

Palladium-mediated CO2 extrusion followed by insertion of ketenes: translating mechanistic studies to develop a one-pot method for the synthesis of ketones
Yang Yang A , Allan J. Canty B and Richard A. J. O’Hair A *
A , Allan J. Canty B and Richard A. J. O’Hair A *
A School of Chemistry, Bio21 Institute of Molecular Science and Biotechnology, The University of Melbourne, Vic. 3010, Australia.
B School of Natural Sciences - Chemistry, University of Tasmania, Private Bag 75, Hobart, Tas. 7001, Australia.
Abstract
Multistage mass spectrometry (MSn) experiments were used to explore extrusion–insertion (ExIn) reactions of the palladium complex [(phen)Pd(O2CPh)]+ (phen, 1,10-phenanthroline). Under collision-induced dissociation (CID) conditions, the organopalladium cation [(phen)Pd(Ph)]+ was formed via decarboxylation and was found to react with phenylmethylketene to yield the enolate [(phen)Pd(CPhMeC(O)Ph)]+ via an insertion reaction. A further stage of CID revealed that the enolate fragments via loss of styrene to form the acyl complex [(phen)Pd(C(O)Ph)]+. Formation of both the coordinated enolate and acyl anions is supported by density functional theory (DFT) calculations. Attempts to develop a palladium-mediated one-pot synthesis of ketones from 2,6-dimethoxybenzoic acid as the key substrate and the ketene substrates R1R2C═C═O (R1 = Ph, R2 = Me; R1 = R2 = Ph) proved challenging owing to low yields and side product formation.
Keywords: collision-induced dissociation, decarboxylation, DFT calculations, extrusion–insertion reactions, insertion of ketene, multistage mass spectrometry, palladium-mediated reactions, reaction mechanisms.
Introduction
Understanding reaction mechanisms underpins the development of new methods in organic synthesis[1] and translation of existing protocols into robust processes for the bulk industrial synthesis of key fine chemicals, e.g. pharmaceuticals and agrochemicals.[2] Unfortunately, mechanistic studies in the area of homogeneous transition metal-catalysed processes in organic synthesis are beset by a number of challenges. With low catalyst loadings, it can be difficult to identify resting states, off-cycle reactions that lead to side products and pathways that lead to deactivation (poisoning) of the catalyst.
Density functional theory (DFT) calculations[3–7] and mass spectrometry experiments[8–14] provide valuable opportunities to interrogate the structure and reactivity of reactive intermediates that are potentially associated with elementary steps of catalytic cycles, allowing a better understanding of existing synthetic processes or the development of new ones. Professor Brian Yates has pioneered the use of DFT calculations to uncover key details for a range of elementary steps in transition metal catalysis. For example, it was demonstrated that the nature of the phosphine ligand can exert an influence on the transmetalation step in the palladium-catalysed Stille coupling reaction.[15]
As part of a longstanding interest in the use of collision-induced dissociation (CID) to initiate decarboxylation reactions of metal carboxylate complexes to unmask organometallic ions in the gas phase,[11,12,16] it occurred to us that ion–molecule reactions of the resultant organometallic ion with an appropriate (hetero)cumulene could be used to generate new coordinated ligands that are isoelectronic with carboxylates. Our initial report on transformation of the anionic copper acetate complex [(CH3)Cu(O2CCH3)]− to the anionic copper(i) dithioacetate complex [(CH3)Cu(S2CCH3)]−[17] convinced us that guided by mechanistic studies, these extrusion–insertion (ExIn) reactions could potentially be harnessed for one-pot organic synthetic methods for a wide range of new species including thioamides, amidines, amides, alkenes and ketones (Eqn 1, Scheme 1a).[18–24] In potential competition with the desired products is the protodecarboxylation side reaction (Eqn 2, Scheme 1a and Cycle B, Scheme 1b).[25,26] Fortunately, comprehensive investigations by Kozlowski’s group into the scope of the palladium-catalysed protodecarboxylation side reaction identified potential solvent and additives that suppress this reaction,[25] which provided a valuable starting point in our solution-phase investigations. We have blended gas-phase studies, DFT calculations and solution-phase studies (one-pot and microwave methods) to examine these ExIn reactions (Scheme 1b).[18,19,22,24] In all cases, stoichiometric (in palladium) variants were demonstrated to occur. Only for the synthesis of amidines and amides could variants that are catalytic in the palladium loading be developed.[19,22] The reason other systems could not be made catalytic in palladium is that the resultant ligand in species 8 (Scheme 1b) binds too strongly to palladium, making the protodepalladation step (Step 3 of Cycle A in Scheme 1b)[27] unfavourable and requiring well-established hydride sources[28] used in depalladation protocols (e.g. formate[29–31] or borohydrides[32,33]) to liberate the organic product and producing palladium(0) (likely via Steps 4 and 5 of Scheme 1b).
(a) Decarboxylation reactions for organic synthesis: extrusion–insertion (ExIn) reactions (Eqn 1) versus protodecarboxylation reaction (Eqn 2); (b) intermediates in variants of the ExIn reaction that are catalytic or stoichiometric in palladium and the competing protodecarboxylation reaction; (c) proposed intermediates in the Ito–Saegusa oxidation of ketones; (d) potential binding modes of palladium-coordinated enolates and examples of structures determined via X-ray crystallography.
