

A one-pot synthesis of oligo(arylene–ethynylene)-molecular wires and their use in the further verification of molecular circuit laws†
Masnun Naher A , Elena Gorenskaia A , Stephen A. Moggach A , Thomas Becker B , Richard J. Nichols C , Colin J. Lambert D and Paul J. Low A *
A *
A School of Molecular Sciences, University of Western Australia, 35 Stirling Highway, Crawley, WA 6009, Australia.
B School of Molecular and Life Sciences, Curtin University, GPO Box U1987, Perth, WA 6845, Australia.
C Department of Chemistry, University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK.
D Department of Physics, Lancaster University, Lancaster LA1 4YB, UK.
Australian Journal of Chemistry 75(9) 506-522 https://doi.org/10.1071/CH21235
Submitted: 15 September 2021 Accepted: 19 November 2021 Published: 23 February 2022
© 2022 The Author(s) (or their employer(s)). Published by CSIRO Publishing. This is an open access article distributed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License (CC BY-NC-ND)
Abstract
A convenient two-step, one-pot synthesis of oligo(arylene–ethynylene) (OAE) type molecular wires in yields of up to 70% via in situ desilylation of protected bis(alkynes) Me3SiC≡CArC≡CSiMe3 (Ar = 2,5-thienyl, 1,4-naphthylene, 9,10-anthrylene) and subsequent Sonogashira cross-coupling with S-(4-iodophenyl) ethanethiolate, 4-iodothioanisole, or 5-bromo-3,3-dimethyl-2,3-dihydrobenzo[b]thiophene is described. The in situ desilylation avoids the manipulation of the sensitive terminal dialkynes (HC≡CArC≡CH), whilst the general approach presented has some advantages over alternative synthetic strategies based on coupling of aryl dihalides (XArX) by avoiding the multi-step preparation and purification of the terminal alkynes S-(4-ethynylphenyl) ethanethiolate, 4-ethynylthioanisole and 5-ethynyl 3,3-dimethyl-2,3-dihydrobenzo[b]thiophene. The molecular conductance of the resulting thiolate or thioether functionalised OAE molecular wires has been determined using scanning tunneling microscope break junction (STM-BJ) methods. The trends in molecular conductance do not track simply with the degree of aromaticity of the molecular core despite the rather similar molecular lengths. Rather, the STM-BJ data are better correlated with the nature of the anchor group, highlighting the important role of electrode–molecule coupling on electron transport in a molecular junction. The experimental conductance data are in good agreement with recently described quantum circuit rules, further highlighting the potential for these relationships to be used as predictive tools in molecular electronics research.
Keywords: molecular electronics, molecule‐electrode coupling, molecular junction, molecular wire, oligo(phenylene–ethynylene), single‐molecule conductance, Sonogashira coupling, STM‐break junction.
Introduction
The field of molecular electronics has matured and evolved over the past decades from an initial focus on the mimicry of solid-state electronic components to emerging applications in molecular materials science.[1,2] Key to all of these advances in the broad field of molecular electronics is the development of methods to create single-molecule[3,4] and ‘large area’[5–8] electrode|molecule(s)|electrode ‘molecular junctions’ that permit the direct measurement of the electrical properties of the molecule(s) or monolayer under an applied bias. Whilst large-area junctions are closer to true device structures,[9–11] single-molecule junctions play a critical role in elucidating the fundamental processes of molecular electronics.[12]
Electron transport through a molecule within a molecular junction, and hence single-molecule conductance, is known to depend on the chemical structure of the molecular backbone and the nature of the anchor groups that bind the molecule to the electrodes, as well as environmental factors such as the solvent.[13] For a simple model junction in which electrons tunnel through a single molecular energy level (or molecular orbital) close to the electrode Fermi level, these concepts are succinctly captured in the Breit–Wigner formula (Eqn 1), which describes the transmission coefficient T of an electron of energy E:
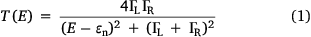
where ΓL and ΓR are terms describing the coupling of the molecular orbital to the left and right electrodes, and εn = En − σ is the energy of the molecular orbital responsible for transmission (En) adjusted for the effects of coupling to the electrodes (σ). The transmission function is maximised when the energy of the transmitting electron is equal to the molecular orbital energy. The coupling terms are strongly dependent on the chemical characteristics of the anchor groups in contact with the electrodes, whilst ɛn carries information from both the molecular backbone and the solvent environment.[14] Based on the simple ideas of the Breit–Wigner expression, studies of transport through molecules with systematically varied molecular backbones and contact groups are readily identified as valuable tools in establishing structure–property relationships governing the electrical characteristics of molecular junctions.
