

Extremely bulky β-diketiminate complexes of calcium(ii) and ytterbium(ii)†
Brant Maitland A , Andreas Stasch A and Cameron Jones A *
A *
A School of Chemistry, PO Box 23, Monash University, Melbourne, Vic., 3800, Australia.
Australian Journal of Chemistry 75(9) 543-548 https://doi.org/10.1071/CH21283
Submitted: 1 November 2021 Accepted: 13 December 2021 Published: 18 February 2022
© 2022 The Author(s) (or their employer(s)). Published by CSIRO Publishing. This is an open access article distributed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License (CC BY-NC-ND)
Abstract
The potassium salt of an extremely bulky β-diketiminate, [K(Ar*Nacnac)] (Ar*Nacnac, [(Ar*NCMe)2CH]−; Ar*, C6H2Me{C(H)Ph2}2-4,2,6) was reacted with either CaI2 or YbI2(THF)2, which afforded [(Ar*Nacnac)MI] (M = Ca or Yb). These are the first examples of structurally characterised, donor solvent-free, N-arene substituted β-diketiminato calcium and ytterbium complexes that incorporate a terminal iodide ligand. Reduction of [(Ar*Nacnac)CaI] with sodium metal gave a complex product mixture, from which a few crystals of the β-diketiminate C–H activated product, [{Ca(μ-Ar*Nacnac-H)}2], were obtained and crystallographically characterised. In an attempt to form a terminal ytterbium hydride compound, treatment of [(Ar*Nacnac)YbI] with K[HBEt3] gave a good yield of the contact ion pair compound [(Ar*Nacnac)Yb(HBEt3)].
Keywords: β‐diketiminate, bulky ligand, calcium, C–H activation, metal hydrides, metal–metal bonding, reduction, ytterbium.
Introduction
Over the past 50 years or so, the β-diketiminate (Nacnac) class of chelating, anionic N-donor ligand has proved invaluable to inorganic and organometallic chemists for the kinetic stabilisation of highly reactive complexes of metals from across the periodic table.[1] Of most relevance to the present study is the use of very bulky β-diketiminates to stabilise low-oxidation-state s-block metal complexes, e.g. [(Nacnac)MM(Nacnac)], from undergoing disproportionation to elemental metal and [M(Nacnac)2], and to prevent β-diketiminato s-block metal hydride compounds, [{(Nacnac)MH}n], from redistributing via the Schlenk equilibrium to saline metal hydrides, MH2, and [M(Nacnac)2].
Most success in low-oxidation-state s-block chemistry has come from dimagnesium(i) systems, the first bulky β-diketiminate coordinated example of which, [{(DipNacnac)Mg}2] (DipNacnac, [(DipNCMe)2CH]−; Dip, 2,6-diisopropylphenyl), was reported in 2007.[2] Since that time, more than 20 other examples have emerged.[3] These display various substitution patterns, including N-aryl substituents that vary in steric bulk from xylyl[4] to 2,4,6-tricyclohexylphenyl (TCHP)[5] or 2,6-di(3-pentyl)phenyl (DiPep).[6] Indeed, the latter substituent has recently been utilised in the stabilisation of the first magnesium(0) complexes, [Na2{(ButDiPepNacnac)Mg}2] and [(ButDiPepNacnac)MgMgMg(ButDiPepNacnac)] (ButDiPepNacnac, [(DiPepNCBut)2CH]−.[7] More than just chemical curiosities, low-oxidation-state magnesium complexes have been widely used around the globe as soluble, selective reducing agents in numerous inorganic and synthetic transformations.[3] Although no β-diketiminate complexes of other Group 2 metals in the 0 or +1 oxidation states have been isolated, it has been suggested that a calcium(i) complex is a transient intermediate in the reduction of N2, which yields the only known examples of homometallic Group 2 dinitrogen complexes, e.g. [{(DiPepNacnac)(THF)Ca}2(μ-N2)] (DiPepNacnac, [(DiPepNCMe)2CH]−).1[8]