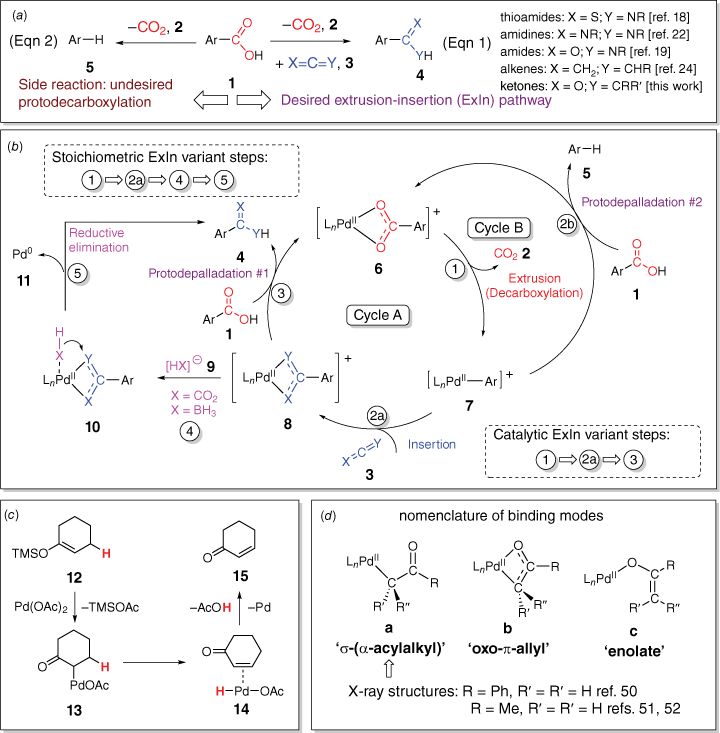
Here, we turn our attention to a new ExIn variant involving decarboxylation followed by insertion of a ketene (for the chemistry of ketenes, see refs [34–36]) to form a coordinated enolate,[37] which then reacts to form a ketone (for examples of metal-mediated ketone synthesis using other organic substrates, see refs [38,39]). We note that insertion of ketenes into Pd–C bonds has been reported[40,41] and that there is a rich literature on palladium enolates (e.g. Scheme 1c).[42–49] Potential binding modes of the coordinated enolate have been identified[44] and several crystal structures have been reported (Scheme 1d).[50–52] Noteworthy is the Ito–Saegusa oxidation of ketones to produce enones (Scheme 1c),[43] in which a silyl enol ether, 12, reacts with palladium acetate to form an enolate, 13, which undergoes beta-hydride transfer to form the palladium hydride intermediate, 14, which can undergo reductive elimination.[47] Thus, if the ExIn product 8 contains a β-hydride, an enone could potentially be formed instead of the desired ketone. Given the known limitations in the substrate scope of the carboxylic acid in palladium-mediated decarboxylation reactions,[25,53] in particular the propensity for C–H bond activation of the proton ortho to the carboxylate, and the propensity of ketenes containing α-hydrides to dimerise,[54] here we examine whether a one-pot method can be developed for the palladium-mediated synthesis of ketones from 2,6-dimethoxybenzoic acid (1a: Ar = 2,6-dimethoxyphenyl) and methylphenylketene (3a: X═O, Y═CMePh) and diphenylketene (3b: X═O, Y═CPh2). These ketenes were chosen because they are relatively stable compounds compared with those containing α-hydrogens, readily prepared via literature methods and related to the other (hetero)cumulenes previously examined in our ExIn studies (e.g. phenylisocyanate, phenylisothiocyanate). The expected product ketones 4a (Ar = 2,6-dimethoxyphenyl, X═O, Y═CMePh) and 4b (Ar = 2,6-dimethoxyphenyl, X═O, Y═CPh2) were also synthesised via an alternative route developed by Joshi et al.[55]
Results and discussion
Gas-phase experiments on decarboxylation of [(phen)Pd(O2CPh)]+ and insertion of methylphenylketene into the Pd–C bond of [(phen)Pd(Ph)]+
The cationic complex [(phen)Pd(O2CPh)]+ was produced under electrospray ionisation (ESI) of a methanolic solution of 1,10-phenanthroline, palladium trifluoroacetate and benzoic acid. As described in past reports,[18,19,22,24] this complex readily undergoes decarboxylation on CID to form [(phen)Pd(Ph)]+ (Eqn 3) as shown in Fig. 1a. The thus-produced organometallic cation [(phen)Pd(Ph)]+ undergoes an ion–molecule reaction (IMR) with methylphenylketene to yield a product ion at m/z 495 (Fig. 1d, Eqn 4). The ion at m/z 483 arises from the IMR between the organometallic cation [(phen)Pd(Ph)]+ (m/z 363) and acetophenone, which is a minor impurity from the synthesis of phenylmethylketene.
Multistage mass spectrometry experiment showing the sequence of reactions involved in the ExIn chemistry: (a) extrusion of CO2 from [(phen)Pd(O2CPh)]+ (m/z 407) under CID conditions at a normalised collision energy of 18 (Eqn 3); (b) ion-molecule reaction (IMR) between the organometallic cation [(phen)Pd(Ph)]+ (m/z 363) and phenylmethylketene at 10 ms reaction time (Eqn 4); (c) CID of the enolate [(phen)Pd(CPhMeC(O)Ph)]+ (m/z 495) (Eqns 5, 6). The concentration of phenylmethylketene in the IMRs is 1.27 × 1010 molecule cm−3; (d) MS5 CID of the acyl complex [(phen)Pd(COPh)]+ (m/z 391) (Eqn 7). The mass-selected ions are denoted by asterisks. The ion at m/z 483 arises from the IMR between the organometallic cation [(phen)Pd(Ph)]+ (m/z 363) and acetophenone, which is a minor impurity from the synthesis of phenylmethylketene.
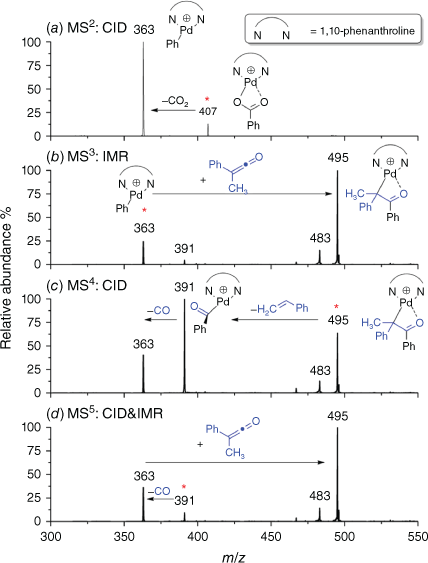
CID of the enolate [(phen)Pd(CPhMeC(O)Ph)]+ (m/z 495) yields products arising from deinsertion (Eqn 5) and styrene loss (Eqn 6). Each of the product assignments was confirmed via deuterium labelling experiments using the deuterated cationic complex [(phen)Pd(O2CC6D5)]+ (Supplementary Fig. S1).
We also examined fragmentation of the acyl cationic complex formed via styrene loss (Fig. 1d). It undergoes a decarbonylation reaction, as expected for such complexes. The regenerated aryl cation complex [(phen)Pd(Ph)]+ reacts with the methylphenylketene in the trap via insertion (Eqn 4).[56] Noting the general focus of our study is on metal-mediated ExIn reactions, it is interesting that the sequence of reactions Eqn 4 → Eqn 6 → Eqn 7 represents a cycle for the decomposition of phenylmethylketene to styrene and CO (Eqn 8).
DFT calculations on mechanistic aspects of insertion of methylphenylketene into the Pd–C bond of [(phen)Pd(Ph)]+ and fragmentation of the resultant coordinated enolate
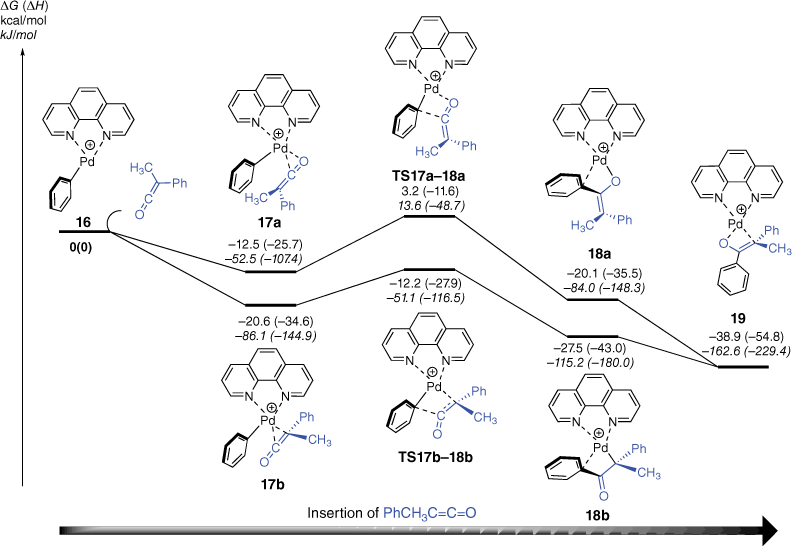
We next used DFT calculations to explore the mechanisms of the insertion reaction between [(phen)Pd(Ph)]+ and methylphenylketene (Fig. 2). Given that ketenes are known to coordinate to metal fragments via both C,C- and C,O-bonding modes,[57] we explored two different pathways for insertion of methylphenylketene into the Pd–C bond of [(phen)Pd(Ph)]+, 16. The DFT calculated energy diagram shown in Fig. 2 highlights that there are indeed two scenarios, which depend on the initial coordination mode of the ketene to the vacant coordination site in 16. Thus, in the initial adducts 17a and 17b, the orientation of the ketene is different. In the former, coordination via the C═O of the ketene occurs, which sets up an insertion pathway via TS17a–18a to yield the O-coordinated enolate, 18a. In contrast, in 17b, coordination via the C═C of the ketene occurs, which leads to the insertion pathway via TS17b–18b to produce the C-coordinated enolate, 18b. Although both pathways are below the separated reactants and are thus expected to occur, it is likely that 18a and 18b rearrange to the more stable O,C-coordinated enolate 19.
