

Differential membrane binding of α/β-peptide foldamers: implications for cellular delivery and mitochondrial targeting
Tzong-Hsien Lee A , James W. Checco B G H , Tess Malcolm A I , Chelcie H. Eller C , Ronald T. Raines B C , Samuel H. Gellman B , Erinna F. Lee D E F , W. Douglas Fairlie D E F and Marie-Isabel Aguilar A *
B C , Samuel H. Gellman B , Erinna F. Lee D E F , W. Douglas Fairlie D E F and Marie-Isabel Aguilar A *
A Department of Biochemistry and Molecular Biology, Monash University, Clayton, Vic. 3800, Australia.
B Department of Chemistry, University of Wisconsin-Madison, Madison, WI 53706, USA.
C Department of Biochemistry, University of Wisconsin-Madison, Madison, WI 53706, USA.
D Department of Biochemistry and Chemistry, School of Agriculture, Biomedicine and Environment, La Trobe Institute for Molecular Science, La Trobe University, Melbourne, Vic. 3086, Australia.
E Cell Death and Survival Laboratory, Olivia Newton-John Cancer Research Institute, Heidelberg, Vic. 3084, Australia.
F School of Cancer Medicine, La Trobe University, Melbourne, Vic. 3086, Australia.
G Present address: Department of Chemistry, University of Nebraska-Lincoln, Lincoln, NE 68588, USA.
H Present address: The Nebraska Center for Integrated Biomolecular Communication (NCIBC), University of Nebraska-Lincoln, Lincoln, NE 68588, USA.
I Present address: School of Chemistry, University of Melbourne, Parkville, Vic. 3052, Australia.
Australian Journal of Chemistry 76(8) 482-492 https://doi.org/10.1071/CH23063
Submitted: 29 March 2023 Accepted: 9 May 2023 Published: 14 June 2023
© 2023 The Author(s) (or their employer(s)). Published by CSIRO Publishing. This is an open access article distributed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License (CC BY-NC-ND)
Abstract
The intrinsic pathway of apoptosis is regulated by the Bcl-2 family of proteins. Inhibition of the anti-apoptotic members represents a strategy to induce apoptotic cell death in cancer cells. We have measured the membrane binding properties of a series of peptides, including modified α/β-peptides, designed to exhibit enhanced membrane permeability to allow cell entry and improved access for engagement of Bcl-2 family members. The peptide cargo is based on the pro-apoptotic protein Bim, which interacts with all anti-apoptotic proteins to initiate apoptosis. The α/β-peptides contained cyclic β-amino acid residues designed to increase their stability and membrane permeability. Dual polarisation interferometry was used to study the binding of each peptide to two different model membrane systems designed to mimic either the plasma membrane or the outer mitochondrial membrane. The impact of each peptide on the model membrane structure was also investigated, and the results demonstrated that the modified peptides had increased affinity for the mitochondrial membrane and significantly altered the structure of the bilayer. The results also showed that the presence of an RRR motif significantly enhanced the ability of the peptides to bind to and insert into the mitochondrial membrane mimic, and provide insights into the role of selective membrane targeting of peptides.
Keywords: apoptosis, Bcl mimetics, dual polarisation interferometry, membrane permeability, membranes, mitochondrial membrane, peptides, peptidomimetics.
Introduction
Apoptosis is the controlled death of cells when exposed to damage or compromising environments and can occur via two pathways: intrinsic and extrinsic. The intrinsic pathway is characterised by the involvement of the Bcl-2 protein family and inhibition of the protein interactions of the Bcl-2 family represents a strategy to induce apoptotic cell death in cancer cells.[1,2] Typically, anti-apoptotic proteins bind to two classes of pro-apoptotic proteins, the BAX/BAK proteins and the BH3-only proteins. BAX and BAK are the essential executioners of apoptosis and are inhibited by direct interactions with the anti-apoptotic family members. The BH3-only proteins initiate the apoptotic cascade by binding to the anti-apoptotic proteins, releasing BAX and BAK, which are then freed to oligomerise and form pores in the mitochondrial membrane. A range of small-molecule compounds (‘BH3-mimetics’) have been developed to bind anti-apoptotic proteins and mimic the action of the BH3-only proteins to induce apoptosis in cancers where Bcl-2 proteins are often overexpressed. These drugs are now being trialled or approved for use in cancer patients and showing significant promise (reviewed in references [1,3,4]).
Previous studies identified truncated, hydrocarbon crosslinked (‘stapled’) α/β-peptide analogues of the BH3-only protein, Bim, could effectively target the anti-apoptotic proteins and induce apoptosis.[5] Targeting of the Bim BH3 analogue to anti-apoptotic proteins releases BAX, thereby promoting mitochondrial outer membrane permeabilisation and consequently apoptosis. However, the impact of binding to the plasma membrane of peptide-based analogues derived from the Bim BH3 is not well documented but can provide new insight into the mechanism of their passage through the membrane.
The aim of this study was to evaluate the membrane interaction properties for selected α/β-peptide analogues of the Bim BH3 domain. The peptides contained modified cyclic β-amino acids with high hydrophobicity designed to increase membrane binding and insertion without the requirement for a hydrocarbon crosslink. The membrane interaction of these peptides with mimics of the plasma membrane and the mitochondrial outer membrane was analysed using dual polarisation interferometry.[6] It was found that both α/β-peptide analogues interacted more strongly with both membrane mimics compared with the natural Bim BH3 sequence, and fluorescent analogues of these peptides appeared to bind to membranes and enter cells, as evaluated by confocal microscopy. These results provide new insight into how peptide modifications can influence membrane binding and cellular entry.
