadendrene using mCPBA produced two diastereoisomeric epoxides, which differed in configuration at the C-10 position, as well as two aldehyde diastereomers as rearrangement products. Likewise, they found that the minor 10R epoxide was more labile than the major 10S epoxide, rapidly isomerising to an aldehyde during column chromatography. Detection of the epoxides 4 and 5, together with their aldehyde and ketone rearrangement products, brings into question their natural product status, given that the extract had been stored at −4°C for 6 months prior to detailed investigation. 1,1-Disubstituted alkenes are known to be unreactive substrates for chemical epoxidation relative to other alkenes.18
Lactols 8 and 9 were isolated from both the mantle and the viscera as an inseparable mixture by NP-HPLC, but the key individual signals could be identified in the 1H NMR spectrum (Table 2); integration of the two sets of signals indicated a 1:2 ratio of 8 and 9 respectively. The colourless oil displayed a sodiated molecular ion peak in the HRESIMS at m/z 357.2045, corresponding to a molecular formula of C20H30O4. Each terpene showed exomethylene signals (8: δH 4.82 (d) and 4.59 (d); 9: δH 4.81 (d) and 4.53 (d)), three quaternary methyl signals (8: δH 0.94 (s, Me-18), 0.96 (s, Me-17) and 0.97 (s, Me-19); 9: δH 0.95 (s, Me-18), 0.97 (s, Me-19) and 1.04 (s, Me-17)) and the diagnostic bridging methine proton (8: δH 2.88 (d, J = 8.9 Hz, H-9); 9: δH 2.70 (d, J = 9.3 Hz, H-9)), indicative of a perhydroazulene ring system. For each terpene, gCOSY connectivities and HMBC correlations were fully consistent with perhydroazulene and 2,8-dioxabicyclo[3.3.0]octane ring systems and with a lactol group at C-15, as observed in the metabolites cheloviolene A and cheloviolene B.19 The relative and absolute configuration of cheloviolene A was recently confirmed by single-crystal X-ray analysis, following an 11-step enantioselective total synthesis carried out by the Overman group. This synthetic work also revised the relative configuration at C-13, C-14 and C-16 of cheloviolene B.20,21
| Position | 15-Dendrillactol (8)B | 15-Epidendrillactol (9) | |||
|---|---|---|---|---|---|
| 13C | 1H, mult. (J, Hz) | 13C | 1H, mult. (J, Hz) | ||
| 1a | 37.6, CH2 | 2.20, m | 37.6, CH2 | 2.37, m | |
| 1b | 1.82, m | 1.88, m | |||
| 2a | 28.5, CH2 | 1.72, m | 28.5, CH2 | 1.73, m | |
| 2b | 1.38, m | 1.38, m | |||
| 3a | 38.4, CH2 | 1.72, m | 38.3, CH2 | 1.61, m | |
| 3b | 1.46, m | 1.27, m | |||
| 4 | 36.2, C | – | 36.2, C | – | |
| 5 | 54.4, CH | 1.72, m | 54.5, CH | 1.87, m | |
| 6a | 26.8, CH2 | 1.74, m | 27.1, CH2 | 1.70, m | |
| 6b | 1.74, m | 1.70, m | |||
| 7a | 37.8, CH2 | 1.72, m | 37.6, CH2 | 1.82, m | |
| 7b | 1.38, m | 1.51, m | |||
| 8 | 46.6, C | – | 47.0, C | – | |
| 9 | 56.2, CH | 2.88, d (8.9) | 57.7, CH | 2.70, d (9.3) | |
| 10 | 153.5, C | – | 153.8, C | – | |
| 11 | 176.2, C | – | 177.0, C | – | |
| 12a | 29.7, CH2 | 2.72, dd (9.3, 18.1) | 31.2, CH2 | 3.26, dd (6.9, 18.4) | |
| 12b | 2.55, dd (9.8, 18.1) | 2.53, dd (10.9, 18.4) | |||
| 13 | 41.9, CH | 3.15, m | 39.1, CH | 3.17 m | |
| 14 | 56.2, CH | 2.35, m | 55.2, CH | 2.16, dd (3.7, 7.0) | |
| 15 | 99.7, CH | 5.63, d (7.0) | 100.3, CH | 5.64, d (3.7) | |
| 16 | 104.1, CH | 6.06, d (4.8) | 107.4, CH | 6.05, d (6.1) | |
| 17 | 24.1, CH3 | 0.96, s | 24.9, CH3 | 1.04, s | |
| 18 | 34.5, CH2 | 0.94, s | 34.5, CH3 | 0.95, s | |
| 19 | 26.0, CH3 | 0.97, s | 26.0, CH2 | 0.97, s | |
| 20a | 114.7, CH2 | 4.82, d (2.4) | 114.2, CH2 | 4.81, d (2.4) | |
| 20b | 4.59, d (2.4) | 4.53, d (2.4) | |||
| 15-OH | – | – | 3.08 br s | ||
Chemical shifts (ppm) for 15-Dendrillactol (8) and 15-Epidendrillactol (9) were referenced to CHCl3 (δH 7.26, δC 77.16) and at 500 MHz. n.d., not detected.
