The mechanism of cleavage of Evans chiral auxiliaries by LiOOH: origins of the different regioselectivities obtained with LiOH, LiOOH, LiOBn and LiSBn
Ka Ho Chow A , Ronald J. Quinn B , Ian D. Jenkins B * and Elizabeth H. Krenske A *
B , Ian D. Jenkins B * and Elizabeth H. Krenske A *
A School of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane, Qld 4072, Australia.
B Griffith Institute for Drug Discovery, Griffith University, Nathan, Qld 4111, Australia.
Abstract
Oxazolidinones are widely used as chiral auxiliaries in asymmetric synthesis. The chiral auxiliary can be selectively removed using LiOOH, LiOBn or LiSBn, but not LiOH, which instead favours endocyclic cleavage with opening of the oxazolidinone ring. Although Evans originally suggested that the regioselectivity of HO−-mediated hydrolysis may be governed by the relative rates of formation of the two tetrahedral intermediates, whereas the regioselectivity of HOO− cleavage might be determined by their relative breakdown rates, it was not clear why this should be the case. Since then, there appear to have been no mechanistic studies that account for the differing selectivities of LiOH v. LiOOH, and the reason for this difference in selectivity has remained somewhat of an enigma in organic chemistry for many years. Herein, we report DFT computations with M06-2X-D3//B3LYP-D3 to examine the origins of the regioselectivities. The computations predict that all four nucleophiles prefer to attack at the endocyclic carbonyl group, which is less hindered. The barrier for decomposition of the initially formed tetrahedral intermediate determines whether or not the reaction continues all the way to the endocyclic cleavage products. For LiOH, the barrier for decomposition of the intermediate is small and the endocyclic cleavage products are formed. For LiOOH, LiOBn and LiSBn, the decomposition barrier is large, and the exocyclic cleavage pathway instead becomes preferred. An alternative mechanism involving formation of a novel cyclic peroxide intermediate was found to involve a relatively high energy pathway and has been excluded.
Keywords: buttressing effect, chiral auxiliaries, Curtin–Hammett, density functional calculations, exocyclic cleavage, lithium hydroperoxide, N-acyloxazolidinones, nucleophilic substitution, reaction mechanisms, regioselectivity, steric acceleration.
Introduction
Chiral oxazolidinones (‘Evans auxiliaries’) are widely used in asymmetric synthesis.1,2 Their most frequent applications involve N-acyl oxazolidinones, which undergo many kinds of diastereoselective transformations on the acyl unit. Once the desired transformation has been achieved, a preferred method of removing the auxiliary involves hydrolysis with lithium hydroperoxide (Scheme 1).3 This mild hydrolytic reagent cleaves off the auxiliary while preserving the integrity of the newly introduced stereocentre(s). The auxiliary can be recovered in high yield after reductive quenching and a simple extraction procedure.
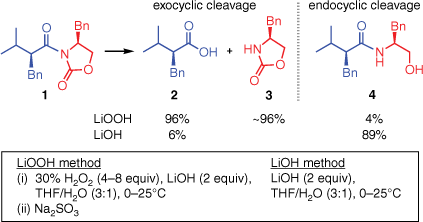
Despite the widespread utility of the LiOOH-mediated hydrolysis, no mechanistic basis for its regioselectivityA has yet been established. Of the two carbonyl groups in the substrate 1, LiOOH preferentially cleaves the bond to the exocyclic carbonyl group, affording the carboxylic acid 2 and the intact oxazolidinone 3. This regiochemical outcome is unexpected, as it appears to indicate a preferential attack by OOH− on the more hindered carbonyl group of 1. The contra-steric selectivity of LiOOH is quite general3,4 (for examples of the use of this reagent in total synthesis, see two papers by Evans et al.5,6) and is obtained even with substrates more hindered than 1. By contrast, hydrolysis by LiOH leads to C–N bond cleavage at the less hindered (endocyclic) carbonyl group, affording products such as 4 in which the oxazolidinone ring has been destroyed.