DFT calculated energy diagram for reaction of [(phen)Pd(C6H5)]+ with phenylmethylketene. The relative Gibbs energies and enthalpies (in parentheses) are given in kcal/mol (kJ/mol) and were calculated at the B3LYP-D3BJ/BS2//M06/BS1 level of theory.
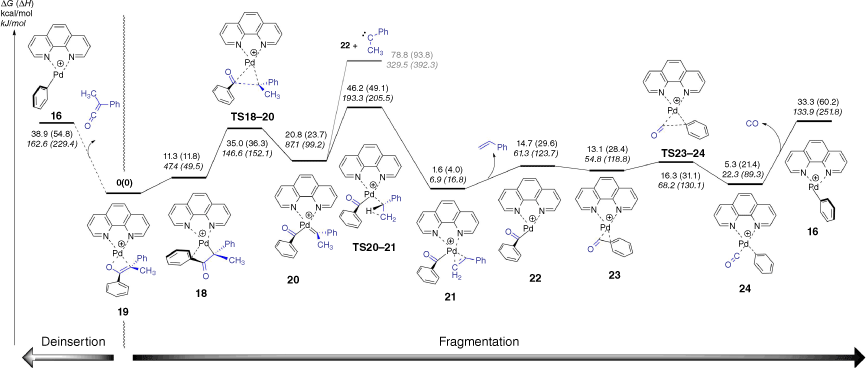
In order to better understand the three fragmentation reactions of the enolate [(phen)Pd(CPhMeC(O)Ph)]+ (Fig. 1c, d, Eqns 5–7), DFT calculations were used to explore mechanistic aspects (Fig. 3). The loss of styrene involves a multistep process. The first step has an energy barrier (relative to the starting complex 19) of 35.0 kcal/mol (1 kcal mol–1 = 4.186 kJ mol–1) and involves cleavage of the C–C bond via insertion of Pd utilising TS18–20 to give the palladium complex 20 containing a coordinated benzoyl anion and the phenylmethylcarbene. Loss of phenylmethylcarbene from 20 is energetically unfavourable compared with rearrangement to a coordinated styrene via TS20–21, involving a 1,2-hydride shift and proceeding over an energy barrier (relative to the starting complex 19) of 46.2 kcal/mol. Although the energy barrier (ΔG) is slightly higher than the energetics of deinsertion, the ΔH are similar and so are predicted to be competitive, which is consistent with the experimental data (Fig. 1c). The DFT calculations suggest that the structure of the ion at m/z 391 is neither 22 nor 23, which contain a coordinated benzoyl anion, but rather the decarbonylated cation [(phen)Pd(Ph)(CO)]+, 24, which can undergo the expected CO loss channel observed in the experiments (Fig. 1c).
DFT calculated energy surface for fragmentation of the enolate-palladium complex to forming [(phen)Pd(C(O)C6H5)]+ and [(phen)Pd(C6H5)]+. The relative Gibbs energies and enthalpies (in parentheses) are given in kcal/mol (kJ/mol) and were calculated at the B3LYP-D3BJ/BS2//M06/BS1 level of theory.
Attempts to develop a ligand-free palladium-mediated one-pot synthesis of ketones from 2,6-dimethoxybenzoic acid and the ketenes R1R2C═C═O (R1 = Ph, R2 = Me; R1 = R2 = Ph)
Encouraged by the gas-phase work described above, we next explored whether a palladium-mediated one-pot method could be developed for the preparation of ketones under ligand-free conditions using DMSO as a solvent, both with and without K2CO3 additive. As previously, the crude reaction mixtures were analysed via GC-MS. Independent synthesis of the ketones (β-methyl-β-phenyl)-2,6-dimethoxyacetophenone (25a) and (β-diphenyl)-2,6-dimethoxyacetophenone (25b) provided information on the GC-MS retention times and mass spectra (Supplementary Figs S2, S5), allowing ready assessment of whether the ketone product had formed in the various attempts listed in Table 1.
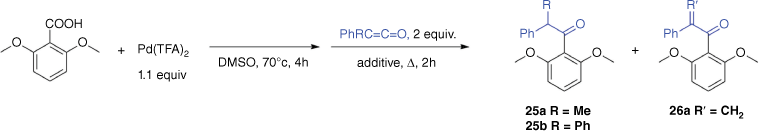 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | R | TemperatureA | Additive | Quench | 25B | 26B | |
| 1 | Me | r. t. | 3 equiv. K2CO3 | 5 equiv. HCl | N.O. | N.O. | |
| 2 | Me | r. t. | – | 5 equiv. HCl | N.O. | N.O. | |
| 3 | Me | 70°C | 3 equiv. K2CO3 | 5 equiv. NaBH4 | Trace | N.O. | |
| 4 | Me | 70°C | 3 equiv. K2CO3 | 5 equiv. HCl | N.O. | Trace | |
| 5 | Me | 70°C | – | 5 equiv. NaBH4 | 12% | Trace | |
| 6 | Me | 70°C | – | 5 equiv. HCl | N.O. | 13% | |
| 7 | Ph | 120°C | – | 5 equiv. HCl | N.O. | N.O. | |
| 8 | Ph | 120°C | – | 5 equiv. NaBH4 | N.O. | N.O. | |
| 9 | Ph | r. t. | 5 equiv. HCl | N.O. | N.O. | ||
| 10 | Ph | r. t. | 3 equiv. K2CO3 | 5 equiv. HCl | N.O. | N.O. | |
Ar.t. = room temperature, BYields were calculated based on the GC/MS spectra using the internal standard, 25a. N.O. = not observed via GC/MS
Only the ketone product 25a was observed at 70°C using no added base and quenching with 5 equiv. NaBH4 (Entry 5 in Table 1). In no cases was the ketone product 25b observed, perhaps resulting from the extra steric bulk of a second phenyl group hindering the insertion reaction. The GC-MS data revealed that a likely explanation for the poor yields is that side products are formed from both the carboxylic acid and ketene substrates. For example, protodepalladation of the arylpalladium intermediate gives rise to 1,3-dimethoxybenzene, 5 (Scheme 1), while the allenes undergo both hydration (to form the carboxylic acid) and dimerisation. The unsaturated ketone product 26a was observed in low yields in three instances (Entries 4–6 in Table 1). Owing to the low yield determined by GC-MS and the small scale of these reactions, attempts to isolate and fully characterise both the saturated and unsaturated ketones using NMR were unsuccessful.