β-Diketiminato Group 2 metal hydride complexes are more widespread than their low-oxidation-state counterparts. A good number of β-diketiminato magnesium[9] and calcium hydride complexes[10] have been reported, and these are finding increasing use in a variety of stoichiometric and catalytic synthetic transformations.[11] To date, there are no crystallographically characterised examples of β-diketiminato beryllium or barium hydrides, though one strontium hydride analogue has been reported.[12] In order to increase the reactivity of β-diketiminato Group 2 metal hydrides, it would be advantageous to have access to three-coordinate, terminal hydride examples, which should be more Lewis acidic and hydridic than their hydride bridged counterparts. There is, however, only one such compound known, [(Ar*Nacnac)MgH] (Ar*Nacnac, [(Ar*NCMe)2CH]−; Ar*, C6H2Me{C(H)Ph2}2-4,2,6), and this is prevented from dimerising through hydride bridges by the imposing steric bulk of its β-diketiminate ligand.[13]
Given the stability of [(Ar*Nacnac)MgH], it seemed that the Ar*Nacnac ligand might be able to stabilise other low-coordinate Group 2 metal hydride systems, or compounds that contain Group 2 metal–metal bonds. In this respect, β-diketiminato dicalcium(i) and terminal calcium(ii) hydride complexes were targeted, as these compound types are unknown. Moreover, given the almost identical ionic radii of calcium(ii) and ytterbium(ii) (1.00 Å and 1.02 Å for six-coordinate complexes, respectively[14]), and the well-documented similar coordination chemistry of the metals,[15] attempts were made to prepare closely related ytterbium(i) and ytterbium(ii) hydride complexes. Although none of the target complexes were accessible, rare examples of terminal iodo complexes of calcium(II) and ytterbium(ii), bearing β-diketiminate ligands, were prepared. The characterisation of these precursor complexes, and the outcomes of their reductions, are described herein.
Results and discussion
As potential precursors to the target low-oxidation-state and hydrido complexes of calcium and ytterbium, β-diketiminato metal(ii) iodides were synthesised. This involved the 1:1 reactions of [K(Ar*Nacnac)] with either CaI2 or YbI2(THF)2 in THF. These reactions afforded compounds 1 and 2 in isolated yields of 70% and 64% respectively, as pale yellow or red crystalline solids, after workup (Scheme 1). Both compounds are thermally stable in solutions of normal organic solvents and in the solid state for weeks at ambient temperature under a dinitrogen atmosphere. They have moderate solubility in arene solvents, but are poorly soluble in aliphatics.
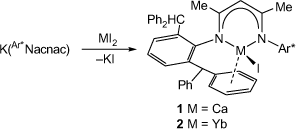
|
NMR spectra of 1 and 2 confirmed that there is no THF coordinated to the metal centres, despite the reactions being carried out in that strong donor solvent. This is unusual for such Lewis acidic metals, and suggests that intramolecular metal–arene coordination, as seen in the solid-state structures, is preferable to THF coordination. However, in C6D6 solutions, the NMR spectra of the two compounds suggest more symmetrical solution state structures than in the solid state. That is, one signal each is seen for the ligand backbone methyl groups, and for the benzhydryl methine protons. One possibility for this observation is that there is fluxional exchange of coordination between the phenyl substituents of the Ar*Nacnac ligand, which is rapid on the NMR timescale. Attempts to probe the nature of this exchange by carrying out variable-temperature NMR spectroscopic studies on d8-toluene solutions of the compounds were thwarted by their poor solubilities in this solvent at temperatures below 0°C. Moreover, no resonance was observed in the 171Yb NMR spectrum of 2.
