

Photochemical generation of the 2-azabicyclo[4.2.0]octa-4,7-diene skeleton
Tyler Fahrenhorst-Jones A , G. Paul Savage B and Craig M. Williams A *
A , G. Paul Savage B and Craig M. Williams A *
A School of Chemistry and Molecular Biosciences, University of Queensland, Brisbane, Qld 4072, Australia.
B CSIRO Manufacturing, Ian Wark Laboratory, Melbourne, Vic. 3168, Australia.
Australian Journal of Chemistry 75(11) 884-887 https://doi.org/10.1071/CH22139
Submitted: 16 June 2022 Accepted: 19 July 2022 Published: 31 August 2022
© 2022 The Author(s) (or their employer(s)). Published by CSIRO Publishing. This is an open access article distributed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License (CC BY-NC-ND)
Abstract
A 2-azabicyclo[4.2.0]octa-4,7-diene derivative was unexpectedly isolated from the photochemical irradiation of 2-vinyl-1,2-dihydropyridine. A cascading 6π–8π–4π electrocyclic rearrangement has been proposed as a possible mechanistic pathway.
Keywords: azabicyclo[4.2.0], azaoctadiene, cascade, dihydropyridine, electrocyclisation.
Introduction
The 2-azabicyclo[4.2.0]octadiene system has seldom been described in the literature. In 1971, Acheson and Wright reported the first example of the 2-azabicyclo[4.2.0]octa-3,7-diene skeleton (i.e. 1), utilising a cycloaddition involving a 1,4-dihydropyridine (2) and dimethyl acetylenedicarboxylate (Scheme 1).[1] A year later, Lehman reported a benzo derivative of the 2-azabicyclo[4.2.0]octa-3,7-diene system (i.e. 3) using the same protocol.[2] However, beyond later work by Acheson et al. in the intervening years,[3,4] further contributions to this area have not been forthcoming.
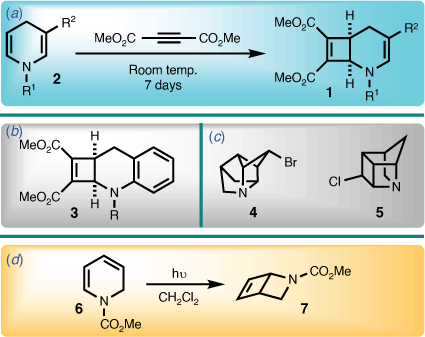
|
In the course of investigating synthetic routes towards highly strained cage bicyclic systems that contain nitrogen (e.g. 4 and 5),[5] a functionalised 1,2-dihydropyridine (6) was viewed as a logical starting point, because they readily undergo photochemically mediated cyclisation to afford the 2-azabicyclo[2.2.0]hex-5-ene ring system (7) (Scheme 1).[6,7] However, introducing extended unsaturation lead to unexpected behaviour, results of which are now disclosed herein.
Results and discussion
The investigation was initiated by synthesising the known 2-vinyl-1,2-dihydropyridine carbamate 10 (Scheme 2). Following Yamaguchi’s procedure,[8] vinyl magnesium bromide (8) reacted regioselectively with pyridine (9) upon ring activation by an alkyl chloroformate. The 2-vinyl-1,2-dihydropyridine methyl carbamate (10) was isolated in 79% yield as a foul-smelling colourless oil, which degraded at room temperature within a day (Scheme 2). It was later discovered that both the methyl- (10) and ethyl- (11) carbamates, derived from the same procedure, were not stable as neat oils, but could instead be stored as dilute solutions at −20°C for a number of days.

|
Interestingly, Tsuchiya et al. also encountered stability problems with several N-alkoxycarbonyl-1,2-dihydropyridine derivatives, and found it was best to handle this system without isolation prior to further transformations.[9] Adopting this method, crude 11 was irradiated at 300 nm in dichloromethane, which afforded the desired 2-azabicyclo[2.2.0]hex-5-ene (12), but only in 2% isolated yield. The major product, isolated in 32% yield, was elucidated as the 2-azabicyclo[4.2.0]octa-4,7-diene 13 (Scheme 2).