DFT calculations on the solution-phase insertion step
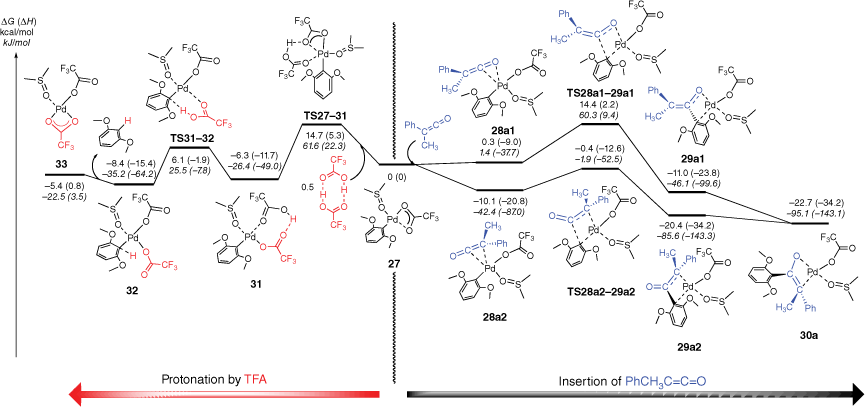
The DFT-determined mechanistic features of the palladium decarboxylation reaction under ligand-free conditions have been previously reported[25,58,59] and so are not discussed here. Instead, we next focused our attention on carrying out DFT calculations to examine the barriers and structural features associated with the insertion reaction of the ketenes (Fig. 4, Supplementary Fig. S7). The arylpalladium complex 27 undergoes coordination with methylphenylketene to form complexes 28a1 and 28a2 (Fig. 4). We were unable to locate transition states for these reactions. The orientation of the ketene is different in complexes 28a1 and 28a2, just as in the gas-phase system (cf. adducts 17a and 17b in Fig. 2). In 28a1, coordination via the C═O of the ketene sets up an insertion pathway via TS28a1–29a1 whereas for 28a2, coordination via the C═C of the ketene leads to the insertion pathway via TS28a2–29a2. Just as in the gas-phase system (Fig. 2), the product enolates coordinate to the Pd centre in a bidentate fashion, with 29a1 coordinating via the enolate O atom and the ipso C atom of the aryl ring while 29a2 coordinates via the enolate C atom and the ipso C atom. The latter structure is thermodynamically favoured over the former, and is similar in energy to a rearranged oxo-π-allyl isomer 30a. The features of the energy diagram for the diphenylketene insertion reactions are similar (Supplementary Fig. S7) and occur via the isomeric ketene complexes 28b1 and 28b2. TS28b2–29b2 is the lowest insertion barrier, but is higher than the analogous TS28a2–29a2, suggesting that the insertion reaction of diphenylketene should be slower than that of methylphenylketene.
DFT calculated energy surface showing protonation (left) and insertion (right) of phenylmethylketene into [(DMSO)(CF3CO2)Pd(Ar)] with comparison of different coordination modes of the enolates. The relative Gibbs energies and enthalpies (in parentheses) are given in kcal/mol (kJ/mol) and were calculated at the B3LYP-D3BJ/BS2//M06/BS1 level of theory in DMSO using the conductor-like polarisable continuum model (CPCM).
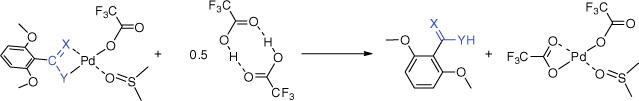
The fact that the energy barriers for TS28a2–29a2 and TS28b2–29b2 are below both barriers TS27–31 and TS31–32 associated with the protonation process does not provide a ready explanation of the low yields of the ketone and the fact 1,3-dimethoxybenzene is also observed in the experiments (Supplementary Fig. S3). Thus, we also calculated the energy required to liberate the ketone in the final protodepalladation reaction (Scheme 1, Step 3) and this is compared with data for the other heterocumulenes in Table 2. These data reveal that the enolate is bound less strongly (i.e. has a lower protodepalladation energy) than coordinated amidine, allyl and amidate ligands. Each of the latter gave much better yields of the liberated organic products than the ketones studied here. Thus, an alternative explanation for the poor yields may be the instability of the ketenes with respect to dimerisation (as evidenced in the GC-MS data).
Conclusions
Having now examined ExIn variants involving decarboxylation followed by insertion of a range of (hetero)cumulenes (Scheme 1), it is worth highlighting the differences between the various systems. The synthesis of amides via insertion of isocyanates is the only system that could be made catalytic. Key factors facilitating this outcome appear to be that the favourable energetics associated with both the insertion barrier (Step 2a of Cycle A) and protodepalladation reaction (Step 3). Although the barriers for insertion are favourable for isothiocyanates, carbodimides and allenes, the resultant anions are all excellent (hetero)-π-allyl ligands and are not readily subject to protodepalladation, requiring instead removal from palladium using hydride sources. This results in the reduction of palladium, making the reaction non-catalytic. In the case of ketenes, a key challenge appears to be the inherent instability of the monomer (which is required for the insertion step) with respect to dimerisation. Indeed, in comparison with the other (hetero)cumulenes, ketenes are the most susceptible to dimerisation.[60] This results in poor yields and higher side product formation as evidenced by the GC-MS analysis of reaction mixtures. Further development of new ExIn variants will need to consider the stability of the heterocumulene with respect to formation of dimers and other higher oligomers and how this will influence yields of the desired ExIn product.
Experimental
Reagents
Reagents, purchased from various commercial sources, were used as received. Chromatographic silica media (Davisil, 40−63 μm) was used as the stationary phase in flash column chromatography.
Gas-phase sample preparation
Ligated palladium cations [(phen)Pd(O2CR)]+, (R = C6H5 or C6D5) were subjected to CID to form arylpalladium cations [(phen)Pd(R)]+, which were then mass-selected for subsequent IMR studies with methylphenylketene reagent. We followed the protocols outlined in previous work.[61,62] For instance, methanolic solutions of palladium(ii) salt (10 mM), carboxylic acid (10 mM) and 1,10-phenanthroline (10 mM) were mixed in the ratio 1:1:2 and then diluted to 10 µM in palladium salt. A syringe pump (flow rate of 5 μL min−1) was used to inject the diluted solution into a modified linear ion-trap mass spectrometer (Thermo Finnigan LTQ) via the ESI source. The modified system allows IMRs between mass-selected ions and neutral molecules such as phenylmethylketene within the linear ion trap.[63,64] Briefly, the phenylmethylketene reagent is injected into the helium bath gas via a gas-tight syringe that is pumped by a syringe pump. As the flow of the helium (1082 cm3 min−1) and phenylmethylketene (3 μL h−1) is known, as is the pressure of the helium in the ion trap (2 × 10−3 Torr), the concentration of the neutral reagent can be determined, allowing measured rates to be converted to rate constants. Full details have been previously described.[64] All spectra data were acquired from between 20 and 100 duplicate spectra with 3–5 microscans in each scan.
Mass spectrometry source conditions
The sheath gas setting was 10 AU (AU, arbitrary units), the auxiliary gas 0.03 AU and sweep gas 0.02 AU; the spray voltage was 3.6 kV, capillary temperature was set to 200 °C, capillary voltage was 2 V, and the tube lens voltage was set to 75 V.