The molecular structures of 1 and 2 are depicted in Fig. 1. The compounds are essentially isostructural, with the metal centres being chelated by the N centres of the Ar*Nacnac ligand, in addition to having a skewed η6-interaction with one of its phenyl groups. As far as we are aware, there are no other examples of donor solvent-free, N-arene substituted β-diketiminato calcium and ytterbium complexes that incorporate a terminal iodide ligand. However, one related THF coordinated triketiminato complex has been recently structurally characterised.[16] Given the very similar ionic radii and coordination chemistry of Ca2+ and Yb2+, it is not surprising that the bond lengths and angles involving both metal centres are very similar, and the majority of bond lengths lie within the known ranges for β-diketiminato calcium(ii) and ytterbium(ii) complexes (as determined from a survey of the Cambridge Crystallographic Database, version 2.0.4, October, 2021).[17] The exceptions here are the metal–iodine distances, which are shorter than in any other examples of this compound class, presumably owing to the fact that the iodide ligands are terminal.
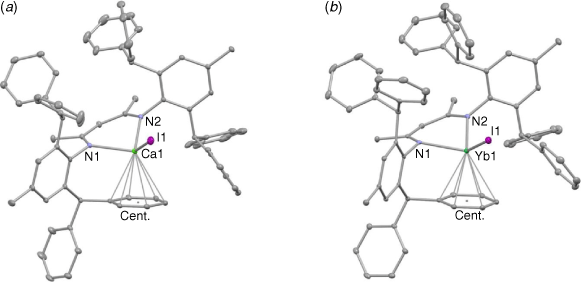
|
With 1 and 2 in hand, their reductions with a variety of reducing agents, e.g. Na metal or KC8, in both coordinating and non-coordinating solvents, were attempted. In each case, complex product mixtures were obtained, as determined by NMR spectroscopic analyses of the reaction solutions. In only one instance were a few crystals of the phenyl C–H activated calcium(ii) product, 3, obtained (Scheme 2). While it cannot be certain how 3 was formed, it seems reasonable that an initial reductive iodide elimination process yields a short-lived radical, (Ar*Nacnac)Ca. Instead of dimerising to give the target Ca–Ca bonded compound, the Ca centre of one radical attacks a phenyl ring of another, leading to loss of dihydrogen, and the formation of 3. It is noteworthy that all previous attempts by us,[17] and others,[8] to prepare calcium(i) compounds bearing Ca–Ca bonds have met with failure.
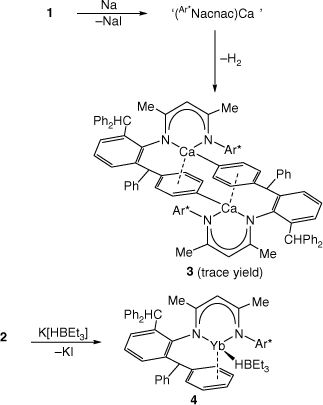
|
Owingto the very low yield of 3, no spectroscopic data could be acquired for the compound. However, its X-ray crystal structure was obtained, and the molecular structure is depicted in Fig. 2. This shows the compound to be centrosymmetric and bimetallic, with each Ca centre N,N-chelated by one deprotonated Ar*Nacnac ligand, while having an η1-interaction with the deprotonated phenyl carbon centre of the opposing ligand. In addition, the calcium atoms are η6-coordinated by the deprotonated phenyl group associated with the Ar*Nacnac-H ligand chelating the metal centre. The Ca–N and Ca–centroid distances in 3 are normal for such interactions, and comparable with the analogous distances found for precursor complex 1. Moreover, the Ca–C(17)′ bond length (2.549(2) Å) is similar to those in related calcium phenyl compounds, e.g. 2.583(3) Å in trans-[CaPh(Br)(THF)4].[19]
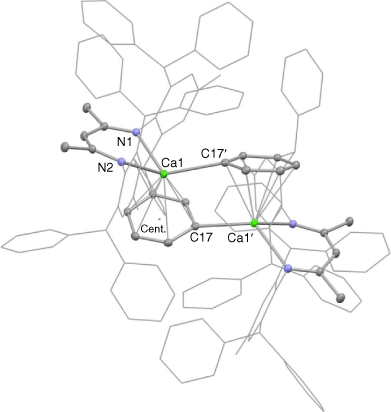
|