The unexpected isolation of 2-azabicyclo[4.2.0]octa-4,7-diene (13) indicated that introduction of the vinyl group on the dihydropyridine skeleton was promoting a more favourable competing photochemical pathway to that of the desired 2-azabicyclo[2.2.0]hex-5-ene (12). Given that 1,2-dihydropyridine carbamates are reported to undergo 6π-electrocyclic ring-opening to 1-azahexatrienes,[10,11] it was reasonable to envisage that the 2-vinyl-1,2-dihydropyridine (11) initially formed the corresponding 1-azaoctatetraene (14) via a 6π-electrocyclic ring-opening. E- to Z-isomerisation of the double bond at position 4 (i.e. to give 15) would then enable an 8π-electrocyclisation to give azacyclooctatriene 16, which is perfectly placed to undergo a final 4π-electrocyclisation leading to the product (i.e. 13) (Scheme 3). Such a mechanistic proposition has analogies with the corresponding hydrocarbon octatetraene, which was observed to undergo 8π-electrocyclic ring closure to cyclooctatriene,[12,13] and subsequently proceeded to give bicyclo[4.2.0]octadiene via a 4π electrocyclic ring closure.[12,14]
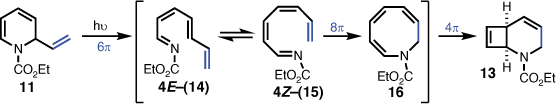
|
To eliminate the notion of an alternate pathway potentially involving the 3-vinyl-2-azabicyclo[2.2.0]hex-5-ene (12), as an intermediate en route to 13, 12 was subjected to repeat photochemical conditions. However, several attempts in a variety of solvents (acetonitrile, toluene, benzene, dichloromethane, and acetone) with and without a photosensitiser (acetone) and at 254 nm, mostly resulted in decomposition. The exception being the isolation of an unstable chlorocyclobutene (17) in 8% yield, which was identified subsequent to irradiation of 12 at 254 nm in dichloromethane (Scheme 4). This observation is in line with the reported exposure of 2-azabicyclo[2.2.0]hex-5-ene carbamates to either hydrochloric acid or chloronium ion, which mediates ring-opening to chlorocyclobutenes via heterolysis of the C–N bond.[7,15] That is, during photolysis dichloromethane degraded producing trace amounts of hydrochloric acid leading to protonation of the carbamate (i.e. 18), and subsequent carbocation formation (i.e. 19), which is trapped by chloride. Considering that the 4π-electrocyclisation involving 1,2-dihydropyridine has been described as endo specific,[16,17] and NOE correlations between the vinyl protons and cyclobutene sp2 protons confirmed the endo-configuration of 12, the resulting relative stereochemistry of 17 is therefore proposed as shown in Scheme 4.
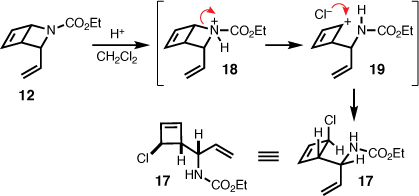
|
Conclusion
In summary, photolysis of 2-vinyl-1,2-dihydropyridine has afforded the first example of a 2-azabicyclo[4.2.0]octa-4,7-diene, the skeleton of which having only been described previously as a potential reaction intermediate over 40 years ago.[18] At this stage, the proposed mechanism is consistent with the observed products, and literature precedents, but nevertheless remains speculative until further experimentation can rigorously exclude alternative pathways.
Experimental
General experimental
Glassware was oven dried (160°C) before use with anhydrous solvents and reagents. Acetonitrile, dichloromethane, benzene, and toluene were freshly distilled to dryness over calcium hydride under an argon atmosphere. Diethyl ether was freshly distilled from elemental sodium/benzophenone under an argon atmosphere. Unless stated otherwise commercially available chemicals were used without further purification. Rayonet (Srinivasen–Griffin) reactors (254 nm or 300 nm) were used for photochemical reactions. NMR spectra were recorded using a Bruker AS500 or AV500 (500, 125 MHz) instruments. Chemical shifts (δ) are reported in parts per million (ppm) and referenced internally according to solvent: DMSO-d6 (1H δ: 2.50 ppm, 13C δ: 39.5 ppm). The following abbreviations are used to report multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad. High resolution electrospray mass spectrometry (HRMS-ESI) measurements were obtained on Bruker MicroOTOF-Q spectrometer or Orbitrap Elite mass spectrometer instruments. Flash column chromatography was undertaken using basic ~150 mesh Brockmann I activated aluminium oxide. TLC was performed with neutral aluminium oxide plates (alumina oxide 60 F254). Fractions were initially visualised using UV irradiation and subsequently by heating TLC plates exposed to phosphomolybdic acid stain.