The 1H NMR data for 8 and 9 did not match that of the cheloviolenes, however, in lactol 8, the coupling constants of H-12a (dd, J = 18.1, 9.3 Hz), H-12b (dd, J = 18.1, 9.8 Hz), H-15 (d, J = 7.0 Hz) and H-16 (d, J = 4.8 Hz) were in accordance with those observed for dendrillolide A (1), suggesting an identical configuration of the 2,8-dioxabicyclo[3.3.0]octane ring system, whereas for 9, the coupling constant of H-15 (d, J = 3.7 Hz) suggested a change in configuration at C-15. Metabolites 8 and 9 were named dendrillactol and 15-epidendrillactol respectively. Following treatment of the lactol mixture 8 and 9 with acetic anhydride and pyridine, the single acetylated product formed was identified as dendrillolide A (1). The specific optical rotation measurement of the reaction product gave a value of [α]21D +26 (c 0.02, CHCl3); for comparison, dendrillolide A (1) isolated from G. coi has [α]21D +61 (c 0.88, CHCl3) compared to a literature value [α]21D +87 (c 0.31, CHCl3).12,13 A detailed molecular modelling and computational study involving DP4/DP+ probability calculations, comparison of experimental vs calculated chemical shifts and of coupling constant values further supported the proposed stereochemistry (see Fig. S20 and Tables S5–S8 Supplementary material for details).
The crude extracts of the two specimens investigated were compared by their 1H NMR spectra. A number of metabolites were found in both the mantle and viscera tissues, including epoxides 2, 4 and 5, the ketone 3, the lactols 8 and 9 plus cheloviolenes A and B, as well as the major metabolite dendrillolide A (1). The only differences between the two tissue types were that the small amounts of dendrillolide C detected were found in the mantle tissue alone, whereas norrisolide, cheloviolene C, macfarlandin D and polyrhaphin C were only detected in the viscera. This general pattern of similar metabolite distribution in both tissue types mirrors that detected in G. hunterae and G. verrieri, and in a previous report on a specimen of G. coi collected from Mackay,9 for all of which there was similar metabolite content in both mantle and visceral tissues. By contrast, in six other species (G. tinctorius, G. tasmaniensis, G. collingwoodi, G. splendidus, G. aureopurpureus and Goniobranchus sp. 1),22 the terpene metabolites detected were predominantly localised in mantle tissues, where they may collectively function as an anti-predator defense. Notably three of these six species contain spongian-16-one metabolites.
Conclusion
Chemical correlation experiments coupled with computational studies led to the determination of the relative configuration of spiroepoxide 4 and of aldehyde 6. The investigation into characterising the mixture of lactols 8 and 9 isolated from G. coi, successfully assigned the relative configuration at C-15 as 15S and 15R respectively, by spectroscopic analysis and chemical conversion into dendrillolide A (1), as well as by modelling and computational analysis. These studies illustrate how combining experimental NMR-derived and computational data can support the configurational analysis of conformationally flexible molecules.