In light of the absence of β-diketiminato calcium or ytterbium terminal hydride complexes in the literature (cf. [(Ar*Nacnac)MgH][13]),[20] it seemed reasonable that 1 and 2 could prove viable precursors to such target complexes. To this end, toluene solutions of either 1 or 2 were treated with the potential hydride transfer reagent K[HBEt3]. Although no product could be isolated from the reaction involving 1, that with 2 gave a good isolated yield (78%) of the purple crystalline ytterbium-borate contact ion pair 4, on workup (Scheme 2). Therefore, the reaction did not proceed via hydride transfer, as initially predicted, but instead ceased after elimination of KI from the mixture of reagents. Although this is similar to the situation in related reactions between less bulky β-diketiminato calcium iodide compounds and K[HBR3],[21] it was thought that the extreme steric bulk of the Ar*Nacnac ligand might force elimination of BEt3, and formation of (Ar*Nacnac)YbH. Clearly, this did not prove to be the case.
Compound 4 is thermally stable in solutions of common organic solvents, and in the solid state, for weeks at ambient temperature under a dinitrogen atmosphere. As was the situation with the precursor complex 2, the NMR spectra for the ytterbium borate species suggest a more symmetrical structure in solution than was observed in the solid state. Similarly to 2, this likely occurs owing to a dynamic exchange between ytterbium coordinated and uncoordinated phenyl groups of the Ar*Nacnac ligand. Variable-temperature NMR spectroscopic studies of 4 were hampered by the low solubility of the compound in non-coordinating deuterated solvents at temperatures below 0°C.
The molecular structure of 4 is shown in Fig. 3. The compound is a contact ion pair, involving a [(Ar*Nacnac)Yb]+ cation and a [HBEt3]− anion that associate through a bridging hydride, H(1). The structure of 4 is closely related to that of the β-diketiminato calcium borate compound [(DipNacnac)(THF)Ca(HBBusec3)],[21] though in that system the metal centre is additionally coordinated by a THF molecule. In contrast, the ytterbium atom in 4 is N,N-chelated by the Ar*Nacnac ligand, while also having what is best described as an η3-interaction with a phenyl substituent of that ligand. The apparently weaker ytterbium–arene interaction in 4, relative to that in 2, probably originates from the greater steric bulk of the HBEt3 unit, relative to iodide, which destabilises the ytterbium–arene interaction in the former.
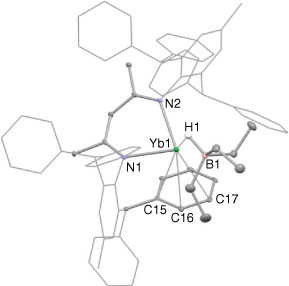
|
Conclusion
In conclusion, reactions of the potassium salt of an extremely bulky β-diketiminate ligand, [K(Ar*Nacnac)], with either CaI2 or YbI2(THF)2 have afforded the isostructural complexes [(Ar*Nacnac)MI] (M = Ca or Yb), which represent the first examples of structurally characterised, donor solvent-free, N-arene substituted β-diketiminato calcium and ytterbium complexes that incorporate a terminal iodide ligand. Alkali metal reductions of both complexes led to complex product mixtures, from one of which a few crystals of a complex in which a C–H activated β-diketiminate ligand is coordinated to a calcium centre, were isolated. Attempts to synthesise terminal metal hydride complexes from [(Ar*Nacnac)MI] were not successful, though reaction of [(Ar*Nacnac)YbI] with K[HBEt3] did give a good yield of the contact ion pair compound [(Ar*Nacnac)Yb(HBEt3)]. We continue to explore the kinetic stabilisation of low-oxidation-state and hydrido complexes of Group 2 and f-block metals.