Evans et al.3,7 suggested that the LiOH- and LiOOH-mediated hydrolyses have different rate-determining steps. For the LiOH-mediated hydrolysis, they proposed that the nucleophilic attack on the substrate by OH− is rate-determining, attack at the endocyclic carbonyl group being faster than attack at the exocyclic carbonyl group. By contrast, for the LiOOH-mediated hydrolysis, it was proposed that the degradation of the tetrahedral intermediate is rate-determining; attack by OOH− on the substrate being reversible. In this scenario, the selectivity is determined by the different rates of decomposition of the endocyclic and exocyclic tetrahedral intermediates.
Beyond this kinetic postulate, the origin of the contra-steric selectivity of the LiOOH-mediated hydrolysis has remained an enigma. Herein, we report an investigation of the hydrolytic mechanisms by means of density functional theory (DFT) calculations. We also examine the related cleavages of N-acyloxazolidinones mediated by LiOBn and LiSBn, which likewise favour exocyclic C–N cleavage.
Results and discussion
LiOH-mediated cleavage
We began by performing computations on the OH−-mediated hydrolysis of a model N-acyloxazolidinone. Typical experimental conditions for the LiOH- and LiOOH-mediated hydrolysis reactions involve the use of 2 equiv. of LiOH and (in the case of LiOOH) 4–8 equiv. of H2O2, in a THF/H2O solvent system. Under these conditions, there are numerous Li-bound species that could plausibly be formed by the N-acyloxazolidinone. Features that may vary include the number and position of bound Li ions, the number and type of solvent molecules coordinated to Li, the extent of aggregation (monomer, oligomer, etc.), and the hydrogen bonding arrangements within these complexes (for insights into the species formed by treatment of N-acyloxazolidinones with Li bases, see Tallmadge and Collum 20158 and Tallmadge et al. 20169). We used the chelate complex 5 (Fig. 1, yellow box) as a model substrate. Complex 5 contains one Li(OH2)2+ unit and serves as a simple representative of the many Li-bound structures likely present in the reaction mixture.
Free energy profile for the reaction of the N-acyloxazolidinone complex 5 with OH−, computed with M06-2X-D3/6-311+G(d,p)//B3LYP-D3/6-31+G(d) in SMD water.
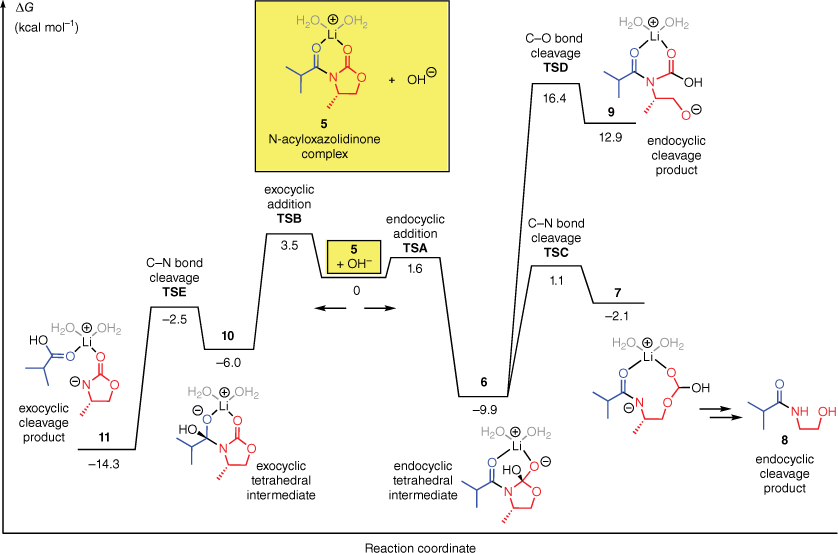
Fig. 1 shows the computed Gibbs free energy profile for the hydrolysis of 5 by OH−. The computational procedure involved geometry optimisations with B3LYP-D3/6-31+G(d)10–14 followed by single-point energy calculations with M06-2X-D3/6-311+G(d,p)14–16 using the SMD implicit solvent model17 of water.