Experimental procedures
Methyl 2-vinylpyridine-1-(2H)-carboxylate (10)
Following the procedure of Yamaguchi et al.[8]: anhydrous pyridine (9) (135 µL, 1.68 mmol) was cooled to −15°C and a tetrahydrofuran solution of vinyl magnesium bromide (8) (1.0 M, 3.3 mL) was added to the stirring solution under an argon atmosphere. Methyl chloroformate (170 µL, 1.75 mmol) was then added slowly such that the reaction temperature did not exceed 0°C. The mixture was stirred for 5 h at −5°C, and at room temperature for 20 min. The solution was subsequently added to an ice cooled aqueous solution of ammonium chloride (1.0 M, 20 mL) in a separatory funnel and shaken vigorously. After extracting with diethyl ether (4 × 10 mL), the combined organic phases were washed with water (4 × 10 mL), and brine (1 × 10 mL), before being dried over sodium sulfate. The solvent was removed in vacuo and the crude oil was purified using basic alumina column chromatography (dichloromethane) to give 10 (219 mg, 79%) as a foul-smelling colourless oil. [Note: Product may be stored at −20°C for 48 h in diethyl ether before degradation starts to occur.] 1H-NMR (300 MHz, DMSO-d6) δ: 6.70–6.68 (m, 1H), 6.00–5.94 (m, 1H), 5.78–5.67 (m, 1H), 5.63–5.58 (m, 1H), 5.29 (br, 1H), 5.15 (br, 1H), 5.09–4.99 (m, 2H), 3.71 (s, 3H). Data reported are consistent with that of Yamaguchi et al.[8]
Ethyl endo-3-vinyl-2-azabicyclo[2.2.0]hex-5-ene-2-carboxylate (12) and ethyl 2-azabicyclo[4.2.0]octa-4,7-diene-2-carboxylate (13)
Adapted from the procedure of Kurita et al.[9]: anhydrous pyridine (9) (5.37 mL, 66.6 mmol) was chilled to −15°C and a tetrahydrofuran solution of vinyl magnesium bromide (8) (1.0 M, 72 mL) was added to the stirring solution under an argon atmosphere. A diethyl ether solution (7 mL) of ethyl chloroformate (6.36 mL, 66.8 mmol) was then added dropwise such that the reaction temperature did not exceed 0°C. The mixture was then stirred for 5 h at −5°C and at room temperature for 20 min. The solution was subsequently added to an ice cooled aqueous solution of ammonium chloride (1.0 M, 200 mL) in a separatory funnel and shaken vigorously. Following extraction with diethyl ether (4 × 60 mL), the combined organic layers were washed with water (4 × 60 mL), and brine (1 × 60 mL), before being dried over sodium sulfate and concentrated in vacuo to approximately a 20 mL solution. The crude solution was added to anhydrous dichloromethane (80 mL) in a pyrex vessel flushed with argon, and irradiated in a Rayonet (Srinivasen–Griffin) photochemical reactor equipped with 300 nm lamps for 46 h. The solution was then concentrated in vacuo and purified via basic alumina column chromatography (50% diethyl ether/hexanes v/v) to give 12 (253 mg, 2%) and 13 (3.85 g, 32%) as yellow oils. 12 – 1H-NMR (500 MHz, DMSO-d6) δ: 6.54 (t, J = 2.4 Hz, 1H), 6.37 (t, J = 2.8 Hz, 1H), 5.81 (ddd, J = 17.0, 10.5, 6.6 Hz, 1H), 5.16–5.12 (m, 2H), 4.73 (br, 1H), 4.60 (br, 1H), 4.00–3.96 (m, 2H), 3.64–3.62 (m, 1H), 1.16–1.10 (m, 3H); 13C-NMR (125 MHz, DMSO-d6) δ (only one rotamer reported): 155.9, 141.7, 141.5, 135.4, 117.0, 63.3, 60.8, 60.1, 43.1, 14.7; HRMS-ESI calcd for C10H13NO2H+ ([M + H])+: 180.1020; found: 180.1021. 13 – 1H-NMR (500 MHz, DMSO-d6) δ: 6.20–6.19 (m, 1H), 6.13 (br, 1H), 5.97–5.94 (m, 1H), 5.78 (br, 1H), 4.97–4.93 (m, 1H), 4.18–4.13 (m, 1H), 4.05 (q, J = 6.6 Hz, 2H), 3.57–3.55 (m, 1H), 3.51–3.39 (m, 1H), 1.19 (t, J = 6.8 Hz, 3H); 13C-NMR (125 MHz, DMSO-d6) δ: 154.5, 139.4, 136.9, 126.8, 123.8, 60.9, 53.2, 43.0, 39.1, 14.6; HRMS-ESI calcd for C10H13NO2H+ ([M + H])+: 180.1020; found: 180.1019.
Ethyl (1-(4-chlorocyclobut-2-en-1-yl)allyl)carbamate (17)
Ethyl endo-3-vinyl-2-azabicyclo[2.2.0]hex-5-ene-2-carboxylate (12) (20 mg, 110 µmol) was dissolved in dichloromethane (20 mL) in a quartz vessel flushed with argon and irradiated in a Rayonet (Srinivasen–Griffin) photochemical reactor equipped with 254 nm lamps for 16 h. Following concentration under a stream of nitrogen the residue was purified by basic alumina column chromatography (50% diethyl ether/hexanes v/v) to give 17 (1.9 mg, 8%) as white crystals. 1H-NMR (500 MHz, DMSO-d6) δ: 7.36–7.16 (m, 1H), 6.27–6.25 (m, 2H), 5.91–5.78 (m, 1H), 5.17–5.05 (m, 3H), 4.19–3.94 (m, 3H), 3.20–3.17 (m, 1H), 1.17–1.13 (m, 3H); 13C-NMR (125 MHz, DMSO-d6) δ: 155.7, 140.7, 138.7, 137.3, 114.8, 59.7, 58.3, 53.4, 55.2, 14.6; HRMS-ESI calcd for C10H14ClNO2H+ ([M + H])+: 216.0786, 218.0757; found: 216.0778, 218.0748.