Materials and methods
Materials
1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE), 1-palymitoyl-2-oleoyl-sn-glycero-3-phospho-l-serine (sodium salt) (POPS), l-α-phosphatidylinositol (soy) (sodium salt) (PI) and 1′,3′-bis[1,2-dioleoyl-sn-glycerol-3-phospho]-sn-glycerol (sodium salt) (TOCL) were purchased from Avanti Polar Lipids (Alabaster, AL, USA). 4-(2-Hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES), HEPES buffered saline (HBS), sodium dodecyl sulfate (SDS), sodium chloride and calcium chloride, all analytical grade, were purchased from Sigma Aldrich (St Louis, MI, USA). Chloroform, methanol and ethanol were all HPLC grade purchased from Merck (Darmstadt, Germany). Hellmanex II was obtained from Hellma (Müllheim, Germany). Water was deionised in a Milli-Q system equipped with UV oxidation to remove organic residue (Millipore, Bedford, MA, USA).
Synthesis of the J-amino acid monomer Fmoc-J5-OH
Mono-pentyl succinate was synthesised based on a previously reported protocol.[7] Succinic anhydride (1 equiv.) was suspended in toluene and N-hydroxysuccinimide (0.3 equiv.), 4-(dimethylamino)pyridine (DMAP, 0.1 equiv.), triethylamine (0.3 equiv.) and 1-pentanol (1 equiv.) were added. The reaction mixture was heated to reflux overnight. On completion of the reaction (as determined by thin layer chromatography (TLC)), the mixture was cooled to room temperature. Ethyl acetate was added, and the organic phase was washed three times with 10% citric acid and one time with brine. The organic phase was dried over magnesium sulfate, and filtered. The solvent was removed by evaporation. The alkyl succinate was then used in the synthesis of J-amino acid monomer without further purification.
The synthesis of the Fmoc-protected J5-amino acid monomer (Fmoc, 9-fluorenylmethoxycarbonyl) for use in solid-phase peptide synthesis (Supplementary Fig. S1) was performed by dissolving Fmoc–APC(Boc)–OH[8] (APC, trans-3-Aminopyrrolidine-4-carboxylic acid; Boc, t-butyloxycarbonyl) in dichloromethane (DCM) with 20% trifluoroacetic acid (TFA). The Boc deprotection reaction was allowed to proceed at room temperature monitored by TLC. After complete deprotection of the Boc group, the solvent was removed by evaporation, and the product was dissolved in DCM and then evaporated to dryness multiple times to fully remove the TFA, until evaporation yielded a grey/white solid.
The coupling of mono-pentyl succinate to Fmoc–APC-OH was performed by dissolving Fmoc–APC–OH (1 equiv.) in DCM, followed by the addition of mono-pentyl succinate (1.1 equiv.) pre-activated with 1.1 equiv. of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI), 1.1 equiv. of 1-hydroxybenzotriazole (HOBt) and 6 equiv. of N,N-diisopropylethylamine (DIEA) in an ice bath. The mixture was allowed to slowly warm from 4°C to room temperature overnight, and was monitored by TLC. After completion of the reaction, and the solvent was removed. The crude mixture was dissolved in ethyl acetate, and the organic layer was extracted three times with 5% NaHSO4, two times with saturated NaHCO3, two times with 1 N HCl, two times with brine, dried over magnesium sulfate, and filtered. The solvent was removed by evaporation. The product was then isolated via column chromatography (6:4 ethyl acetate/hexane, 5% acetic acid). The fractions containing the product were combined, and the solvent was removed by evaporation to give the final Fmoc-protected amino acid for use in solid-phase peptide synthesis. Characterisation was carried by electrospray ionisation mass spectrometry (ESI-MS) (Supplementary Fig. S1) and 1H-NMR spectroscopy (Supplementary Fig. S2).
Peptide synthesis and purification
α-Peptides and α/β-peptides were synthesised by microwave-assisted solid-phase peptide synthesis on NovaPEG Rink Amide resin, as previously described.[9] Fmoc-protected amino acids (with standard compatible side chain protecting groups; 4 equiv.) were activated with a solution of PyBOP (benzotriazol‐1‐yloxytripyrrolidinophosphonium hexafluorophosphate) (4 equiv.), 0.1 M HOBt and DIEA (8 equiv.) in 1-methyl-2-pyrrolidone (NMP) and added to the resin. For the coupling of Fmoc-J5-OH, 3 equiv. of monomer (with 3 equiv. PyBOP and 6 equiv. DIEA) was often used to conserve monomer. Coupling reactions were irradiated by microwave to 70°C for 6 min (2 min ramp, 4 min hold) with a 5 min cool-down for α-residues, or for 14 min (2 min ramp, 12 min hold) with a 5 min cool-down for cyclic β-residues. Fmoc deprotection reactions were carried out with a solution of 20% piperidine in N,N-dimethylformamide (DMF), irradiated by microwave at 80°C for 4 min (2 min ramp, 2 min hold). After assembly of the linear chain, oligomers that were not fluorophore-labelled were acetylated by stirring the resin in a solution of 8:2:1 DMF:DIEA:acetic anhydride for ~10 min at room temperature. For oligomers labelled with 6-carboxyfluorescein (6-FAM) at the N-terminus, resin was treated with 4 equiv. of 6-FAM (AnaSpec AS-81004) under the above coupling conditions at room temperature overnight, protected from light.
Once the oligomer had been fully assembled on resin, peptides and α/β-peptides were cleaved using a solution of 95% TFA, 2.5% water and 2.5% tri-isopropylsilane (TIS), concentrated under a stream of nitrogen, and then precipitated with cold diethyl ether. Crude peptide mixtures were then purified by reversed-phase HPLC. After purification, final peptide purity was assessed by analytical HPLC and identity confirmed by MALDI-TOF-MS (Matrix‐assisted laser desorption ionization–time‐of‐flight mass spectrometry) (Supplementary Figs S3–S6).