The computations predict that the preferred site of attack on 5 by OH− is the endocyclic carbonyl group. The computed barrier for endocyclic attack (TSA, ΔG‡ = 1.6 kcal mol−1) is 2 kcal mol−1 lower than the barrier for exocyclic attack (TSB, 3.5 kcal mol−1). In both cases, attack takes place at the top face of the substrate (as drawn), which is not blocked by the methyl substituent on the oxazolidinone. The tetrahedral intermediate of the favoured endocyclic attack (6) can degrade by two different pathways, leading to C–N or C–O bond cleavage (TSC and TSD respectively). The C–N bond cleavage is strongly preferred (by 15 kcal mol−1). It leads to the ring-opened intermediate 7. Subsequent decarboxylation would lead to β-hydroxyamide 8, the major product obtained experimentally in this type of reaction.
The computed energy profile of the OH−-mediated hydrolysis supports Evans’ proposal3,7 that the initial nucleophilic addition step determines the regioselectivity. In the computations, the endocyclic addition is indeed kinetically favoured and, once it has occurred, endocyclic C–N bond cleavage is facile.
LiOOH-mediated cleavage
We next examined the corresponding OOH−-mediated hydrolysis. The computed energy profile is shown in Fig. 2.
Free energy profile for the reaction of the N-acyloxazolidinone complex 5 with OOH− in water.
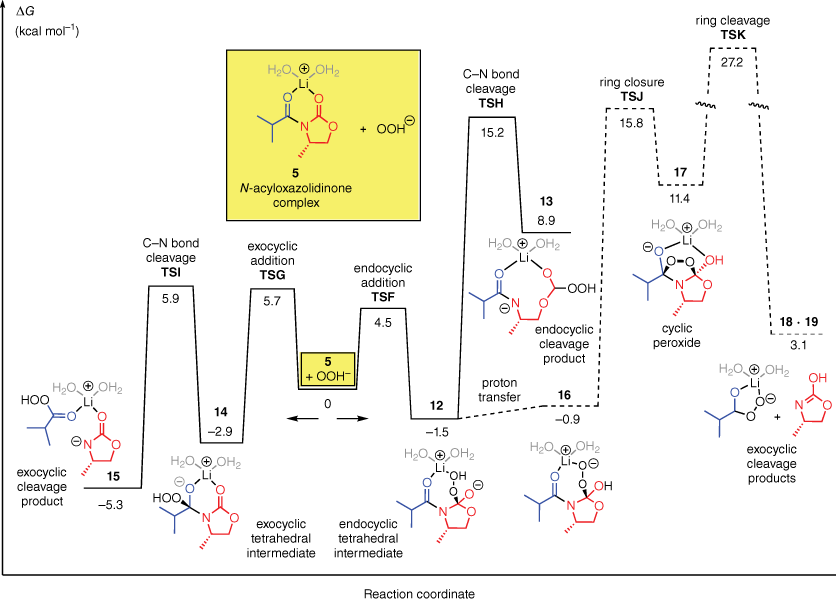
Similar to the reaction of 5 with OH−, the reaction of 5 with OOH− favours attack at the endocyclic carbonyl group. The respective transition states (TSF and TSG) differ by 1.2 kcal mol−1. However, once the tetrahedral intermediate of the favoured endocyclic attack (12) has been formed, the reaction energy profile differs significantly from that of OH−. The most important difference is that the C–N bond cleavage of the endocyclic intermediate becomes much higher in energy. For OOH−, the C–N bond cleavage has a barrier of 15 kcal mol−1 (TSH), as compared with 3 kcal mol−1 for OH−. The transition state energies for the two steps of the exocyclic cleavage pathway (TSG and TSI), by comparison, are much smaller, at 6 kcal mol−1 each. Therefore, even though OOH− prefers to attack the endocyclic carbonyl group, the high barrier for subsequent degradation of the resulting intermediate means that the intermediate prefers to revert to the reactants. Reaction by the exocyclic pathway is preferred overall. The computations on the OOH−-mediated hydrolysis agree with Evans’ original proposal that the degradation of the intermediate determines the regioselectivity.
We also considered a different mechanism that could, in principle, account for the exocyclic cleavage by OOH−. In this mechanism, shown by the dashed lines at the right hand side of Fig. 2, the endocyclic intermediate 12 undergoes an intramolecular nucleophilic addition by the hydroperoxy group onto the exocyclic carbonyl group, leading to a five-membered cyclic peroxide (17). The peroxide then degrades to the exocyclic cleavage products 18 + 19. Computations predict, however, that this pathway is relatively high in energy compared to the direct exocyclic attack pathway (barrier = 27 kcal mol−1).