Data availability
The data that support this study are available in the article and accompanying online supplementary material.
Declaration of funding
The Australian Research Council (DP180103004, FT110100851), the University of Queensland (UQ), and the Commonwealth Scientific and Industrial Research Organisation (CSIRO), are gratefully acknowledged for financial support. T. F.-J gratefully acknowledges the Australian Government and UQ for Research Training Program (RTP) and Tuition Fee Offset Scholarships.
Conflicts of interest
The authors declare no conflicts of interest.
Supplementary material
Copies of 1H, 13C and 2D NMR spectral data (PDF). Supplementary material is available online.
References
[1] RM Acheson, ND Wright, J Chem Soc D 1971, 1421.| Crossref | GoogleScholarGoogle Scholar |
[2] PG Lehman, Tetrahedron Lett 1972, 4863.
[3] RM Acheson, ND Wright, PA Tasker, J Chem Comm Perkin Trans 1 1972, 2918.
| Crossref | GoogleScholarGoogle Scholar |
[4] RM Acheson, G Paglietti, J Chem Comm Perkin Trans 1 1979, 591.
| Crossref | GoogleScholarGoogle Scholar |
[5] T Fahrenhorst-Jones, PV Bernhardt, GP Savage, CM Williams, Org Lett 2022, 24, 903.
| Crossref | GoogleScholarGoogle Scholar |
[6] FW Fowler, J Org Chem 1972, 37, 1321.
| Crossref | GoogleScholarGoogle Scholar |
[7] P Beeken, JN Bonfiglio, I Hasan, JJ Piwinski, B Weinstein, KA Zollo, FW Fowler, J Am Chem Soc 1979, 101, 6677.
| Crossref | GoogleScholarGoogle Scholar |
[8] R Yamaguchi, Y Nakazono, T Matsuki, E Hata, M Kawanisi, Bull Chem Soc Jpn 1987, 60, 215.
| Crossref | GoogleScholarGoogle Scholar |
[9] J Kurita, K Iwata, H Sakai, T Tsuchiya, Chem Pharm Bull 1985, 33, 4572.
| Crossref | GoogleScholarGoogle Scholar |
[10] T Kumagai, S Saito, T Ehara, Tetrahedron Lett 1991, 32, 6895.
| Crossref | GoogleScholarGoogle Scholar |
[11] DF Maynard, WH Okamura, J Org Chem 1995, 60, 1763.
| Crossref | GoogleScholarGoogle Scholar |
[12] OL Chapman, GW Borden, RW King, B Winkler, J Am Chem Soc 1964, 86, 2660.
| Crossref | GoogleScholarGoogle Scholar |
[13] BE Thomas, JD Evanseck, KN Houk, J Am Chem Soc 1993, 115, 4165.
| Crossref | GoogleScholarGoogle Scholar |
[14] RL Grange, MJ Gallen, H Schill, JP Johns, L Dong, PG Parsons, PW Reddell, VA Gordon, PV Bernhardt, CM Williams, Chem Eur J 2010, 16, 8894.
| Crossref | GoogleScholarGoogle Scholar |
[15] GR Krow, GC Cannon, 2-Azabicyclo[2.2.0]hex-5-enes and 2-Azabicyclo[2.2.0]hexanes. A Review. Heterocycles 2004, 64, 577.
| 2-Azabicyclo[2.2.0]hex-5-enes and 2-Azabicyclo[2.2.0]hexanes. A Review.Crossref | GoogleScholarGoogle Scholar |
[16] GR Krow, YB Lee, WS Lester, N Liu, J Yuan, J Duo, SB Herzon, Y Nguyen, D Zacharias, J Org Chem 2001, 66, 1805.
| Crossref | GoogleScholarGoogle Scholar |
[17] GR Krow, G Lin, F Yu, J Org Chem 2005, 70, 590.
| Crossref | GoogleScholarGoogle Scholar |
[18] (a) RM Acheson, G Paglietti, PA Tasker, J Chem Soc Perkin Trans 1 1974, 2496.
| Crossref | GoogleScholarGoogle Scholar |
(b) B Weinstein, L-CC Lin, FW Fowler, J Org Chem 1980, 45, 1657.
| Crossref | GoogleScholarGoogle Scholar |