Experimental
General experimental procedure
Specific rotations were measured at 23°C on a Jasco P-2000 polarimeter for solutions in CHCl3 using a 1-mL cell (10-cm path length). NMR spectral data were recorded on a Bruker Avance 500 spectrometer using a 5-mm SEI probe or a Bruker Avance DRX 700-MHz spectrometer with a 5-mm TXI Zgrad probe for solutions in CDCl3 at 298 K. Heteronuclear single quantum correlation (HSQC) and heteronuclear multiple bond correlation (HMBC) data were acquired using a 1JC–H of 145 Hz, whereas HMBC spectra were acquired using nJC–H of 8 Hz. Positive and negative ion electrospray mass spectra were determined using either a Bruker Esquire HCT instrument for low-resolution electrospray ionisation mass spectrometry or a MicrOTOF-Q or an Orbitrap Elite instrument for high-resolution electrospray ionisation mass spectrometry with MeOH as solvent. NP-HPLC was undertaken using a Waters 515 pump connected to a Gilson 132 series refractive index detector with a Waters μPorasil (10 μm, 7.8 × 300 mm2) or a Phenomenex Luna (5 μm, 10 × 250 mm2) column and using isocratic elution conditions at flow rates between 1 and 2 mL min–1. Silica gel 60 G and silica TLC plates F254 were purchased from Merck. Solvents were either distilled or were of HPLC grade.
Biological material
Two nudibranch specimens were collected using SCUBA at depths between 2 and 15 m. A single specimen of G. coi was collected from Percy Isles, north Queensland, in October 2015. The other specimen of G. coi was collected from Coolum, south Queensland, in February 2017. Both samples were stored at −20°C until extraction. Photographs of the specimens (Goniobranchus coi numbers 1426 and 1647) are held at the School of Chemistry and Molecular Biosciences, The University of Queensland.
Extraction and purification
Prior to extraction, each specimen was dissected into viscera and mantle body segments. Individual body segments were finely chopped and extracted with acetone (3 × 2 mL). The extracts were then filtered and reduced to an aqueous suspension before partitioning between H2O (2 mL) and EtOAc (4 × 2 mL). The organic extracts were dried over anhydrous Na2SO4, filtered and concentrated under N2 to yield a yellow oil (29.3 and 24.1 mg) from the mantle tissues and an orange oil (51.1 and 43.4 mg) from the viscera tissues. Based on their 1H NMR profiles, the extracts of the mantle were combined, and the viscera extracts likewise combined. Each extract was subjected to normal-phase flash column chromatography with a gradient elution of 100% hexanes to 100% MeOH from CH2Cl2 and EtOAc. For the mantle, fractions eluting from hexanes/CH2Cl2 (1:1) to CH2Cl2 (100%) were screened by 1H NMR and were separated by NP-HPLC (25% EtOAc in hexanes) to provide 15-dendrillactol (8) and 15-epidendrillactol (9) (7.11 mg inseparable mixture), dendrillolide C (0.2 mg), dendrillolide A (21.1 mg), macfarlandin C (0.9 mg), macfarlandin E (0.4 mg), 12-desacetoxypolyrhaphin A (2 mg), aplyviolene (0.8 mg), 12-desacetoxyshahamin C (0.6 mg), 5,9-epoxydendrillolide A (0.8 mg), 10-oxonordendrillolide A (1.2 mg) and 10,20-epoxydendrillolide A (4: 0.2 mg). The viscera fractions eluting from hexanes/CH2Cl2 (1:1) to CH2Cl2/EtOAc (4:1) were separated by NP-HPLC (25% EtOAc in hexanes) yielding 15-dendrillactol (8) and 15-epidendrillactol (9) (together 4.86 mg), dendrillolide A (14.5 mg), macfarlandin E (0.6 mg), macfarlandin C (0.6 mg), 12-desacetoxypolyrhaphin A (1.2 mg), 12-desacetoxyshahamin C (0.5 mg), macfarlandin D (0.4 mg), aplyviolene (1.0 mg), polyrhaphin C (0.2 mg), cheloviolene C (0.1 mg), norrisolide (0.2 mg), 5,9-epoxydendrillolide A (0.6 mg), aldehyde product (6: 0.55 mg), 10-oxonordendrillolide A (3: 0.6 mg) and 10,20-epoxydendrillolide A (2: 0.2 mg).