In a recent study of the LiOOH-mediated hydrolysis of an N-acyloxazolidinone, Beutner and coworkers18 detected, by mass spectrometry, the formation of a lithium percarboxylate RC(O)O2Li. This salt would be readily formed by ligand dissociation from a complex similar to 15. Beutner and coworkers showed that the lithium percarboxylate decomposed by a disproportionation reaction mediated by the excess H2O2, and not (as had previously been thought) by reduction during workup. They also showed that the rate-determining step of the overall hydrolysis reaction occurred prior to the decomposition of the percarboxylate. Our computations are consistent with these results.
LiOBn- and LiSBn-mediated cleavages
Two other reagents, LiOBn and LiSBn, cleave N-acyloxazolidinones with a preference for exocyclic cleavage (Scheme 2).19,20 Reaction of an N-acyloxazolidinone with LiOBn affords benzyl carboxylates (21),19 except in very hindered cases3 where undesired oxazolidinone ring-cleavage products analogous to 4 (Scheme 1) are obtained instead. Reaction with LiSBn affords benzyl thioesters (22), even for hindered substrates.20
To understand these selectivities, we computed the additions of BnO− and BnS− to the N-acyloxazolidinone complex 5 (Fig. 3). For both nucleophiles, the computations predict that the reaction energy profile is broadly analogous to that of OOH−, in that the endocyclic carbonyl group is the preferred site of attack but the degradation of the endocyclic intermediate has a very high energy barrier. Therefore, the exocyclic pathway is favoured overall.
Free energy profiles for the reactions of the N-acyloxazolidinone complex 5 with (a) BnO− and (b) BnS− in water.
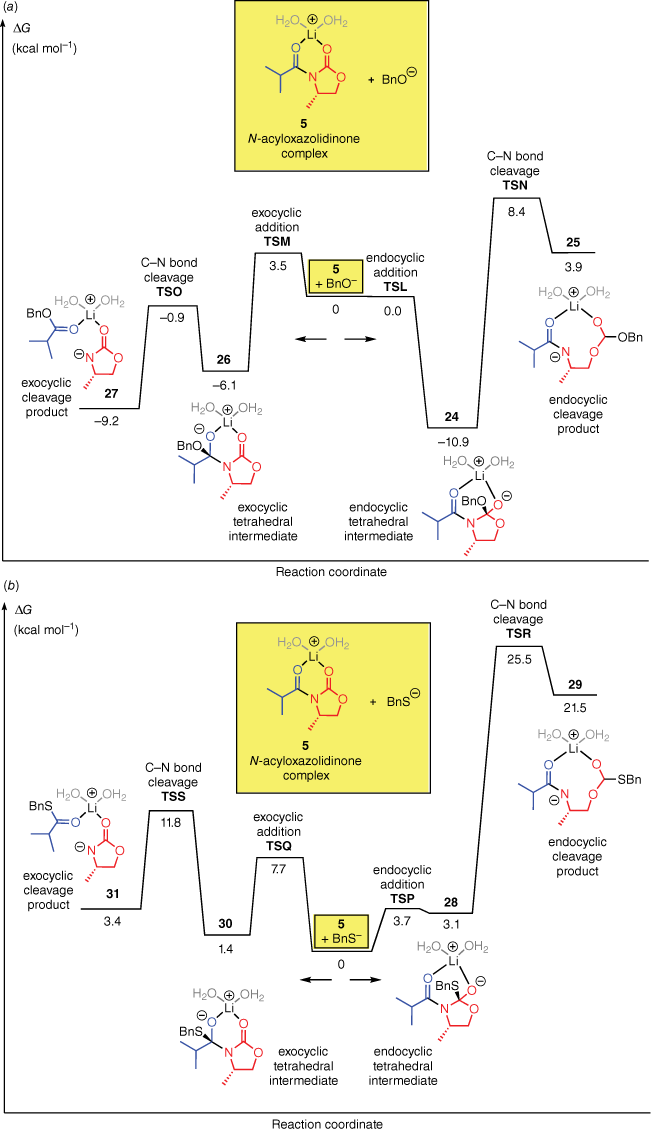
The computed regioselectivities, based on the difference between the rate-determining barrier heights for the endocyclic and exocyclic pathways (ΔΔG‡), are 5 kcal mol−1 for BnO− and 14 kcal mol−1 for BnS−. Qualitatively, this result is consistent with the higher selectivity obtained experimentally with LiSBn, as compared to LiOBn.3,19,20
It is interesting to note that for each of the four nucleophiles, the barrier for exocyclic C–N bond cleavage is always lower than for the corresponding endocyclic C–N bond cleavage. We attribute this to steric acceleration, resulting from the ‘buttressing effect’ of the neighbouring isopropyl substituent (or the R group in the N-acyloxazolidinones 20).