Liposome preparation
Stock solutions of POPC, POPE, POPS, PI and TOCL were prepared at 2 mM in chloroform. Sufficient amounts of POPC were set aside and the appropriate volume of POPC, POPE, POPS, PI and TOCL were combined to form a POPC-POPE-POPS-PI-TOCL solution with a molar ratio of 54:30:2:10:4. Aliquots containing 0.8 μmol of each lipid mixture (POPC, POPC-POPE-POPS-PI-TOCL (Mito)) were dried using a gentle stream of N2 gas in a Pyrex test tube, and vacuum-dried overnight to form lipid films. These were then hydrated with 10 mM HEPES buffer, 150 mM NaCl, pH 7.4 buffer at 37°C in a shaker-incubator for 1 h. The samples were then ultrasonicated in a bath-type sonicator for 30 min, ideally resulting in a clear solution. The resulting solution was extruded 21 times through a 100 nm polycarbonate membrane using an Avestin Liposofast extruder (Avestin, ON, Canada).
Dual polarisation interferometry (DPI)
The Analight Bio2000 containing a silicone oxynitride FB80 AnaChip was used to perform all dual polarisation interferometry measurements. An independent Harvard Apparatus PHD2000 programmable syringe pump was used to control the flow rate of the bulk buffer. The bulk buffer was 10 mM HEPES, 150 mM NaCl, pH 7.4 for all experiments. The chip was cleaned at 28°C by rinsing once with 10% Hellmanex II, followed by twice with 2% (w/v) SDS and finally twice with 100% ethanol. The temperature was then decreased to 20°C and the optical properties of the chip calibrated using 80% (w/w) ethanol followed by bulk calibration using water. Temperature was then adjusted to 28°C. The final liposome solution (200 μL) was then injected at 20 μL/min, forming a stable bilayer in the presence of 1 mM CaCl2. The divalent cation Ca2+ is important in the formation of the bilayer via direct liposome adsorption on a planar solid support. Addition of CaCl2 is required to ensure reproducible deposition of the liposomes and formation of the phospholipid bilayer. Without Ca2+, the resulting lipid bilayer is of inconsistent geometric structure and has liposomes present. The concentration of Ca2+ required is lipid composition- and concentration-dependent and also varied according to the type of substrate.
The bilayer was then left to stabilise for 30 min before being cooled to 20°C. When the signal from the chip had stabilised, 160 μL of a peptide solution was injected at 40 μL/min. Bim BH3 and α/β peptides were dissolved in bulk buffer and injected at concentrations of 2, 5, 10 and 20 μM onto each bilayer type in four separate experiments. Between experiments, the surface was cleaned with 10% Hellmanex II, 2% (w/v) SDS and 100% ethanol and a fresh bilayer produced for each new experiment. Consecutive peptide injections were performed on a single bilayer for both bilayer types. Starting with the lowest peptide concentration, 2 μM, peptide was injected and left for 30 min to equilibrate before the next highest concentration, 5 μM, was injected, and the remaining peptide concentrations, 10 and 20 μM, were then introduced sequentially with a 30 min waiting time after each injection.
Two orthogonal polarisations were passed through the sensor chip, creating two different waveguide modes, namely transverse electric (TE) and transverse magnetic (TM) waveguide modes. Each mode generated an evanescent field from the top-sensing waveguide surface interacting with materials coming into contact with the sensor surface and resulting in a change in refractive index (RI). Thus, birefringence could be obtained with DPI by calculating the difference between two effective refractive indices, namely the RI of TM waveguide mode (nTM) and RI of TE waveguide mode (nTE).
The mass per unit area for an adsorbed anisotropic layer was calculated using the de Feijter formula[10] for mlipid, the mass of the lipid in the bilayer and mpeptide, the mass of peptide bound to the bilayer:
where df is the thickness of the bilayer, and niso is the average RI of the bilayer calculated from the experimentally obtained RI values nTM and nTM using the formula:
where nbuffer is the RI of the buffer experimentally determined.
Circular dichroism (CD) spectropolarimetry
Circular dichroism experiments were performed using a Jasco 815 spectropolarimeter using a 0.1 cm path length quartz cell. All spectra were obtained at 20°C. POPC and mitochondrial outer membrane (POPC-POPE-POPS-PI-CL 54:30:2:10:4) small unilamellar vesicles were prepared by hydrating lipid in 10 mM sodium phosphate buffer, pH 7.4, and were extruded through a polycarbonate filter with 100 nm pore diameter. Bim BH3, (see Fig. 1) DPI-5-5 and DPI-5-5-RRR peptides were added separately to both 1 mM POPC and mitochondrial outer membrane solutions at final concentrations of 20 μM, resulting in peptide-to-lipid ratios of 1:50. The peptide–liposome solution was mixed by inversion and incubated for 2 min. Each spectrum was obtained by averaging five scans in the 190–260 nm wavelength range. All CD spectra are reported as mean residue ellipticity (MRE) [θ] (deg cm2 dmol−1).
Live-cell confocal microscopy
HeLa cells were plated in an eight-well microscopy slide at 50 000 cells/well in 200 µL DMEM (Dulbecco’s Modified Eagle Medium) medium containing 10% (v/v) foetal bovine serum (FBS) supplemented with penicillin and streptomycin. Cells were allowed to recover overnight at 37°C, 5% CO2. Before an experiment, the medium was removed and replaced with 198 µL of fresh DMEM with 0% or 10% FBS. Fluorophore-labelled peptides in DMSO were added (final DMSO concentration 1%) and the plates were incubated at 37°C, 5% CO2 for 2–5.5 h. After incubation, the medium was removed, the cells were washed with PBS, and then fresh DMEM was added, followed by the addition of wheat germ agglutinin–Alexa Fluor 488 conjugate (WGA488) (Life Technologies) to each well at 5 µg/mL and incubation for 10–20 min. Subsequently, Hoechst 33342 (Life Technologies) was added to stain nuclei for 5 min. The medium was then removed and the wells were washed two times with PBS, and then fresh PBS was added for imaging. Images were captured on an Eclipse TE2000-U laser scanning confocal microscope (Nikon) equipped with an Axio Camdigital camera (Carl Zeiss). All images were contrast-adjusted uniformly across all conditions.