Experimental
General considerations
All manipulations were carried out using standard Schlenk and glove box techniques under an atmosphere of high-purity dinitrogen. THF, toluene and hexane were distilled over molten potassium. 1H and 13C{1H} NMR spectra were recorded on a Bruker Avance III 400 spectrometer and are referenced to the resonances of the solvent used. The chemical shifts δ are reported in parts per million (ppm). Mass spectra were collected using an Agilent Technologies 5975D inert MSD with a solid-state probe. FTIR spectra were collected for solid samples, as Nujol mulls, on an Agilent Cary 630 attenuated total reflectance (ATR) spectrometer. Microanalyses were carried out at the Science Centre, London Metropolitan University. Melting points were determined in sealed glass capillaries under dinitrogen and are uncorrected. [K(Ar*Nacnac)] was prepared by the literature procedure.[13] All other reagents were used as received from commercial sources.
Preparation of [(Ar*Nacnac)CaI] (1)
A suspension of [K(Ar*Nacnac)] (3.20 g, 3.09 mmol) and CaI2 (1.00 g, 3.40 mmol) in THF (80 mL) was stirred overnight at room temperature. The resultant reaction mixture was dried under vaccuum, and the residue extracted with toluene (20 mL). Hexane (5 mL) was slowly added to the orange extract until an off-white solid began to precipitate. The mixture was then stored overnight at 4°C to give pale yellow crystals of 1 (2.38 g, 70%). Mp 104–107°C. 1H NMR (400 MHz, 303 K, C6D6): δ 1.03 (s, 6H, NCCH3), 1.87 (s, 6H, ArCH3), 4.54 (s, 1H, CCHC), 5.74 (s, 4H, Ph2CH), 6.77 (t, 3J 7.2 Hz, 4H, Ar–H), 7.03–7.08 (m, 8H, Ar–H), 7.12 (t, 3J 7.2 Hz, 8H, Ar–H), 7.17–7.22 (m, 16H, Ar–H), 7.48 (d, 3J 8 Hz, 8H, Ar–H). 13C{1H} NMR (100 MHz, 303 K, C6D6): δ 19.8 (NCCH3), 21.6 (Ar–CH3), 51.4 (Ph2CH), 94.5 (CCHC), 125.5, 125.6, 127.3, 128.4, 129.0, 129.0, 129.1, 132.4, 137.7, 141.1, 143.5, 144.9 (Ar-C), 167.0 (NCCH3). IR (Nujol, cm−1): 1597 w, 1514 w, 1030 w, 924 w, 856 w, 834 w, 774 w, 744 m. MS (electron impact, EI) (m/z, %): 942.4 (Ar*NacnacH+, 100). Elemental analysis for C77H75N2CaI (1.hexane): calc.: 77.36; H, 6.32; N, 2.34%; found: C, 77.46; H, 6.45; N, 2.38%.