The title compound was obtained as a colourless oil (0.21 mg); [α]21D +44 (c 0.021, CHCl3); 1H NMR and 13C NMR (CDCl3, 700 MHz) data are presented in Table 1; HRESIMS m/z [M + Na]+ calculated for C22H32NaO6, 415.2091; found: 415.2083.
The title compound was obtained as a colourless oil (0.55 mg); [α]21D +16 (c 0.055, CHCl3); 1H NMR and 13C NMR (CDCl3, 700 MHz) data are presented in Table 1; HRESIMS m/z [M + Na]+ calculated for C22H32NaO6, 415.2091; found 415.2092.
The title compound was obtained as a colourless oil (7.11 mg); 1H NMR and 13C NMR (CDCl3, 500 MHz) data are presented in Table 2; HRESIMS m/z [M + Na]+ calculated for C20H30NaO4, 357.2036; found: 357.2045. Isolated as an epimeric mixture with 9.
The title compound was obtained as a colourless oil (7.11 mg); 1H NMR and 13C NMR (CDCl3, 500 MHz) data are presented in Table 2; HRESIMS m/z [M + Na]+ calculated for C20H30NaO4, 357.2036; found: 357.2045. Isolated as an epimeric mixture with 8.
Procedure for epoxidation of dendrillolide A (1)
A solution of mCPBA (1.5 mg, 0.00638 mmol) in CH2Cl2 (1.5 mL) was washed with phosphate buffer (pH 7.6, 100 mmol, 2 × 2 mL). Fresh buffer (pH 7.6, 2 mL) was then added and the reaction mixture cooled to 0°C and added to a solution of dendrillolide A (1) (2.0 mg, 376.2 g mol–1, 0.00532 mmol) in DCM (0.5 mL). The biphasic solution mixture was left at room temperature (25°C) for 3 days and monitored by TLC (100% DCM; visualised UV 254 nm and vanillin dip). The phosphate buffer layer was removed and the organic layer washed with a saturated aqueous solution of sodium thiosulfate (2 × 2 mL), then with distilled H2O (1 × 2 mL). The organic layer was then dried over MgSO4, filtered through cotton wool and dried under nitrogen. 2-D NMR analysis was carried out on the crude reaction mixture of 4 and 5. The sample was then purified by NP-HPLC (25% EtOAc in hexanes) to yield aldehyde 6 (0.42 mg) and aldehyde 7 (0.90 mg). Epoxides 4 and 5 degraded before purification could be achieved.
The title compound was obtained as a colourless oil (0.42 mg); [α]21D +11.8 (c 0.04, CHCl3); 1H NMR and 13C NMR (CDCl3, 700 MHz) data are presented in Table 1; HRESIMS m/z [M + K]+ calculated for C22H32KO6, 431.1830; found: 431.2030.
The title compound was obtained as a colourless oil (0.90 mg); 1H NMR and 13C NMR (CDCl3, 700 MHz) data are presented in Table 1. The sample degraded before specific optical rotation could be measured.