Cell-killing assays
For CellTiter-Glo assays, wild-type (wt), mcl-1−/−, or bax−/−/bak−/− double knock-out (DKO) mouse embryonic fibroblasts (MEFs) were cultured in DMEM, supplemented with 10% (v/v) FCS, 250 µM l-asparagine and 50 µM 2-mercaptoethanol. On the day of experiments, cells were plated in opaque 96-well culture plates (~2000 cells/well) in DMEM, supplemented with 1% FCS, 250 µM asparagine and 50 µM 2-mercaptoethanol, and allowed to recover for 4 h. α/β-Peptides in DMSO were then diluted into media to make 2× stock solutions of α/β-peptides, and added to cells. Cells were incubated at 37°C, 5% CO2 for 24–48 h, and cell viability was determined by the addition of CellTiter-Glo chemiluminescence reagent (Promega). The percentage of viable cells was determined by dividing the luminescence signal from experimental wells by the signal obtained from control wells treated with DMSO. Similar protocols were followed for the analysis of cancer cell lines.
Results
Peptide design
The aim of this study was to design a peptide that would carry these pro-apoptotic peptides into the cell to act at the mitochondria. Several strategies have been used to enhance the cell entry of peptides that include the incorporation of hydrocarbon cross-links, alkyl chains and/or positively charged residues.[11,12]
Three peptides were synthesised with the sequences listed in Fig. 1. Bim BH3 contains the naturally occurring sequence derived from the Bim BH3 domain.[13] The aim of the present study was to design α/β-peptides that both mimic the α-helical structure of parent proteins targeted by Bim BH3 domain and have enhanced cell penetration. We previously developed a strategy to generate a family of α/β-peptides that closely mimic side chain presentation by a long α-helix by the incorporation of a number of novel cyclic β-amino acids at strategic positions in the parent sequence.[5,14–19] This strategy was applied to the design of the two α/β-peptides described in the present study and extended to enhance cell permeation of the Bim BH3 peptide.
Peptide sequences used in this study. The structures of the three modified β-amino acids (X = ACPC, Z = APC and J5) incorporated into the peptides are also shown. Ac indicates an acetylated N terminus; –NH2 indicates an amidated C terminus.

An analogue of the cyclic β-amino acid APC[13,17] was designed featuring an alkyl chain linked through a succinyl ester (J5, Fig. 1). This residue was designed to be hydrolysed to APC residues once inside the cell by intracellular esterases. Therefore, it was hypothesised that the J5 residue may be used to replace an APC residue in an optimised α/β-peptide, allowing the release of this optimised α/β-peptide in the cell. This strategy was also designed to eliminate the need for hydrocarbon cross-links or other moieties to enable cell entry.[5] DPI-5-5 contains the ACPC (trans-2-aminocyclopentane carboxylic acid) residue at positions 2 and 16, the ACP residue at position 9 and the J5 residue bearing a pentyl chain at positions 6 and 13 (Fig. 1).
Many peptides that are able to enter cells contain an overall net positive charge, often from multiple arginine residues.[11] DPI-5-5 has a net charge of +1, which may be insufficient to allow membrane permeability even in the presence of the alkyl esters. To test the hypothesis that increasing positive charge may facilitate more efficient cell entry, the peptide DPI-5-5-RRR, bearing three additional arginine residues at the C-terminus, was also synthesised.
Peptide conformation determined by CD
The conformation of each peptide in buffer or in each of the model lipid systems was determined by CD (Fig. 2). Bim BH3 adopted very little structure in buffer and in POPC, but demonstrated significant α-helical structure in the mitochondrial membrane mixture (Mito). DPI-5-5 adopted little structure in all three solutions although the minima at approximately 208 nm in both lipid mixtures suggest the existence of a low level of stabilised secondary structure. The spectra for DPI-5-5-RRR showed a stronger minimum at 208 nm in all three conditions, which is consistent with CD spectra and structures previously reported for α/β-peptides.[20] These results indicate that the presence of the conformationally constrained β-amino acids has resulted in the adoption of a stable secondary structure, irrespective of the solution conditions.
Secondary structure of Bim BH3, DPI-5-5 and DPI-5-5-RRR in HBS buffer (red line), plasma membrane environment (POPC, blue line) and mitochondrial membrane environment (Mito, black line). Peptides are 20 μM in the buffer solution, 10 μM in the plasma membrane environment and 20 μM in the mitochondrial membrane environment. Spectra were obtained from 190 to 260 nm, and all values are plotted as MRE × 103 (deg cm2 dmol−1).
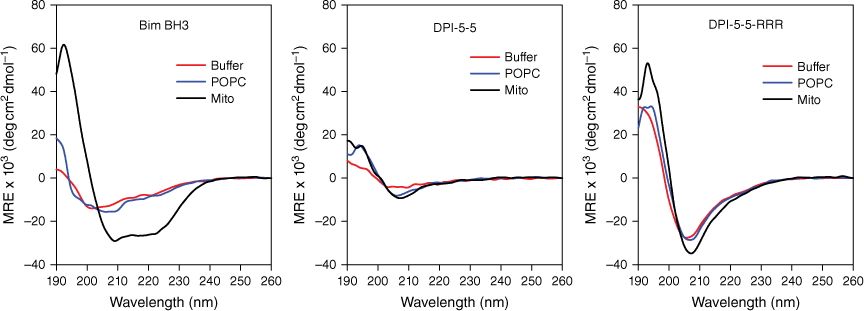
Peptide binding to lipid bilayers
The structural properties of each model membrane used in this study were analysed prior to peptide injection, including the mass of lipid deposited, and the thickness and birefringence of the bilayers (Table 1). The thickness values obtained for POPC and for the model mitochondrial membrane (Mito) are consistent with previous studies[21] and indicate that a single bilayer was generated in each case. The birefringence values for each bilayer were 0.0205 and 0.0210, similar in magnitude to other previously analysed membrane mimics.[21–23]
| Thickness (nm) | Birefringence | Mass (ng/mm2) | |
|---|---|---|---|
| POPC | 4.44 ± 0.07 | 0.0205 ± 0.0008 | 4.45 ± 0.06 |
| Mito (POPC/POPE/POPS/PI/TOCL = 54/30/2/10/4) | 4.66 ± 0.06 | 0.0210 ± 0.0009 | 4.67 ± 0.06 |
Data represent the average ± s.d. of 8–10 measurements.