Preparation of [(Ar*Nacnac)YbI] (2)
A solution of [K(Ar*Nacnac)] (0.800 g, 0.819 mmol) in THF (10 mL) was added to a solution of YbI2(THF)2 (0.514 g, 0.9 mmol) in THF (20 mL) at 0°C. The mixture was warmed to room temperature and stirred for 12 h, before being dried under vaccuum. The residue was extracted with toluene (20 mL) and the extract cooled to −30 °C to afford dark red crystals of 2 (0.650 g, 64%). Crystals for the X-ray diffraction experiment were grown from benzene. Mp 206–210°C. 1H NMR (400 MHz, 303 K, C6D6): δ 1.03 (s, 6H, NCCH3), 1.86 (s, 6H, ArCH3), 4.56 (s, 1H, CCHC), 5.78 (s, 4H, Ph2CH), 6.73 (s, 4H, Ar–H), 6.90–7.30 (m, 32H, Ar–H), 7.50 (s, 8H, Ar–H). 13C{1H} NMR (100 MHz, 303 K, C6D6): δ 19.8 (NCCH3), 23.1 (Ar–CH3), 51.3 (Ph2CH), 95.0 (CCHC), 125.3, 125.4, 127.3, 128.0, 128.8, 129.0, 129.1, 132.3, 137.7, 141.0, 142.9, 144.7 (Ar-C), 166.7 (NCCH3). No signal was observed in the 171Yb NMR spectrum of the compound. IR (Nujol, cm−1): 1516 w, 1493 m, 1030 m, 924 w, 861 w, 764 w, 745 m, 734 m. MS/EI (m/z, %): 1242.7 (M+, 1), 942.4 (Ar*NacnacH+, 26). Elemental analysis for C78H69N2YbI (2.toluene): calc.: C, 70.21; H 5.21; N, 2.10%; found: C, 70.44; H, 5.21; N, 2.38.
Preparation of [(Ar*Nacnac)Yb(HBEt3)] (4)
A solution of K[HBEt3] in THF (1.00 M, 0.290 mmol, 0.29 mL) was added to a solution of 2 (0.300 g, 0.242 mmol) in toluene (15 mL) at −30°C. The reaction mixture was warmed to room temperature and then stirred for 1 h. The mixture was then filtered and volatiles removed under vaccum, leaving a purple reside. A hexane extract of this residue (25 mL) was cooled to −40°C to give 4 as purple crystals (0.230 g, 78%). Mp 199–202°C. 1H NMR (400 MHz, 303 K, C6D6): δ 0.30–0.42 (m, 6H, BCH2), 0.78 (s, 6H, NCCH3), 1.00 (m, 9H, BCH2CH3), 1.95 (s, 6H, ArCH3), 4.38 (s, 1H, CCHC), 5.87 (s, 4H, Ph2CH), 6.86–6.94 (m, 4H, Ar–H), 6.96–7.15 (m, 20H, Ar–H), 7.16–7.28 (m, 12H, Ar–H), 7.44 (d, 3J 6.8 Hz, 8H, Ar–H). The BH resonance was not observed. 13C{1H} NMR (100 MHz, 303 K, C6D6): δ 12.3 (BCH2), 15.4 (BCH2CH3), 19.8 (NCCH3), 22.8 (ArCH3), 51.7 (Ph2CH), 94.5 (CCHC), 125.3, 125.7, 127.3, 127.9, 128.9, 131.8, 137.0, 141.7, 143.4, 144.2 (Ar-C), 166.7 (NCCH3). IR (Nujol, cm−1): 1912 m, 1494 m, 1030 m, 1014 m, 922 m, 764 s, 742 s, 733 s, 697 s. MS/EI (m/z, %): 942.8 (Ar*NacnacH+, 100). Elemental analysis for C77H77BN2Yb: calc.: C, 76.16; H, 6.39; N, 2.31%; found: C, 76.30; H, 6.34; N, 2.39.
X-ray crystallography
Crystals suitable for X-ray structural determination were mounted in silicone oil. Crystallographic measurements were made with either an Xcalibur Gemini Ultra diffractometer with Mo Kα radiation (λ 0.71073 Å), or the MX1 beamline of the Australian Synchrotron (λ 0.71090 Å). The software package Blu-Ice[22] was used for synchrotron data acquisition, while the program XDS[23] was employed for synchrotron data reduction. All structures were solved by direct methods and refined on F2 by full matrix least squares (SHELX-16[24]) using all unique data. Hydrogen atoms are typically included in calculated positions (riding model). Crystal data, details of data collections and refinements for all structures can be found in their CIF files and are summarised in Supplementary Table S1. Crystallographic data (CIF files) for all structures have been deposited with the Cambridge Crystallographic Data Centre (CCDC no. 2118520–2118523).
Data availability
The data that support this study are available in the article and accompanying online supplementary material.