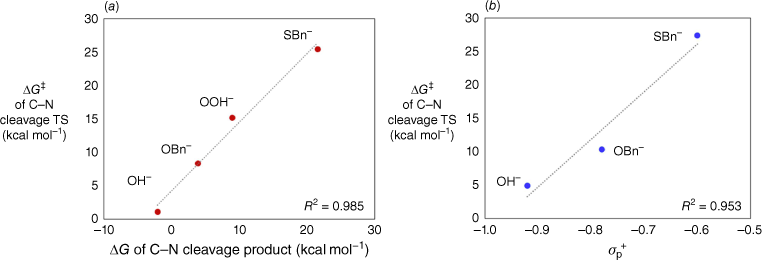
A comparison of the four nucleophiles, LiOH, LiOOH, LiOBn and LiSBn, reveals obvious differences between the barriers for nucleophilic addition and those for degradation of the tetrahedral intermediates. In particular, the barriers for the endocyclic nucleophilic additions of the four nucleophiles are very similar, 0–5 kcal mol−1, whereas those for the decomposition of the endocyclic intermediates vary widely, 1–26 kcal mol−1. Why do the decomposition barriers vary so substantially? There appears to be a strong thermodynamic contribution. In Fig. 4a, the transition state energies for decomposition of the intermediates are plotted against the energies of the resulting ring-opened products. A strong linear correlation is observed (R2 = 0.99). The more stable the product, the lower the barrier for ring opening. The most stable ring-opened product is that obtained from OH−. A plot of the transition state energies against the electrophilic substituent constants σp+ is also linear (Fig. 4b; the plot includes the three derivatives for which suitable reference values are available in the literature21). This supports the idea that the barrier for ring opening is determined by the ability of the substituent (i.e. the incorporated nucleophile) to stabilise the nascent carbonyl functionality through resonance.
Plots showing the relationships between the barrier for decomposition of the endocyclic tetrahedral intermediate and (a) the energy of the ring-opened product, or (b) the electrophilic substituent constant σp+ of the substituent. The σp+ values of OMe and SMe were used to represent OBn and SBn respectively.
Conclusion
Density functional theory calculations explain why the reactions of N-acyloxazolidinones with LiOH favour cleavage of the endocyclic C–N bond while the corresponding reactions with LiOOH, LiOBn and LiSBn favour cleavage of the exocyclic C–N bond. For the LiOH- and LiOOH-mediated hydrolyses, our calculations provide support for Evans’ original kinetic proposals.3,7 In the case of LiOH, the nucleophilic addition of OH− to the substrate favours attack at the endocyclic carbonyl group, and the resulting tetrahedral intermediate has a low barrier to decomposition, meaning that the endocyclic cleavage pathway is favoured overall. For LiOOH, the nucleophilic addition step likewise favours attack on the endocyclic carbonyl group, but the resulting tetrahedral intermediate has a very high barrier to decomposition, meaning that overall, the exocyclic cleavage pathway is preferred. High barriers for decomposition of the endocyclic intermediates are also found in the reactions of LiOBn and LiSBn, which similarly favour exocyclic cleavage. For LiOOH, LiOBn and LiSBn, it is essentially a Curtin–Hammett scenario where the exocyclic and endocyclic addition steps are both reversible and the exocyclic pathway overall represents the kinetically favoured mechanism. Owing to the likelihood of multiple Li-bound structures being present in the reaction mixtures, and due to the limitations associated with modelling reactions involving highly polar species in solution,22 certain aspects of the reaction energetics cannot be accurately estimated with the DFT-based approach used here. Nonetheless, the computations permit a determination that the key factor responsible for the selectivity is the ability of the nucleophile to eventually stabilise the ring-opened endocyclic product. If strong (OH), then endocyclic cleavage predominates; if weak (OOH, OBn and SBn), then exocyclic cleavage predominates. The reaction rates and selectivities of these cleavage reactions are influenced by multiple other variables, including solvent, water content, base, temperature and amount of H2O2.18 Although it is not possible to model all the species present in these reaction mixtures, our calculations nonetheless provide a satisfactory explanation for the inherent regioselectivities of the four different nucleophiles OH−, OOH−, BnO− and BnS−.