The simultaneous measurement of multiple parameters for membrane bilayers by DPI analysis allows examination of peptide-induced changes in membrane structure, which can be monitored in real time. This allows investigation of the structural and dynamic changes occurring within the lipid matrix during peptide interaction, specifically in regards to the analysis of molecular orientation order. For each experiment, consecutive peptide concentrations of 2, 5, 10 and 20 μM were injected onto the same supported lipid bilayer. The accumulative binding of each peptide was first characterised by the TM and TE phase changes as a function of time and was subsequently resolved into the mass of membrane-bound peptide and birefringence for both lipid bilayers as previously described.[6,24] Plots of mass changes versus time for each peptide are shown in Fig. 3 for POPC (plasma membrane mimic) and POPC/POPE/POPS/PI/TOCL (Mito mimic) and birefringence versus time in Fig. 4.
Real-time peptide mass on membrane mimics (ng/mm2) injected at 30-min intervals at concentrations of 2, 5, 10 and 20 μM. Mass vs time for (a, d) Bim BH3; (b, e) DPI-5-5; and (c, f) DPI-5-5-RRR injected on the plasma membrane mimic POPC or the mitochondrial outer membrane mimic (POPC/POPE/POPS/PI/TOCL = 54/30/2/10/4).

Real-time changes in membrane disorder, measured as the birefringence (Δnf) on peptide binding following injections at 30-min intervals at concentrations of 2, 5, 10 and 20 μM. Birefringence vs time for (a, d) Bim BH3; (b, e) DPI-5-5; and (c, f) DPI-5-5-RRR injected on the plasma membrane mimic POPC or the mitochondrial outer membrane mimic (POPC/POPE/POPS/PI/TOCL = 54/30/2/10/4).

The amount of bound Bim BH3 peptide increased with increasing peptide concentration and was largely reversible on both membranes (Fig. 3a, d). However, the binding behaviour of the two analogues on each supported lipid bilayer was quite different from Bim BH3. On POPC, DPI-5-5 binding increased with higher concentrations, but at 20 μM, induced a significant mass loss (i.e. a drop below the baseline of the fresh bilayer), most likely due to loss of membrane material (Fig. 3b). On the Mito mimic, DPI-5-5 also caused significant mass loss at 20 μM (as seen on POPC), but this was not preceded by high level of peptide binding (Fig. 3e). In contrast, DPI-5-5-RRR bound at higher levels than Bim BH3 on POPC and the amount bound increased significantly at higher concentrations (Fig. 3c). On the Mito mimic, DPI-5-5-RRR also exhibited very high binding with a significant proportion of peptide remaining bound after the end of the dissociation phase (Fig. 3f).
The binding characteristics of each analogue were further evaluated by the relative changes in the molecular organisation of the bilayer as a function of membrane-bound peptide mass. The molecular order of a lipid bilayer is measured by the anisotropic opto-geometrical parameter, birefringence (Δnf), and the influence of peptide binding on this parameter can be obtained by DPI.[6,24] The changes in birefringence as a function of time for each peptide are shown in Fig. 4 for POPC and for the Mito mimic. Relatively small changes in birefringence were observed for Bim BH3 on both model membranes. In contrast, both DPI-5-5 and DPI-5-5-RRR caused much larger changes in the birefringence of both membranes, indicating large changes in the bilayer structure during peptide binding.
More insight into the changes in membrane structure was obtained through analysis of the plots of birefringence versus mass of peptide bound (Fig. 5). On POPC, the membrane order decreased during the binding of Bim BH3 and the extent of this drop increased with higher concentrations. However, the bilayer disorder returned to the initial conditions at the end of each dissociation (Fig. 5a). This result indicates that Bim BH3 binds and may partially insert itself, but then dissociates, allowing the bilayer to recover. In contrast, DPI-5-5 caused a large drop in bilayer order, which was reversible at 2, 5 and 10 μM. However, at 20 μM, this peptide caused a large drop in bilayer order at the point of mass loss, but the bilayer order of the remaining membrane recovered during dissociation. DPI-5-5-RRR also caused a large drop in bilayer order that was irreversible at 2, 5 and 10 μM. At 20 μM, there was a further drop in bilayer order, but this returned to the level observed for the 10 μM injection after dissociation of the peptide.
The effect of (a, d) Bim-BH3; (b, e) DPI-5-5; and (c, f) DPI-5-5-RRR on the molecular order of the plasma membrane mimic POPC (a, c, e) or the mitochondrial outer membrane mimic (POPC/POPE/POPS/PI/TOCL = 54/30/2/10/4) (b, d, f), determined by the optical birefringence (Δnf) as a function of membrane-bound peptide mass.