Moving to the more general case of non-resonant transport, the current flowing through a molecular junction is better described by the expressions drawn from the Landauer model (Eqn 2)
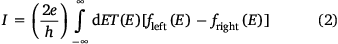
where T(E) is the transmission coefficient for electrons passing from one electrode to the other, and fleft(E) and fright(E) describe the energy distribution of electrons entering the junction from the left or right electrodes.[13,14] The Landauer model, implemented within the framework of DFT and non-equilibrium Green’s function calculations,[15] has proven to be an exceptionally robust tool through which to explore and rationalise the electrical properties of molecular junctions.[16]
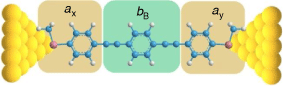
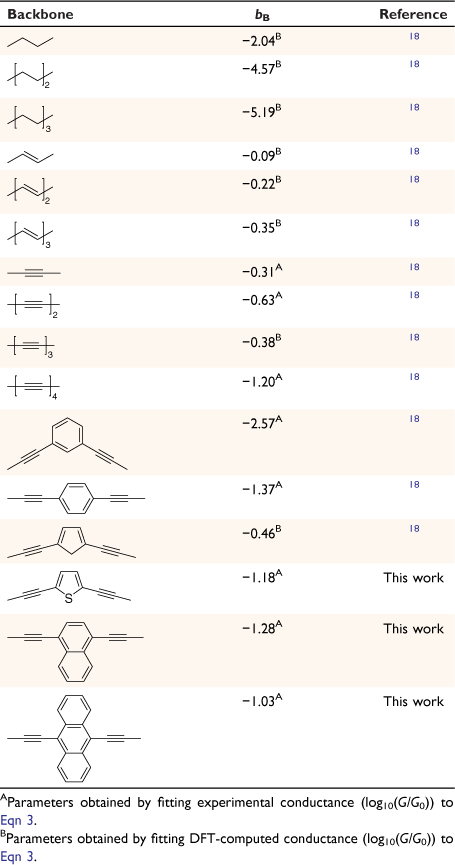
In looking beyond atomistic modelling of the complete junction, attention is now being directed to the description of entire molecular circuits, and the laws that govern the underlying structure–property relationships.[17] As part of such efforts, a ‘circuit rule’ that partitions the molecular junction into a series of weakly coupled scattering regions, each described by an independent numerical parameter, has been proposed.[18] The rules most usefully apply to non-resonant tunnel junctions (i.e. the Fermi energy of the electrodes falls near the middle of the molecular HOMO–LUMO gap) formed from a molecule of general form X–B–Y (where X and Y are the anchor groups that bind the molecule to the electrodes and B is the molecular backbone, Fig. 1), with conductance dominated by coherent tunneling (i.e. molecules less than ca. 3 nm in length).
|
When the scattering regions defined by X, B and Y are weakly coupled, the conductance of the junction, GXBY, can be written (Eqn 3)
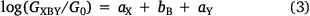
where G0 is the quantum of conductance, aX, bB and aY are independent parameters describing the anchor X, the bridge B and anchor Y, respectively, suggesting that the logarithm of the junction conductance is a sum of transferrable parameters.[18] A number of these parameters have been determined from both computational and experimental conductance data, providing the basis of a ‘tool kit’ for exploring molecular structure–electrical property relationships and molecular design strategies (Table 1, 2).[18,19] However, before the potential of the circuit laws can be fully realised, more such parameters need to be determined from conductance data of systematically varied molecular structures.