Computational methods
Density functional theory calculations were performed in Gaussian 16 (ver. C.01, see https://gaussian.com/gaussian16/).23 Geometry optimisations used the B3LYP-D3 functional10–14 and 6-31+G(d) basis set, with the SMD implicit solvent model17 of water. Vibrational frequency calculations confirmed the nature of each stationary point (energy minimum or transition state) and were used to obtain thermochemical data. Errors in low-frequency vibrations introduced by the harmonic oscillator approximation were treated by application of Truhlar’s approach,24 raising all harmonic frequencies below 100 cm−1 to exactly 100 cm−1 before evaluation of the vibrational component of the thermal contribution to entropy. Intrinsic reaction coordinate (IRC) calculations25,26 identified the bond-forming or bond-breaking processes occurring in key transition states. Single-point energies were computed at the B3LYP-D3 optimised geometries using the M06-2X-D3 functional14,15 and 6-311+G(d,p) basis set, in SMD water. Gibbs free energies were calculated by adding the corrected B3LYP-D3 thermochemical quantities to the M06-2X-D3 electronic potential energies and are reported at a standard state of 298.15 K and 1 mol L–1.
Supplementary material
Coordinates and energies of computed structures are available on the journal’s website. Supplementary material is available online.
Declaration of funding
The authors thank the Australian Research Council (DP180103047 to E. H. Krenske) for funding. Computational resources were provided by the National Computational Infrastructure through the National Computational Merit Allocation Scheme (supported by the Australian Government’s National Collaborative Research Infrastructure Strategy, NCRIS) and by the University of Queensland Research Computing Centre.
References
1 Heravi MM, Zadsirjan V. Oxazolidinones as chiral auxiliaries in asymmetric aldol reactions applied to total synthesis. Tetrahedron Asymmetry 2013; 24: 1149-1188.
| Crossref | Google Scholar |
2 Ager DJ, Prakash I, Schaad DR. 1,2-Amino alcohols and their heterocyclic derivatives as chiral auxiliaries in asymmetric synthesis. Chem Rev 1996; 96: 835-876.
| Crossref | Google Scholar |
3 Evans DA, Britton TC, Ellman JA. Contrasteric carboximide hydrolysis with lithium hydroperoxide. Tetrahedron Lett 1987; 28: 6141-6144.
| Crossref | Google Scholar |
4 Evans DA, Carter PH, Dinsmore CJ, Barrow JC, Katz JL, Kung DW. Mild nitrosation and hydrolysis of polyfunctional amides. Tetrahedron Lett 1997; 38: 4535-4538.
| Crossref | Google Scholar |
5 Evans DA, Wood MR, Trotter BW, Richardson TI, Barrow JC, Katz JL. Total syntheses of vancomycin and eremomycin aglycons. Angew Chem Int Ed 1998; 37: 2700-2704.
| Crossref | Google Scholar |
6 Evans DA, Kim AS, Metternich R, Novack VJ. General strategies toward the syntheses of macrolide antibiotics. The total syntheses of 6-deoxyerythronolide B and oleandolide. J Am Chem Soc 1998; 120: 5921-5942.
| Crossref | Google Scholar |
7 Evans DA, Britton TC, Ellman JA, Dorow RL. The asymmetric synthesis of α-amino acids. Electrophilic azidation of chiral imide enolates, a practical approach to the synthesis of (R)- and (S)-α-azido carboxylic acids. J Am Chem Soc 1990; 112: 4011-4030.