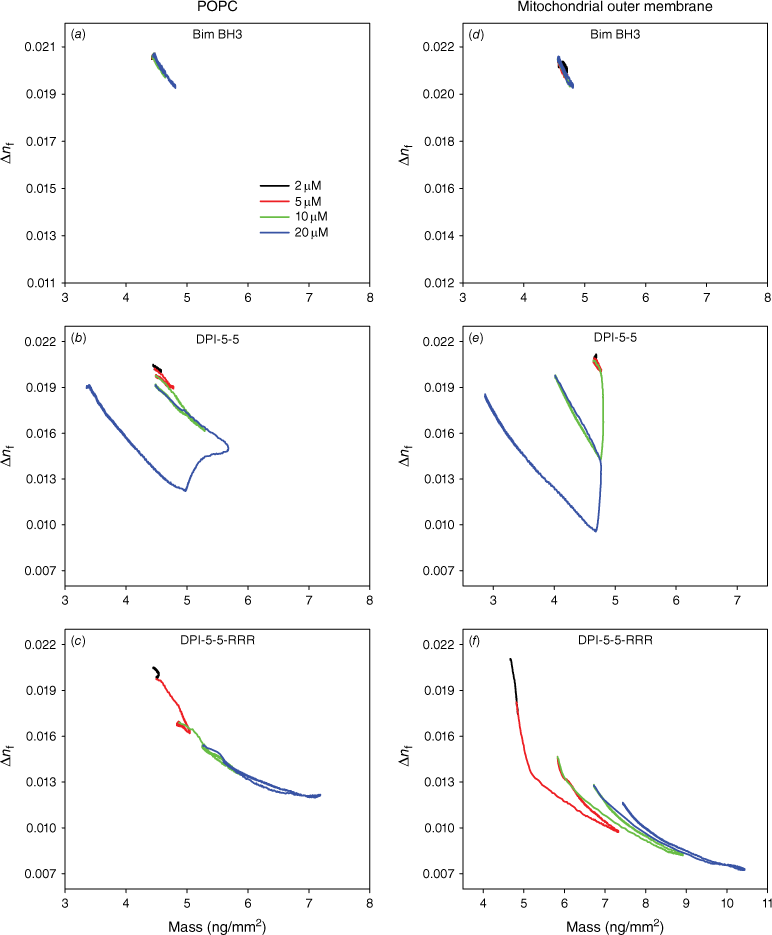
While similar dependencies of birefringence on mass were observed on the Mito mimic for Bim BH3, significant changes were again observed for DPI-5-5 and DPI-5-5-RRR. DPI-5-5 caused a small reversible drop in Mito birefringence at 2 and 5 μM, but a larger drop in mass at 10 and 20 μM with the birefringence returning to just under the initial birefringence value at each concentration. The binding of DPI-5-5-RRR to Mito caused a large decrease in birefringence at 5, 10 and 20 μM, and the birefringence values only recovered to a small proportion of the initial value.
In summary, the mass–birefringence profiles shown illustrate quite distinct behaviours of the three peptides on each of the model membranes. Bim BH3 showed very similar behaviour on both POPC and Mito, indicating low binding and minimal impact on the membrane bilayer. DPI-5-5 showed ‘reverse rectangular’ lines, reflecting significant binding at high concentrations accompanied by membrane loss and subsequent bilayer structure recovery after dissociation. Finally, DPI-5-5-RRR exhibited overlapping downward-trending lines that indicate an increasing amount of irreversibly bound peptide that is associated, with only partial recovery of the membrane bilayer structure.
Evaluation of cellular uptake by confocal microscopy
To gain insight into how DPI-5-5 and DPI-5-5-RRR interact with cells, fluorescein-labelled analogues of each peptide (Flu-DPI-5-5, and Flu-DPI-5-5-RRR, respectively) were synthesised and cellular interactions were monitored by live-cell confocal microscopy. When HeLa cells were incubated with Flu-DPI-5-5, a dose-dependent increase in green fluorescence in the confocal images was observed (Supplementary Fig. S7). In the absence of serum, most of this green fluorescence appeared on the outside of the cell at a higher concentration (≥10 µM), which suggest the presence of aggregated peptide strongly associated with the cell membrane that remained bound after multiple washing steps. In the presence of 10% serum, substantially less of the Flu-DPI-5-5 was observed on the outside of the cells as there was significant intracellular green fluorescence, suggesting that Flu-DPI-5-5 entered the HeLa cells under these conditions. The substantial difference in the localisation of fluorescence in the images obtained in the presence versus absence of serum may have resulted from the association of the hydrophobic Flu-DPI-5-5 with serum proteins, reducing the tendency for the peptide to aggregate.
When HeLa cells were treated with Flu-DPI-5-5-RRR, a significant amount of intracellular fluorescence was observed in the confocal microscopy images, suggesting that this peptide is able to enter cells (Supplementary Fig. S8). The increased amount of intracellular fluorescence for Flu-DPI-5-5-RRR versus Flu-DPI-5-5 indicates that the additional C-terminal arginine residues increase the efficiency of cellular uptake via a non-specific charge mechanism of interactions through the plasma membrane. Overall, the microscopy experiments suggest that α/β-peptides containing the J5 residue associate with cells and, under some conditions, are able to cross the cell membrane. However, it is not clear from these experiments if these α/β-peptides can gain access to the cytosol (to antagonise their target proteins), or remain trapped in intracellular vesicles.
Evaluation of activity by cell-based killing assays
To evaluate the ability of these peptides to induce apoptosis, a variety of cell types were treated with each peptide and cell viability measured using a CellTiter-Glo luminescent viability assay, which measures ATP levels. When non-cancerous MEFs were treated with DPI-5-5 and DPI-5-5-RRR, which each feature two J5 amino acid residues, there was a loss of cell viability at the highest concentration (20 µM). However, these peptides also killed bax−/−/bak−/− DKO cells, which lack the critical regulators of apoptosis BAX and BAK, and should not be susceptible to death through antagonism of Bcl-2 family protein–protein interactions. The observation that these peptides killed the DKO MEFs indicates that this loss in cell viability is due to an off-target effect rather than to antagonism of Bcl-2 protein–protein interactions.