DFT studies
The Gaussian 16 suite of programs was used to fully optimise all reactants, intermediates, transition states and products at the M06 level of DFT.[65,66] The effective-core potential of Hay and Wadt with a double-ξ valence basis set (LANL2DZ) was used to describe Pd[67,68] and the 6-31G(d) basis set was chosen for the other atoms.[69] In addition, a polarisation function (ξf = 1.472) was added solely for Pd.[70,71] BS1 is used to designate this combination of basis sets. In order to account for the solvation effects of DMSO on the optimised structures, the CPCM model was used.[72] Frequency calculations were carried out at the same level of theory as those for the structural optimisation. The Berny algorithm was used to locate each of the transition structures. To establish that transition states connected to minima, intrinsic reaction coordinate (IRC) calculations were carried out.[73,74]
The energies were further refined using single-point energy calculations. Thus, the energies of the structures obtained from the M06/BS1 calculations were recalculated with a larger basis set (BS2) at the B3LYP-D3BJ or CAM- B3LYP-D3BJ level of theory.[75–78] BS2 utilises def2-TZVP11 for all atoms along with the effective core potential including scalar relativistic effects for Pd.[79] The solvation effect of DMSO was also considered in the single-point calculations using the CPCM model. Relative enthalpy, ΔH, and Gibbs energies, ΔG, at the BS2 level of theory were calculated using the correction values calculated from M06/BS1. Based on the method reported by Okuno, extra corrections for entropy calculations were considered in the solvent system.[80] When DMSO participates in the equilibrium of a certain transformation step, an additional correction was considered based on the concentration of the DMSO using the method proposed by Keith and Carter (eqn 6 of their paper was used).[81] Unless otherwise stated, all the enthalpy and Gibbs free energies were calculated and corrected from the B3LYP-D3BJ/BS2//M06/BS1 level of theory.
Synthetic procedures
1H NMR and 13C{1H} NMR spectra were recorded on a Jeol 400 MHz NMR spectrometer at 298 K. Chemical shifts are reported in parts per million (ppm) and referenced to the residual solvent peak (d6-DMSO: 1H NMR 2.50 ppm, 13C NMR 39.52 ppm). Coupling constants (J) are reported in hertz (Hz). Standard abbreviations indicating multiplicity used are as follows: m, multiplet; quint, quintet; q, quartet; t,triplet; d, doublet; s, singlet; br, broad. High-resolution electrospray ionisation mass spectra (ESI-HRMS) were collected on a ThermoScientific Exactive Plus Orbitrap mass spectrometer (Thermo, Bremen, Germany).
GC analyses were carried out using an Agilent 7890A/5975C GC-MS instrument with an HP-5 ms capillary column (Agilent Technologies, phenylmethyl siloxane, 30 m × 0.25 mm × 0.25 μm) and a time program beginning with 5 min at 70°C, followed by 15 °C/min ramp to 300°C, then 10 min at this temp.
General procedure for the preparation of 2-phenyl propionic acid
To a stirred −78°C solution of diisopropylamine (40 mmol) in 120 mL dry THF under Ar was added 20 mL n-BuLi (2 M in cyclohexane) dropwise. The solution was stirred at −78°C for 5 min and 2-phenylacetic acid (20 mmol) in 20 mL dry THF was added in dropwise over 10 min. The reaction mixture was stirred at 0°C for 1 h and recooled to −78°C, and a single portion of MeI (30 mmol) was added. After stirring the mixture overnight at room temperature, the reaction was quenched with water and concentrated under vacuum. The residue was dissolved in water and extracted with Et2O before and after acidification (pH 1) yielded 2.40 g (80%) as a brown oil.
General procedure for the preparation of ketenes
In a flame-dried flask under nitrogen gas flow, the carboxylic acid (50 mmol) was dissolved in dry DCM (150 mL) and was treated with oxalyl chloride (55 mmol). The solution was then stirred for 20 min in an ice-water bath and then treated with dry DMF (0.1 mL). After 30 min stirring at 0°C and another 2 h at room temperature, the reaction was quenched with 0.5 M HCl and extracted with DCM (3 × 100 mL) and dried over Na2SO4 anhydride. The solvent was removed under reduced pressure. The acid chlorides were then stored in a vial for the subsequent steps or in a sealed vial in a freezer for 1 month.
Acid chlorides (10 mmol) were dissolved in dry Et2O (20 mL, 0.5 M) under N2 before cooling to 0°C. Triethylamine (10 mmol) was added dropwise over 10 min, and the reaction was stirred overnight at 0°C. The solution was filtered through a sintered adaptor into a second flask and concentrated. The crude oil was then purified by distillation.
Prepared according to the general procedure from 2-phenylpropanic acid (1.68 g, 10 mmol), oxalyl chloride (0.94 mL, 11 mmol) and distilled Et3N (1.39 mL, 10 mmol). Distillation of the crude oil (60–80°C, 1 mbar) yielded 0.55 g, 42%, as a bright yellow-orange oil. 1H NMR (CDCl3, 400 MHz) δ 7.40–7.28 (5H, m), 1.61 (3H, s). 13C NMR (CDCl3, 400MHz) δ 175.71, 137.57, 129.21, 128.32, 128.05, 57.56. MS(EI) (relative intensity), m/z 132.1 (100%), 134.1 (79%), 78.1 (58%)
Prepared according to the general procedure from 2,2-diphenylacetic acid (2.12 g, 10 mmol), oxalyl chloride (0.94 mL, 11 mmol) and distilled Et3N (1.39 mL, 10 mmol). Distillation of the crude oil (100–120°C, 1 mbar) yielded 1.22 g, 63%, as bright orange oil. 1H NMR (CDCl3, 400 MHz) δ 7.38 (5H, m), 7.24 (5H, m). 13C NMR (CDCl3, 400MHz) δ 167.65, 137.01, 129.36, 127.8, 126.33, 57.96. MS(EI) (relative intensity) m/z 194.1 (42%), 165.1 (100%), 82.4 (10%)
Ketone synthesis using the method developed by Joshi et al.
The ketone species were prepared independently following a literature procedure in which 1,3-dimethoxybenzene was reacted with n-butyllithium and methylphenylacetyl chloride or diphenylacetyl chloride.[55]
The compound was prepared using method of Joshi et al.: yield 27% as a white solid. Column chromatography (silica gel, petroleum spirit/ethyl acetate 1:10). 1H NMR (400 MHz, d-chloroform, ppm) δ 7.25–7.13 (m, 6H), δ 6.43 (d, J = 8.4 Hz, 2H), 4.20 (q, J = 7.2 Hz, 1H), 3.62 (s, 6H), 1.52 (d, J = 7.1 Hz, 3H). 13C NMR (400 MHz, d-chloroform, ppm) δ 204.73, 156.75, 140.19, 130.63, 128.67, 128.02, 126.71, 120.15, 103.94, 55.77, 53.94, 16.88. HR-ESMS(ESI) m/z [M + H]+ calcd. for C17H19O3 271.13287, found 271.13211.
The compound was prepared using method of Joshi et al.: yield 17% as a white solid. Column chromatography (silica gel, petroleum spirit/ethyl acetate:1/10). 1H NMR (400 MHz, d-chloroform, ppm) δ 7.36–7.15 (m, 11H), 6.45 (d, J = 8.4 Hz, 2H), 5.63 (s, 1H), 3.68 (s, 6H). 13C NMR (400 MHz, d-chloroform, ppm) δ 201.38, 157.03, 139.22, 131.84, 129.78, 128.63, 127.27, 120.08, 104.95, 64.86, 56.34. HR-ESMS(ESI) m/z [M + H]+ calcd. for C22H21O3 333.14852, found 333.14866.