| Crossref | Google Scholar |
8 Tallmadge EH, Collum DB. Evans enolates: solution structures of lithiated oxazolidinone-derived enolates. J Am Chem Soc 2015; 137: 13087-13095.
| Crossref | Google Scholar |
9 Tallmadge EH, Jermaks J, Collum DB. Structure–reactivity relationships in lithiated Evans enolates: influence of aggregation and solvation on the stereochemistry and mechanism of aldol additions. J Am Chem Soc 2016; 138: 345-355.
| Crossref | Google Scholar |
10 Lee C, Yang W, Parr RG. Development of the Colle–Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 1988; 37: 785-789.
| Crossref | Google Scholar |
11 Becke AD. A new mixing of Hartree–Fock and local density-functional theories. J Chem Phys 1993; 98: 1372-1377.
| Crossref | Google Scholar |
12 Becke AD. Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 1993; 98: 5648-5652.
| Crossref | Google Scholar |
13 Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J Phys Chem 1994; 98: 11623-11627.
| Crossref | Google Scholar |
14 Grimme S, Antony J, Ehrlich S, Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J Chem Phys 2010; 132: 154104.
| Crossref | Google Scholar |
15 Zhao Y, Truhlar DG. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem Acc 2008; 120: 215-241.
| Crossref | Google Scholar |
16 Goerigk L, Grimme S. A thorough benchmark of density functional methods for general main group thermochemistry, kinetics, and noncovalent interactions. Phys Chem Chem Phys 2011; 13(14): 6670.
| Crossref | Google Scholar |
17 Marenich AV, Cramer CJ, Truhlar DG. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J Phys Chem B 2009; 113: 6378-6396.
| Crossref | Google Scholar |
18 Beutner GL, Cohen BM, DelMonte AJ, Dixon DD, Fraunhoffer KJ, Glace AW, Lo E, Stevens JM, Vanyo D, Wilbert C. Revisiting the cleavage of Evans oxazolidinones with LiOH/H2O2. Org Process Res Dev 2019; 23: 1378-1385.
| Crossref | Google Scholar |
19 Evans DA, Ennis MD, Mathre DJ. Asymmetric alkylation reactions of chiral imide enolates. A practical approach to the enantioselective synthesis of α-substituted carboxylic acid derivatives. J Am Chem Soc 1982; 104: 1737-1739.
| Crossref | Google Scholar |
20 Damon RE, Coppola GM. Cleavage of N-acyl oxazolidones. Tetrahedron Lett 1990; 31: 2849-2852.
| Crossref | Google Scholar |
21 Brown HC, Okamoto Y. Electrophilic substituent constants. J Am Chem Soc 1958; 80: 4979-4987.
| Crossref | Google Scholar |
22 Plata RE, Singleton DA. A case study of the mechanism of alcohol-mediated Morita Baylis–Hillman reactions. The importance of experimental observations. J Am Chem Soc 2015; 137: 3811-3826.
| Crossref | Google Scholar |
23 Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich AV, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams-Young D, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark MJ, Heyd JJ, Brothers EN, Kudin KN, Staroverov VN, Keith TA, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, Fox DJ. Gaussian 16, Revision C.01. Wallingford, CT, USA: Gaussian, Inc.; 2019.
24 Zhao Y, Truhlar DG. Computational characterization and modeling of buckyball tweezers: density functional study of concave–convex π⋯π interactions. Phys Chem Chem Phys 2008; 10: 2813-2818.
| Crossref | Google Scholar |
25 Gonzalez C, Schlegel HB. An improved algorithm for reaction path following. J Chem Phys 1989; 90: 2154-2161.
| Crossref | Google Scholar |
26 Gonzalez C, Schlegel HB. Reaction path following in mass-weighted internal coordinates. J Phys Chem 1990; 94(14): 5523-5527.
| Crossref | Google Scholar |
Footnotes
1 Strictly speaking, the preference for the exocyclic C–N bond cleavage pathway is an example of chemoselectivity, as it reflects a competition between the reactions of the amide and carbamate carbonyl groups. However, the LiOOH-mediated hydrolysis also displays similar selectivity with substrates such as 32, in which both carbonyl groups are amides.3 We use the more general term ‘regioselectivity’ here to cover both types of cases.