Similar toxicity levels were observed for a variety of cancer cell lines tested: HeLa (cervical cancer), KM12 (colon cancer), MDA-MB-435 (breast cancer) and SK-MEL-2 (malignant melanoma) (Supplementary Fig. S10). For both DPI-5-5 and DPI-5-5-RRR, which showed some degree of cell killing, toxicity was not effectively blocked by the caspase inhibitor Quinoline–Val–Asp–Difluorophenoxymethylketone (Q-VD-OPh). Because caspases are critical for apoptosis, the lack of inhibition by Q-VD-OPh strongly suggests that any cell death observed for these α/β-peptides is due to a non-specific cytotoxic effect. The only change came from MDA-MB-435 cells treated with DPI-5-5 (Supplementary Fig. S9d), which appeared to show caspase-dependent cell death (attenuated by Q-VD-OPh) at 20 µM.
Discussion
The ability to design peptides that reach their specific intracellular target is an ongoing challenge. Although high-affinity ligands have been reported for a large number of intracellular protein targets, they often have low or no biological activity owing to their inability to cross the plasma membrane. Previous studies have reported the design of high-affinity α/β-peptides that target Bcl-2 protein–protein interactions.[5] In order to explore the molecular features that influence membrane permeability and thus facilitate cell entry of this class of peptides, a novel cyclic β-amino acid residue was designed and incorporated into the parent Bim peptide sequence. Here, we used DPI to evaluate the membrane binding and disruption of the peptide analogues.
In the present study, the membrane binding properties of Bim BH3 (control), DPI-5-5 and DPI-5-5-RRR were analysed in order to understand the structural features that influence the ability of the peptides to associate with membranes. The DPI results indicated that Bim BH3 binds reversibly to the plasma membrane mimic (POPC) and the mitochondrial membrane mimic (Mito) and also induces small reversible changes in birefringence, suggesting it binds to the surface of both membranes and then readily dissociates, irrespective of the secondary structure (extended in POPC and helical in Mito).
In comparison, DPI-5-5 binds weakly at low concentrations, but very strongly at higher concentrations to both the plasma membrane mimic (POPC) and the mitochondrial membrane mimic to the point where it causes large changes in the order of the bilayer structure. This effect occurs at a lower concentration on the Mito than on POPC suggesting it has a higher affinity for the mitochondrial membrane than POPC. Significantly, the loss of birefringence of the membrane was largely recovered in the membrane that remained on the surface for both POPC and Mito. In contrast, DPI-5-5-RRR binds partially reversibly to POPC with no membrane loss or damage. However, this peptide binds much more strongly than DPI-5-5 to the mitochondrial membrane mimic, indicating that the positively charged RRR motif significantly increases its affinity for the negatively charged mitochondrial mimic and at the same time diminishes the damage to the membrane.
Overall, the results indicate that the RRR motif enhances the ability of the peptide to bind and partially insert into the plasma membrane without destroying the POPC membrane. It also substantially enhances the affinity of the peptide to bind to the mitochondrial membrane and insert into it without destroying it. DPI-5-5 and DPI-5-5-RRR adopted a similar secondary structure in both liposomes. Hence, the mitochondrial membrane can accommodate the RRR-peptide via lipid bilayer structural changes that do not disrupt the overall bilayer integrity.
The data were correlated with cell entry properties of fluorescently labelled versions of DPI-5-5 and DPI-5-5-RRR and cell toxicity. Although the fluorescent analogues of both peptides were shown to enter cells, neither peptide exhibited any anti-pro-apoptotic activity, instead exhibiting non-specific toxic effects through an unknown off-target effect. It is possible that the target may be the mitochondrial membrane or that its improved affinity for the mitochondrial membrane has hijacked its target binding to a Bcl-2 protein. As the cellular uptake is increased by the RRR motif but there is no change in cellular toxicity between the peptides, the results indicate that the peptides may not be able to escape endosomes to act on their target. Future studies could focus on the specific impact of the J5 side chain using analogues with ACPC, and on the ability to escape endosome or delivery to the mitochondria.
Nevertheless, the strategy to incorporate a hydrophobic amino acid increased the membrane binding and, together with the RRR motif, resulted in successful passage through the plasma membrane. The lipidation of bioactive molecules is an important strategy to enhance cellular permeabilisation and drug uptake[12] and the presence of hydrophobic amino acids in these peptides resulted in strong binding to both model membranes and live cells. In summary, these insights into the mechanism of binding of these peptides to the plasma membrane and the mitochondrial membrane reveal further properties that should be considered in the design of peptides with intracellular protein targets.
Data availability
The data that support this study are available in the article and accompanying online supplementary material.
Declaration of funding
This research was funded by the National Health and Medical Research Council, grant nos APP1084648 and APP1142750. Research at UW-Madison was supported in part by NIH grants R01 GM056414 (S. H. G.) and R01 GM044783 (R. T. R.). J. W. C. was supported in part by a Biotechnology Training Grant from NIH (T32 GM008349).
Acknowledgements
MIA would like to dedicate this manuscript to her friend and colleague Professor Edouard Nice who has been a constant inspiration in all things bioanalysis including HPLC, optical biosensors, proteomics and life in general.
References
1 Lee EF, Fairlie WD. Discovery, development and application of drugs targeting BCL-2 pro-survival proteins in cancer. Biochem Soc Trans 2021; 49: 2381-95.
| Crossref | Google Scholar |
2 Lee EF, Harris TJ, Tran S, Evangelista M, Arulananda S, John T, Ramnac C, Hobbs C, Zhu H, Gunasingh G, Segal D, Behren A, Cebon J, Dobrovic A, Mariadason JM, Strasser A, Rohrbeck L, Haass NK, Herold MJ, Fairlie WD. BCL-XL and MCL-1 are the key BCL-2 family proteins in melanoma cell survival. Cell Death Dis 2019; 10: 342.
| Crossref | Google Scholar |
3 Diepstraten ST, Anderson MA, Czabotar PE, Lessene G, Strasser A, Kelly GL. The manipulation of apoptosis for cancer therapy using BH3-mimetic drugs. Nat Rev Cancer 2022; 22: 45-64.
| Crossref | Google Scholar |
4 Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol 2014; 15: 49-63.