2,6-Dimethoxybenzoic acid (0.2 mmol, 36.4 mg) and Pd(TFA)2 (0.22 mmol, 72.6 mg) were dissolved in anhydrous DMSO (2 mL). The solution was heated at 70°C over 4 h followed by addition of 2 equiv. ketene (0.4 mmol). After heating for another 2 h, the reaction was cooled to room temperature and stirred for 30 min after adding NaBH4 (5 equiv., 1 mmol, 38 mg). The mixture was then quenched with 100 mL water and extracted with ethyl acetate (50 mL × 3). The organic phase was washed with 100 mL water and 100 mL brine, and dried with the drying agent MgSO4. The reaction mixture was analysed by GC-MS.
Acknowledgements
We thank Professor Uta Wille for access to the GC-MS. We thank Professor Jonathan White for the X-ray crystallographic structure determination of compound 25a (Supplementary Fig. S12, Supplementary Table S1). We thank the Bio21 Mass Spectrometry and Proteomics Facility for access to the ThermoScientific Exactive Plus Orbitrap mass spectrometer. YY thanks The University of Melbourne for the award of a PhD scholarship.
References
2 Wei CS, Simmons EM, Hsaio Y, Eastgate MD. Development of Robust, Scaleable Catalytic Processes through Fundamental Understanding of Reaction Mechanisms. Top Catal 2017; 60(8): 620-630.
| Crossref | Google Scholar |
3 Dedieu A. Theoretical studies in palladium and platinum molecular chemistry. Chem Rev 2000; 100(2): 543-600.
| Crossref | Google Scholar |
4 Sperger T, Sanhueza IA, Kalvet I, Schoenebeck F. Computational Studies of Synthetically Relevant Homogeneous Organometallic Catalysis Involving Ni, Pd, Ir, and Rh: An Overview of Commonly Employed DFT Methods and Mechanistic Insights. Chem Rev 2015; 115(17): 9532-9586.
| Crossref | Google Scholar |
5 Sperger T, Sanhueza IA, Schoenebeck F. Computation and Experiment: A Powerful Combination to Understand and Predict Reactivities. Acc Chem Res 2016; 49(6): 1311-1319.
| Crossref | Google Scholar |
6 Xiong Q, Chen HH, Zhang T, Shan CH, Bai RP, Lan Y. On the Mechanism of Palladium-Catalyzed Unsaturated Bond Transformations: A Review of Theoretical Studies. Asian J Org Chem 2019; 8: 1194-1206.
| Crossref | Google Scholar |
7 Xue L, Lin Z. Theoretical aspects of palladium-catalysed carbon-carbon cross-coupling reactions. Chem Soc Rev 2010; 39(5): 1692-1705.
| Crossref | Google Scholar |
8 Böhme DK, Schwarz H. Gas-phase catalysis by atomic and cluster metal ions: The ultimate single-site catalysts. Angew Chem Int Ed 2005; 44(16): 2336-2354.
| Crossref | Google Scholar |
9 O’Hair RAJ. The 3D quadrupole ion trap mass spectrometer as a complete chemical laboratory for fundamental gas-phase studies of metal mediated chemistry. Chem Commun 2006; 14: 1469-1481.
| Crossref | Google Scholar |
10 O’Hair RAJ. Mass spectrometry based studies of gas phase metal catalyzed reactions. Int J Mass Spectrom 2015; 377: 121-129.
| Crossref | Google Scholar |
11 O’Hair RAJ. Organometallic Gas-Phase Ion Chemistry and Catalysis: Insights into the Use of Metal Catalysts to Promote Selectivity in the Reactions of Carboxylic Acids and Their Derivatives. Mass Spectrom Rev 2021; 40(6): 782-810.
| Crossref | Google Scholar |
12 O’Hair RAJ, Rijs NJ. Gas phase studies of the Pesci decarboxylation reaction: synthesis, structure, and unimolecular and bimolecular reactivity of organometallic ions. Acc Chem Res 2015; 48(2): 329-340.
| Crossref | Google Scholar |
13 O’Hair RAJ. Gas-phase studies of metal catalyzed decarboxylative cross-coupling reactions of esters. Pure Appl Chem 2015; 87(4): 391-404.
| Crossref | Google Scholar |
14 Schwarz H. Ménage à trois: single-atom catalysis, mass spectrometry, and computational chemistry. Catal Sci Technol 2017; 7(19): 4302-4314.
| Crossref | Google Scholar |
15 Ariafard A, Yates BF. Subtle balance of ligand steric effects in Stille transmetalation. J Am Chem Soc 2009; 131(39): 13981-13991.
| Crossref | Google Scholar |
17 Li J, Khairallah GN, O’Hair RAJ. Dimethylcuprate-Mediated Transformation of Acetate to Dithioacetate. Organometallics 2015; 34(2): 488-493.
| Crossref | Google Scholar |
18 Noor A, Li J, Khairallah GN, Li Z, Ghari H, Canty AJ, Ariafard A, Donnelly PS, O’Hair RAJ. A one-pot route to thioamides discovered by gas-phase studies: palladium-mediated CO2 extrusion followed by insertion of isothiocyanates. Chem Commun 2017; 53(27): 3854-3857.
| Crossref | Google Scholar |
19 Yang Y, Canty AJ, McKay AI, Donnelly PS, O’Hair RAJ. Palladium-Mediated CO2 Extrusion Followed by Insertion of Isocyanates for the Synthesis of Benzamides: Translating Fundamental Mechanistic Studies To Develop a Catalytic Protocol. Organometallics 2020; 39: 453-467.
| Crossref | Google Scholar |
20 Yang Y, Canty AJ, O’Hair RAJ. Gas-phase studies of copper(i)-mediated CO2 extrusion followed by insertion of the heterocumulenes CS2 or phenylisocyanate. J Mass Spectrom 2020; 56(4): e4579.
| Crossref | Google Scholar |
21 Yang Y, Canty AJ, O’Hair RAJ. Why does the synthesis of N-phenylbenzamide from benzenesulfinate and phenylisocyanate via the palladium-mediated extrusion–insertion pathway not work? A mechanistic exploration. Aust J Chem 2023; 76(1): 49-57.
| Crossref | Google Scholar |
22 Yang Y, Noor A, Canty AJ, Ariafard A, Donnelly PS, O’Hair RAJ. Synthesis of Amidines by Palladium-Mediated CO2 Extrusion Followed by Insertion of Carbodiimides: Translating Mechanistic Studies to Develop a One-Pot Method. Organometallics 2019; 38: 424-435.
| Crossref | Google Scholar |
23 Yang Y, Spyrou B, Donnelly PS, Canty AJ, O’Hair RAJ. The role of silver carbonate as a catalyst in the synthesis of N-phenylbenzamide from benzoic acid and phenyl isocyanate: a mechanistic exploration. Aust J Chem 2022; 75(9): 495-505.
| Crossref | Google Scholar |
24 Yang Y, Spyrou B, White JM, Canty AJ, Donnelly PS, O’Hair RAJ. Palladium-Mediated CO2 Extrusion Followed by Insertion of Allenes: Translating Mechanistic Studies to Develop a One-Pot Method for the Synthesis of Alkenes. Organometallics 2022; 41(13): 1595-1608.