Conflicts of interest
The authors declare no conflicts of interest.
Declaration of funding
This research was supported by the Australian Research Council.
Supplementary material
Crystal data, details of data collections and refinement for all structures are available in the Supplementary material online.
Acknowledgements
Part of this research was undertaken on the MX1 beamline at the Australian Synchrotron, part of the Australian Nuclear Science and Technology Organisation (ANSTO).
References
[1] (a) L Bourget-Merle, MF Lappert, JR Severin, Chem Rev 2002, 102, 3031.| Crossref | GoogleScholarGoogle Scholar | 12222981PubMed |
(b) Y-C Tsai, Coord Chem Rev 2012, 256, 722.
| Crossref | GoogleScholarGoogle Scholar |
(c) RL Webster, Dalton Trans 2017, 46, 4483.
| Crossref | GoogleScholarGoogle Scholar |
(d) M Asay, C Jones, M Driess, Chem Rev 2011, 111, 354.
| Crossref | GoogleScholarGoogle Scholar |
(e) C Camp, J Arnold, Dalton Trans 2016, 45, 14462.
| Crossref | GoogleScholarGoogle Scholar |
[2] S Green, C Jones, A Stasch, Science 2007, 318, 1754.
| Crossref | GoogleScholarGoogle Scholar | 17991827PubMed |
[3] (a) C Jones, Nat Rev Chem 2017, 1, 0059.
| Crossref | GoogleScholarGoogle Scholar |
(b) A Stasch, C Jones, Dalton Trans 2011, 40, 5659.
| Crossref | GoogleScholarGoogle Scholar |
(c) C Jones, Commun Chem 2020, 3, 159.
| Crossref | GoogleScholarGoogle Scholar |
(d) C Jones, A Stasch, Top Organomet Chem 2013, 45, 73.
| Crossref | GoogleScholarGoogle Scholar |
[4] SJ Bonyhady, D Collis, N Holzmann, AJ Edwards, RO Piltz, G Frenking, A Stasch, C Jones, Nat Commun 2018, 9, 3079.
| Crossref | GoogleScholarGoogle Scholar | 30082681PubMed |
[5] K Yuvaraj, I Iskander, DDL Jones, L Maron, C Jones, Chem Sci 2020, 11, 3516.
| Crossref | GoogleScholarGoogle Scholar | 34109023PubMed |
[6] TX Gentner, B Rösch, G Ballmann, J Langer, H Elsen, S Harder, Angew Chem, Int Ed 2019, 58, 607.
| Crossref | GoogleScholarGoogle Scholar |
[7] (a) B Rösch, TX Gentner, J Eyselein, J Langer, H Elsen, S Harder, Nature 2021, 592, 717.
| Crossref | GoogleScholarGoogle Scholar | 33911274PubMed |
(b) C Jones, Nature 2021, 592, 687.
| Crossref | GoogleScholarGoogle Scholar |
[8] B Rösch, TX Gentner, J Langer, G Färber, J Eyselein, L Zhao, C Ding, G Frenking, S Harder, Science 2021, 371, 1125.
| Crossref | GoogleScholarGoogle Scholar | 33707259PubMed |
[9] (a) SP Green, C Jones, A Stasch, Angew Chem Int Ed 2008, 47, 9079.
| Crossref | GoogleScholarGoogle Scholar |
(b) R Lalrempuia, CE Kefalidis, SJ Bonyhady, B Schwarze, L Maron, A Stasch, C Jones, J Am Chem Soc 2015, 137, 8944.
| Crossref | GoogleScholarGoogle Scholar |
(c) CN deBruin-Dickason, CA Rosengarten, GB Deacon, C Jones, Chem Commun 2021, 57, 1599.
| Crossref | GoogleScholarGoogle Scholar |
[10] (a) ASS Wilson, MS Hill, MF Mahon, C Dinoi, L Maron, Science 2017, 358, 1168.