Procedure for the acetylation of lactol diastereomers
Lactol diastereomers 8 and 9 (324.2 g mol–1, 1.0 mg, 0.00296 mmol) were dissolved in a mixture of anhydrous pyridine (200 µL) and acetic anhydride (200 µL). After 18 h at 25°C, the reaction mixture was quenched with Milli-Q H2O (1 mL). The aqueous layer was extracted with EtOAc (4 × 1 mL), the combined organic extracts were washed with aqueous HCl (1 mL, 0.1 M) and with Milli-Q H2O (1 mL), then dried over anhydrous Na2SO4, filtered and concentrated under nitrogen to yield a clear oil, which was further purified by normal phase flash chromatography (1:1 DCM:EtOAc) (1.09 mg) and finally by NP-HPLC (25% EtOAc in hexanes) to give (+)-dendrillolide A (1) as a colourless oil (0.2 mg, 20% yield); [α]21D + 26 (c 0.02, CHCl3); [α]21D + 87 (c 0.31, CHCl3)12; 1H NMR (CDCl3, 500 MHz) data were in accord with the literature data23,24; LRESIMS m/z 399.2 [M + Na]+.
Supplementary material
An image of Goniobranchus coi (Fig. S2); copies of 1-D and 2-D NMR spectra of compounds 4–9 (Fig. S5–S19); details of the molecular modelling and computational calculations (Tables S1–S8) are available as Supplementary material. The Supplementary material (Fig. S1–S20, Tables S1–S8) is available online.
Data availability
The data that support this study are available in the article and accompanying online Supplementary material. Further details can be shared upon reasonable request to the corresponding author.
Declaration of funding
The authors thank the School of Chemistry and Molecular Biosciences and The University of Queensland for financial support.
Acknowledgements
The assistance of Dr T. Le (NMR studies) and P. Josh (MS) is acknowledged. The nudibranch specimens were provided by Associate Professor K. Cheney (The University of Queensland). Mary J. Garson was the recipient of the 2023 Distinguished Fellowship of the Royal Australian Chemical Institute.
References
1 Rudman WB, Bergquist PR. A review of feeding specificity in the sponge-feeding Chromodorididae (Nudibranchia: Mollusca). Molluscan Res 2007; 27: 60-88.
| Crossref | Google Scholar |
2 Mullins DA. Goniobranchus coi species profile. In: Nudibranch Domain; 2024. Available at https://nudibranchdomain.org/product/goniobranchus-coi/
3 Rudman BW. Comment on Chromodoris coi & sponges by Asther Lau. In: The Sea Slug Forum, 15 December 2005. Sydney, NSW, Australia: Australian Museum; 2005. Available at http://www.seaslugforum.net/find/15477 [Verified 20 May 2024].
4 Bobzin SC, Faulkner DJ. Diterpenes from the Pohnpeian marine sponge Chelonaplysilla sp. J Nat Prod 1991; 54: 225-232.
| Crossref | Google Scholar |
5 Bobzin SC, Faulkner DJ. Novel rearranged spongian diterpenes from the Palauan sponge Dendrilla sp.: reassessment of the structures of dendrillolide A and dendrillolide B. J Org Chem 1989; 54: 5727-5731.
| Crossref | Google Scholar |
6 Karuso P, Bergquist PR, Cambie RC, Buckleton JS, Clark GR, Rickard CEF. Terpenoid constituents of morphologically similar sponges in the family Aplysillidae. Aust J Chem 1986; 39: 1643-1653.
| Crossref | Google Scholar |
7 Hambley TW, Poiner A, Taylor WC. Diterpene metabolites of the marine sponge chelonaplysilla violacea: Aplyviolene and aplyviolacene. Tetrahedron Lett 1986; 27: 3281-3282.
| Crossref | Google Scholar |
8 Buckleton JS, Bergquist PR, Cambie RC, Clark GR, Karuso P, Rickard CE. Structure determination of aplyviolene from Chelonaplysilla violacea. Acta Crystallogr C 1986; 42: 1846-1848.
| Crossref | Google Scholar |
9 Dewi AS, Pierens GK, Cheney KL, Blanchfield JT, Garson MJ. Chromolactol, an oxygenated diterpene from the Indo-Pacific nudibranch Goniobranchus coi: spectroscopic and computational Studies. Aust J Chem 2018; 71: 798-803.