| Crossref | Google Scholar |
25 Dickstein JS, Curto JM, Gutierrez O, Mulrooney CA, Kozlowski MC. Mild aromatic palladium-catalyzed protodecarboxylation: kinetic assessment of the decarboxylative palladation and the protodepalladation steps. J Org Chem 2013; 78(10): 4744-4761.
| Crossref | Google Scholar |
26 Dickstein JS, Mulrooney CA, O’Brien EM, Morgan BJ, Kozlowski MC. Development of a catalytic aromatic decarboxylation reaction. Org Lett 2007; 9(13): 2441-2444.
| Crossref | Google Scholar |
27 O’Duill ML, Engle KM. Protodepalladation as a Strategic Elementary Step in Catalysis. Synthesis 2018; 50(24): 4699-4714.
| Crossref | Google Scholar |
28 Tsuji J, Mandai T. Palladium-Catalyzed Hydrogenolysis of Allylic and Propargylic Compounds with Various Hydrides. Synthesis 1996; 1996(01): 1-24.
| Crossref | Google Scholar |
29 Hey H, Arpe H-J. Removal of Allyl groups by Formic Acid Catalyzed by (Triphenylphosphane)palladium. Angew Chem Int Ed Engl 1973; 12(11): 928-929.
| Crossref | Google Scholar |
30 Oshima M, Sakamoto T, Maruyama Y, Ozawa F, Shimizu I, Yamamoto A. Synthesis and properties of (η3-1-methylallyl)palladium(ii) formates as models of intermediates in the palladium-catalyzed reductive cleavage of allylic carboxylates and carbonates with formic acid. Bull Chem Soc Jpn 2000; 73: 453-464.
| Crossref | Google Scholar |
31 Oshima M, Yamazaki H, Shimizu I, Nisar M, Tsuji J. Palladium-Catalyzed Selective Hydrogenolysis of Alkenyloxiranes with Formic-Acid. Stereoselectivity and Synthetic Utility. J Am Chem Soc 1989; 111: 6280-6287.
| Crossref | Google Scholar |
32 Hutchins RO, Learn K. Regio- and Stereoselective Reductive Replacement of Allylic Oxygen, Sulfur, and Selenium Functional Groups by Hydride Via Catalytic Activation by Palladium(0) Complexes. J Org Chem 1982; 47: 4380-4382.
| Crossref | Google Scholar |
33 Hutchins RO, Learn K, Fulton RP. Reductive Displacement of Allylic Acetates by Hydride Transfer Via Catalytic Activation by Palladium(0) Complexes. Tetrahedron Lett 1980; 21: 27-30.
| Crossref | Google Scholar |
34 Allen AD, Tidwell TT. Ketenes and Other Cumulenes as Reactive Intermediates. Chem Rev 2013; 113(9): 7287-7342.
| Crossref | Google Scholar |
35 Allen AD, Tidwell TT. Recent advances in ketene chemistry. ARKIVOC 2016; 2016(1Spec.Issue): 415-490.
| Crossref | Google Scholar |
38 Culkin DA, Hartwig JF. Palladium-catalyzed α-arylation of carbonyl compounds and nitriles. Acc Chem Res 2003; 36(4): 234-245.
| Crossref | Google Scholar |
39 Dieter RK. Reaction of acyl chlorides with organometallic reagents: A banquet table of metals for ketone synthesis. Tetrahedron 1999; 55(14): 4177-4236.
| Crossref | Google Scholar |
40 Mitsudo T, Kadokura M, Watanabe Y. Palladium-complex-catalyzed reactions of ketenes with allylic carbonates or acetates. Novel syntheses of α-allylated carboxylic esters and 1,3-dienes. J Org Chem 2002; 52(9): 1695-1699.
| Crossref | Google Scholar |
41 Mitsudo T, Kadokura M, Watanabe Y. Novel synthesis of α,β-unsaturated ketones by the palladium-catalyzed arylation of ketenes with aroyl chlorides or the decarbonylative cross-condensation of acyl halides. J Org Chem 2002; 52(15): 3186-3192.
| Crossref | Google Scholar |
42 Diao T, Pun D, Stahl SS. Aerobic dehydrogenation of cyclohexanone to cyclohexenone catalyzed by Pd(DMSO)2(TFA)2: evidence for ligand-controlled chemoselectivity. J Am Chem Soc 2013; 135(22): 8205-8212.
| Crossref | Google Scholar |
43 Ito Y, Hirao T, Saegusa T. Synthesis of α,β-unsaturated carbonyl compounds by palladium(ii)-catalyzed dehydrosilylation of silyl enol ethers. J Org Chem 1978; 43(5): 1011-1013.
| Crossref | Google Scholar |
44 Ito Y, Nakatsuka M, Kise N, Saegusa T. Preparation of Pd(ii) enolate complexes and their reactions. Tetrahedron Lett 1980; 21(30): 2873-2876.
| Crossref | Google Scholar |
45 Minami I, Takahashi K, Shimizu I, Kimura T, Tsuji J. New synthetic methods for α,β -unsaturated ketones, aldehydes, esters and lactones by the palladium-catalyzed reactions of silyl enol ethers, ketene silyl acetals, and enol acetates with allyl carbonates. Tetrahedron 1986; 42(11): 2971-2977.
| Crossref | Google Scholar |
46 Muzart J. One-Pot Syntheses of α,β-Unsaturated Carbonyl Compounds through Palladium-Mediated Dehydrogenation of Ketones, Aldehydes, Esters, Lactones and Amides. Eur J Org Chem 2010; 2010(20): 3779-3790.
| Crossref | Google Scholar |
47 Porth S, Bats JW, Trauner D, Giester G, Mulzer J. Insight into the Mechanism of the Saegusa Oxidation: Isolation of a Novel Palladium(0)–Tetraolefin Complex. Angew Chem Int Ed 1999; 38(13-14): 2015-2016.
| Crossref | Google Scholar |
48 Shimizu I, Minami I, Tsuji J. Palladium-catalyzed synthesis of α,β-unsaturated ketones from ketones via allyl enol carbonates. Tetrahedron Lett 1983; 24(17): 1797-1800.
| Crossref | Google Scholar |
49 Theissen RJ. Preparation of α,β-unsaturated carbonyl compunds. J Org Chem 1971; 36(6): 752-757.
| Crossref | Google Scholar |
50 Falvello LR, Garde R, Miqueleiz EM, Tomás M, Urriolabeitia EP. Evidence of C═H activation of acetone by a platinum(ii) complex. Synthesis and structural characterization of [Pt(CH2COCH3)Cl(bipy)] (bipy = 2,2′-bipyridyl). Inorg Chim Acta 1997; 264(1–2): 297-303.
| Crossref | Google Scholar |
51 Veya P, Floriani C, Chiesi-Villa A, Rizzoli C. Terminal and bridging bonding modes of the acetophenone enolate to palladium(ii): the structural evidence and the insertion of isocyanides. Organometallics 2002; 12(12): 4899-4907.
| Crossref | Google Scholar |
52 Vicente J, Abad JA, Chicote M-T, Abrisqueta M-D, Lorca J-A, Ramírez de Arellano MC. Synthesis of New Ketonyl Palladium(ii) and Platinum(ii) Complexes with Nitrogen-Donor Ligands. Crystal Structure of [Pt{CH2C(O)Me}2(bpy)]. Organometallics 1998; 17(8): 1564-1568.