| Crossref | GoogleScholarGoogle Scholar | 29191904PubMed |
(b) S Harder, J Brettar, Angew Chem Int Ed 2006, 45, 3474.
| Crossref | GoogleScholarGoogle Scholar |
[11] (a) See, for example: In: S Harder, editor. Alkaline-Earth Metal Compounds: Oddities and Applications. Heidelberg: Springer; 2013
(b) MS Hill, DJ Liptrot, Chem Soc Rev 2016, 45, 972.
| Crossref | GoogleScholarGoogle Scholar |
(c) B Mukherjee, D Scuhknecht, J Okuda, Angew Chem Int Ed 2018, 57, 9590.
| Crossref | GoogleScholarGoogle Scholar |
[12] B Rosch, TX Gentner, H Elsen, CA Fischer, J Langer, M Wiesinger, S Harder, Angew Chem Int Ed 2019, 58, 5396.
| Crossref | GoogleScholarGoogle Scholar |
[13] M Arrowsmith, B Maitland, G Kociok-Kohn, A Stasch, C Jones, MS Hill, Inorg Chem 2014, 53, 10543.
| Crossref | GoogleScholarGoogle Scholar | 25203490PubMed |
[14] RD Shannon, Acta Crystallogr 1976, A32, 751.
| Crossref | GoogleScholarGoogle Scholar |
[15] C Ruspic, J Spielmann, S Harder, Inorg Chem 2007, 46, 5320.
| Crossref | GoogleScholarGoogle Scholar | 17530842PubMed |
[16] GG Skvortsov, GK Fukin, AV Cherkasov, TA Kovylina, AA Trifonov, Inorg Chem Acta 2020, 508, 119623.
| Crossref | GoogleScholarGoogle Scholar |
[17] SJ Bonyhady, C Jones, S Nembenna, A Stasch, AJ Edwards, GJ McIntyre, Chem Eur J 2010, 16, 938.
| Crossref | GoogleScholarGoogle Scholar | 19950340PubMed |
[18] C Jones, S Nembenna, A Stasch, J Chem Crystallogr 2011, 41, 1490.
| Crossref | GoogleScholarGoogle Scholar |
[19] R Fischer, M Gärtner, H Görls, M Westerhausen, Organometallics 2006, 25, 3496.
| Crossref | GoogleScholarGoogle Scholar |
[20] For examples of hydride bridged β-diketiminato ytterbium(II) compounds see ref. [15], and GM Richardson, I Douair, SA Cameron, J Bracegirdle, RA Keyzers, MS Hill, L Maron, MD Anker, Nat Commun 2021, 12, 3147.
| Crossref | GoogleScholarGoogle Scholar | 34035284PubMed |
[21] SP Sarish, A Jana, HW Roesky, T Schulz, M John, D Stalke, Inorg Chem 2010, 49, 3816.
| Crossref | GoogleScholarGoogle Scholar |
[22] TM McPhillips, SE McPhillips, HJ Chiu, AE Cohen, AM Deacon, PJ Ellis, E Garman, A Gonzalez, NK Sauter, RP Phizackerley, SM Soltis, P Kuhn, J Synchrotron Rad 2002, 9, 401.
| Crossref | GoogleScholarGoogle Scholar |
[23] W Kabsch, J Appl Cryst 1993, 26, 795.
| Crossref | GoogleScholarGoogle Scholar |
[24] Sheldrick GM. SHELX-16. University of Göttingen; 2016.
† This invited submission is associated with the special issue of The Australian Journal of Chemistry that celebrates the 85th birthday of Professor Glen B. Deacon.
1 N.B. one formally calcium(i) complex which does not contain a β‐diketiminate ligand has been reported. See: Krieck S, Görls H, Yu L, Reiher M, Westerhausen M. J. Am. Chem. Soc. 2009, 131, 2977. https://doi.org/10.1021/ja808524y