| Crossref | Google Scholar |
5 Checco JW, Lee EF, Evangelista M, Sleebs NJ, Rogers K, Pettikiriarachchi A, Kershaw NJ, Eddinger GA, Belair DG, Wilson JL, Eller CH, Raines RT, Murphy WL, Smith BJ, Gellman SH, Fairlie WD. α/β-Peptide Foldamers Targeting Intracellular Protein–Protein Interactions with Activity in Living Cells. J Am Chem Soc 2015; 137: 11365-75.
| Crossref | Google Scholar |
6 Lee TH, Hirst DJ, Kulkarni K, Del Borgo MP, Aguilar MI. Exploring Molecular–Biomembrane Interactions with Surface Plasmon Resonance and Dual Polarization Interferometry Technology: Expanding the Spotlight onto Biomembrane Structure. Chem Rev 2018; 118: 5392-487.
| Crossref | Google Scholar |
7 Guzzo PR, Miller MJ. Catalytic, asymmetric synthesis of the carbacephem framework. J Org Chem 1994; 59: 4862-7.
| Crossref | Google Scholar |
8 Lee HS, LePlae PR, Porter EA, Gellman SH. An efficient route to either enantiomer of orthogonally protected trans-3-aminopyrrolidine-4-carboxylic acid. J Org Chem 2001; 66: 3597-9.
| Crossref | Google Scholar |
9 Horne WS, Price JL, Gellman SH. Interplay among side chain sequence, backbone composition, and residue rigidification in polypeptide folding and assembly. Proc Natl Acad Sci U S A 2008; 105: 9151-6.
| Crossref | Google Scholar |
10 de Feijter JA, Benjamins J, Veer FA. Ellipsometry as a tool to study the adsorption behaviour of synthetic and biopolymers at the air–water interface. Biopolymers 1978; 17: 1759-72.
| Crossref | Google Scholar |
11 Hao M, Zhang L, Chen P. Membrane Internalization Mechanisms and Design Strategies of Arginine-Rich Cell-Penetrating Peptides. Int J Mol Sci 2022; 23: 9038.
| Crossref | Google Scholar |
12 Menacho-Melgar R, Decker JS, Hennigan JN, Lynch MD. A review of lipidation in the development of advanced protein and peptide therapeutics. J Control Release 2019; 295: 1-12.
| Crossref | Google Scholar |
13 Peterson-Kaufman KJ, Haase HS, Boersma MD, Lee EF, Fairlie WD, Gellman SH. Residue-Based Preorganization of BH3-Derived α/β-Peptides: Modulating Affinity, Selectivity and Proteolytic Susceptibility in α-Helix Mimics. ACS Chem Biol 2015; 10: 1667-75.
| Crossref | Google Scholar |
14 Boersma MD, Haase HS, Peterson-Kaufman KJ, Lee EF, Clarke OB, Colman PM, Smith BJ, Horne WS, Fairlie WD, Gellman SH. Evaluation of diverse α/β-backbone patterns for functional α-helix mimicry: analogues of the Bim BH3 domain. J Am Chem Soc 2012; 134: 315-23.
| Crossref | Google Scholar |
15 Checco JW, Gellman SH. Targeting recognition surfaces on natural proteins with peptidic foldamers. Curr Opin Struct Biol 2016; 39: 96-105.
| Crossref | Google Scholar |
16 Checco JW, Gellman SH. Iterative Non-proteinogenic Residue Incorporation Yields α/β-Peptides with a Helix–Loop–Helix Tertiary Structure and High Affinity for VEGF. Chembiochem 2017; 18: 291-9.
| Crossref | Google Scholar |
17 Checco JW, Kreitler DF, Thomas NC, Belair DG, Rettko NJ, Murphy WL, Forest KT, Gellman SH. Targeting diverse protein–protein interaction interfaces with α/β-peptides derived from the Z-domain scaffold. Proc Natl Acad Sci U S A 2015; 112: 4552-7.
| Crossref | Google Scholar |
18 Horne WS, Boersma MD, Windsor MA, Gellman SH. Sequence-based design of α/β-peptide foldamers that mimic BH3 domains. Angew Chem Int Ed 2008; 47: 2853-6.
| Crossref | Google Scholar |
19 Johnson LM, Mortenson DE, Yun HG, Horne WS, Ketas TJ, Lu M, Moore JP, Gellman SH. Enhancement of α-helix mimicry by an α/β-peptide foldamer via incorporation of a dense ionic side-chain array. J Am Chem Soc 2012; 134: 7317-20.
| Crossref | Google Scholar |
20 Horne WS, Price JL, Keck JL, Gellman SH. Helix bundle quaternary structure from α/β-peptide foldamers. J Am Chem Soc 2007; 129: 4178-80.
| Crossref | Google Scholar |
21 Andreu-Fernández V, Genoves A, Lee TH, Stellato M, Lucantoni F, Orzáez M, Mingarro I, Aguilar MI, Pérez-Payá E. Peptides derived from the transmembrane domain of Bcl-2 proteins as potential mitochondrial priming tools. ACS Chem Biol 2014; 9: 1799-811.
| Crossref | Google Scholar |
22 Hirst DJ, Lee TH, Kulkarni K, Wilce JA, Aguilar MI. The impact of cell-penetrating peptides on membrane bilayer structure during binding and insertion. Biochim Biophys Acta 2016; 1858: 1841-9.
| Crossref | Google Scholar |
23 Hirst DJ, Lee TH, Swann MJ, Unabia S, Park Y, Hahm KS, Aguilar MI. Effect of acyl chain structure and bilayer phase state on binding and penetration of a supported lipid bilayer by HPA3. Eur Biophys J 2011; 40: 503-14.
| Crossref | Google Scholar |
24 Lee TH, Hirst DJ, Aguilar MI. New insights into the molecular mechanisms of biomembrane structural changes and interactions by optical biosensor technology. Biochim Biophys Acta 2015; 1848: 1868-85.
| Crossref | Google Scholar |