| Crossref | Google Scholar |
53 Myers AG, Tanaka D, Mannion MR. Development of a decarboxylative palladation reaction and its use in a Heck-type olefination of arene carboxylates. J Am Chem Soc 2002; 124(38): 11250-11251.
| Crossref | Google Scholar |
54 Farnum DG, Johnson JR, Hess RE, Marshall TB, Webster B. Aldoketene Dimers and Trimers from Acid Chlorides. A Synthesis of Substituted 3-Hydroxycyclobutenones. J Am Chem Soc 1965; 87(22): 5191-5197.
| Crossref | Google Scholar |
55 Joshi BS, Dabholkar KDM, Gawad DH. Reaction of furanopyrones with aluminium chloride in benzene. Indian J Chem 1972; 10(6): 567-570.
| Google Scholar |
56 Lesslie M, Yang Y, Canty AJ, Piacentino E, Berthias F, Maitre P, Ryzhov V, O’Hair RAJ. Ligand-induced decarbonylation in diphosphine-ligated palladium acetates [CH3CO2Pd((PR2)2CH2)]+ (R = Me and Ph). Chem Commun 2018; 54(4): 346-349.
| Crossref | Google Scholar |
57 Grotjahn DB, Collins LSB, Wolpert M, Bikzhanova GA, Lo HC, Combs D, Hubbard JL. First direct structural comparison of complexes of the same metal fragment to ketenes in both C,C- and C,O-bonding modes. J Am Chem Soc 2001; 123(34): 8260-8270.
| Crossref | Google Scholar |
58 Xue L, Su W, Lin Z. A DFT study on the Pd-mediated decarboxylation process of aryl carboxylic acids. Dalton Trans 2010; 39(41): 9815-9822.
| Crossref | Google Scholar |
59 Zhang S-L, Fu Y, Shang R, Guo Q-X, Liu L. Theoretical Analysis of Factors Controlling Pd-Catalyzed Decarboxylative Coupling of Carboxylic Acids with Olefins. J Am Chem Soc 2010; 132(2): 638-646.
| Crossref | Google Scholar |
61 Woolley M, Ariafard A, Khairallah GN, Kwan KH, Donnelly PS, White JM, Canty AJ, Yates BF, O’Hair RAJ. Decarboxylative-Coupling of Allyl Acetate Catalyzed by Group 10 Organometallics, [(phen)M(CH3)]+. J Org Chem 2014; 79(24): 12056-12069.
| Crossref | Google Scholar |
62 Woolley MJ, Khairallah GN, da Silva G, Donnelly PS, Yates BF, O’Hair RAJ. Role of the Metal, Ligand, and Alkyl/Aryl Group in the Hydrolysis Reactions of Group 10 Organometallic Cations, [(L)M(R)]+. Organometallics 2013; 32: 6931-6944.
| Crossref | Google Scholar |
63 Donald WA, McKenzie CJ, O’Hair RAJ. C—H Bond Activation of Methanol and Ethanol by a High-Spin FeIVO Biomimetic Complex. Angew Chem Int Ed 2011; 50(36): 8379-8383.
| Crossref | Google Scholar |
64 Brydon SC, da Silva G, O’Hair RAJ, White JM. Experimental and Theoretical Investigations into the Mechanisms of Haliranium Ion π-Ligand Exchange Reactions with Cyclic Alkenes in the Gas Phase. Phys Chem Chem Phys 2021; 23(45): 25572-25589.
| Crossref | Google Scholar |
65 Frisch MJT, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich AV, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams-Young D, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery Jr JA, Peralta JE, Ogliaro F, Bearpark MJ, Heyd JJ, Brothers EN, Kudin KN, Staroverov VN, Keith TA, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, Fox DJ. Gaussian 16 Rev. C.01. Wallingford, CT; 2016.
66 Zhao Y, Truhlar DG. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem Acc 2008; 120(1): 215-241.
| Crossref | Google Scholar |
67 Andrae D, Häußermann U, Dolg M, Stoll H, Preuß H. Energy-adjustedab initio pseudopotentials for the second and third row transition elements. Theor Chim Acta 1990; 77(2): 123-141.
| Crossref | Google Scholar |
68 Dolg M, Wedig U, Stoll H, Preuss H. Energy‐adjusted ab initio pseudopotentials for the first row transition elements. J Chem Phys 1987; 86(2): 866-872.
| Crossref | Google Scholar |
69 Hariharan PC, Pople JA. The influence of polarization functions on molecular orbital hydrogenation energies. Theor Chim Acta 1973; 28(3): 213-222.
| Crossref | Google Scholar |
70 Ehlers AW, Böhme M, Dapprich S, Gobbi A, Höllwarth A, Jonas V, Köhler KF, Stegmann R, Veldkamp A, Frenking G. A set of f-polarization functions for pseudo-potential basis sets of the transition metals Sc–Cu, Y–Ag and La–Au. Chem Phys Lett 1993; 208(1): 111-114 A set of f-polarization functions for pseudo-potential basis sets of the transition metals ScCu, YAg and LaAu.
| Crossref | Google Scholar |
71 Höllwarth A, Böhme M, Dapprich S, Ehlers AW, Gobbi A, Jonas V, Köhler KF, Stegmann R, Veldkamp A, Frenking G. A set of d-polarization functions for pseudo-potential basis sets of the main group elements Al–Bi and f-type polarization functions for Zn, Cd, Hg. Chem Phys Lett 1993; 208(3): 237-240.
| Crossref | Google Scholar |
72 Barone V, Cossi M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J Phys Chem A 1998; 102(11): 1995-2001.
| Crossref | Google Scholar |
73 Fukui K. Formulation of the reaction coordinate. J Phys Chem 1970; 74(23): 4161-4163.
| Crossref | Google Scholar |
74 Fukui K. The path of chemical reactions – the IRC approach. Acc Chem Res 1981; 14(12): 363-368.
| Crossref | Google Scholar |
75 Becke AD. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev A 1988; 38(6): 3098-3100.
| Crossref | Google Scholar |
76 Becke AD. Density functional thermochemistry. III. The role of exact exchange. J Chem Phys 1993; 98(7): 5648-5652.
| Crossref | Google Scholar |
77 Lee C, Yang W, Parr RG. Development of the Colle–Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 1988; 37(2): 785-789.
| Crossref | Google Scholar |
78 Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J Phys Chem 1994; 98(45): 11623-11627.
| Crossref | Google Scholar |
79 Weigend F, Furche F, Ahlrichs R. Gaussian basis sets of quadruple zeta valence quality for atoms H–Kr. J Chem Phys 2003; 119(24): 12753-12762.
| Crossref | Google Scholar |
80 Okuno Y. Theoretical Investigation of the Mechanism of the Baeyer–Villiger Reaction in Non-polar Solvents. Chem Eur J 1997; 3(2): 212-218.
| Crossref | Google Scholar |
81 Keith JA, Carter EA. Quantum Chemical Benchmarking, Validation, and Prediction of Acidity Constants for Substituted Pyridinium Ions and Pyridinyl Radicals. J Chem Theory Comput 2012; 8(9): 3187-3206.
| Crossref | Google Scholar |