| Crossref | Google Scholar |
10 Ermanis K, Parkes KEB, Agback T, Goodman JM. The optimal DFT approach in DP4 NMR structure analysis – pushing the limits of relative configuration elucidation. Org Biomol Chem 2019; 17: 5886-5890.
| Crossref | Google Scholar |
11 Forster LC, Pierens GK, Clegg JK, Garson MJ. Dynamic NMR and Computational studies inform the conformational description of dendrillane terpenes from the nudibranch Goniobranchus coi. J Nat Prod 2020; 83: 714-719.
| Crossref | Google Scholar |
12 Sullivan B, Faulkner DJ. Metabolites of the marine sponge Dendrilla sp. J Org Chem 1984; 49: 3204-3206.
| Crossref | Google Scholar |
13 Molinski TF, Faulkner DJ, He CH, Van Duyne GD, Clardy J. Three new rearranged spongian diterpenes from Chromodoris macfarlandi: reappraisal of the structures of dendrillolides A and B. J Org Chem 1986; 51: 4564-4567.
| Crossref | Google Scholar |
14 Grimblat N, Gavin JA, Daranas AH, Sarotti AM. Combining the power of J coupling and DP4 analysis on stereochemical assignments. The J-DP4 methods. Org Lett 2019; 21: 4003-4007.
| Crossref | Google Scholar |
15 Li S, Shi Y, Li P, Xu J. Nucleophilic organic base DABCO-mediated chemospecific rearrangement of terminal epoxides into methyl ketones. J Org Chem 2019; 84: 4443-4450.
| Crossref | Google Scholar |
16 Meinwald J, Labana SS, Chadha MS. Peracid reactions. III. The oxidation of bicyclo[2.2.1]heptadiene. J Am Chem Soc 1963; 85: 582-585.
| Crossref | Google Scholar |
17 Bombarda I, Raharivelomanana P, Ramanoelina PAR, Faure R, Bianchini JP, Gaydou EM. Spectrometric identifications of sesquiterpene alcohols from niaouli (Melaleuca quinquenervia) essential oil. Anal Chem Acta 2001; 447: 113-123.
| Crossref | Google Scholar |
18 Allain EJ, Hager LP, Deng L, Jacobsen EN. Highly enantioselective epoxidation of disubstituted alkenes with hydrogen peroxide catalyzed by chloroperoxidase. J Am Chem Soc 1993; 115: 4415-4416.
| Crossref | Google Scholar |
19 Bergquist PR, Bowden BF, Cambie RC, Craw PA, Karuso P, Poiner A, Taylor WC. The constituents of marine sponges. VI. Diterpenoid metabolites of the New Zealand sponge Chelonaplysilla violacea. Aust J Chem 1993; 46: 623-632.
| Crossref | Google Scholar |
20 Garnsey MR, Slutskyy Y, Jamison CR, Zhao P, Lee J, Rhee YH, Overman LE. Short enantioselective total syntheses of cheloviolenes A and B and dendrillolide C via convergent fragment coupling using a tertiary carbon radical. J Org Chem 2018; 83: 6958-6976.
| Crossref | Google Scholar |
21 Slutskyy Y, Jamison CR, Zhao P, Lee J, Rhee YH, Overman LE. Versatile construction of 6-substituted cis-2,8-dioxabicyclo[3.3.0]octan-3-ones: short enantioselective total syntheses of cheloviolenes a and b and dendrillolide c. J Am Chem Soc 2017; 139: 7192-7195.
| Crossref | Google Scholar |
22 Winters AE, White AM, Dewi AS, Mudianta IW, Wilson NG, Forster LC, Garson MJ, Cheney KL. Distribution of defensive metabolites in nudibranch molluscs. J Chem Ecol 2018; 44: 384-396.
| Crossref | Google Scholar |
24 Allred TK, Dieskau AP, Zhao P, Lackner GL, Overman LE. General access to concave-substituted cis-dioxabicyclo[3.3.0]octanones: enantioselective total syntheses of marfarlandin C and dendrillolide A. J Org Chem 2020; 85: 15532-15551.
| Crossref | Google Scholar |