|
|
Due to the almost ubiquitous use of gold electrodes in the formation of molecular junctions to design candidate molecules through which to both further experimentally test the circuit laws and expand the range of available molecular parameters, compounds featuring anchors well-suited to gold electrodes are readily identified as initial targets. For gold-electrode based molecular junctions, thiol-based anchor groups are frequently used in both single molecules and large area junctions due to the strong gold–sulfur bond, the often high junction formation probability and the propensity of thiols (as thiolates) to form well-packed self-assembled monolayers (SAMs) with a high stability on gold.[20,21] Indeed, the thiolate–gold electrode bond (RS–Au) strength is very similar to the gold–gold (Au–Au) bond, and therefore thiols (as thiolate) form a robust interaction and strongly couple electronically with the gold electrodes. These concepts are of paramount importance in stabilising the molecule within the molecular junction and strongly influence the electronic characteristics of the assembly.[22]
However, despite the advantages of thiol(ate) anchor groups in molecular electronics research, free thiols and surface-anchored thiolates in films readily undergo side reactions including oxidative disulfide formation, and other oxidation reactions.[23–25] To avoid handling free thiols, molecules designed to be attached within molecular junctions by thiolate anchors are commonly introduced as the acetyl protected thioacetate (RSAc). The thioacetate group can be easily unmasked by deprotection in situ in the junction, to give the free thiol and in turn forming a thiyl radical (RS˙) at the surface of the gold electrode where it binds through covalent interaction.[22]
In much the same way that thio(ate) has served as the most common anchor group for securing molecules within gold-electrode junctions, oligo(arylene–ethynylene)-type molecules (OAEs) have been used as the ‘fruit flies’ of organic backbone structures.[26–29] OAE-based structures of defined length and various anchor groups are available,[30–32] and offer the possibility to introduce pendant groups and to vary the electronic nature of the arylene core(s).[33] This synthetic versatility has permitted the use of OAE structures in the exploration of an increasingly wide range of electrical transport and thermoelectrical phenomena, in both single-molecule junctions and ‘large area’ or ensemble molecular junctions (Fig. 1).[24,34–37] The backbone parameters bB for such diethynylarylene cores can therefore be identified as useful in the development of structure–property relationships and discovery in molecular electronics.
Given the role that SAc functionalised OAE compounds continue to play in the development of the field of molecular electronics, and the availability of the anchor group parameter for the arylthiolate group, which in turn allows calculations of the arylene backbone parameter from experimental conductance data, the development of simple and straight-forward routes to synthesise thioacetate functionalised OAE structures is of interest. The Sonogashira coupling reaction is one of the most frequently applied methods for synthesising OAE compounds using strategies such as those outlined in Scheme 1, but despite the general utility of this approach, limitations remain.[38–40] The key reagent S-(4-ethynylphenyl) ethanethiolate is prepared in a multi-step reaction sequence, beginning with the reduction of iodobenzylsulfonylchloride to the thiolate salt which is trapped by acetylation to give S-(4-iodophenyl) ethanethioate.[41] The Sonogashira cross-coupling reactions of S-(4-iodophenyl) ethanethioate with trimethylsilylacetylene and subsequent removal of the silyl protecting group produces the building block S-(4-ethynylphenyl) ethanethiolate in 87% overall yield.[42]

|
Whilst S-(4-ethynylphenyl) ethanethiolate can be cross-coupled with a wide range of dihalo arylenes (XArX; X = I, Br, OTf) to give SAc-functionalised OAE compounds (Scheme 1, Route 1),[33,41,43–45] the low tolerance of the thioacyl functional group towards nucleophiles can result in inadvertent deprotection of the thioacetate reagent in the reaction mixture, resulting in reduced yields.[35] Furthermore, attempts to couple S-(4-ethynylphenyl) ethanethiolate with un-activated dihaloarylenes result in a cyclotrimerisation reaction rather than entering into the cross-coupling cycle.[46]
Alternatively, the corresponding diethynyl arylenes (HC≡CArC≡CH) can be cross-coupled with S-(4-iodophenyl) ethanethiolate under similar Sonogashira conditions (Scheme 1, Route 2).[43,47–50] However, whilst stepwise protection/deprotection sequences can be effective routes to the preparation of the key diethynylarylene reagents,[48,51,52] the limited thermal and chemical stability of many diethynyl arylenes such as 9,10-diethynyl anthracene[53,54] and 2,5-diethynyl thiophene[55] limits their practical use as cross-coupling partners. To overcome these various inconveniences, a number of procedures have been developed for the preparation of OAE compounds based on the in situ deprotection of bis(trimethylsilylethynyl) arylenes (Me3SiC≡CArC≡CSiMe3) prior to coupling with an arylhalide.[53,56–59] Various desilylating agents have been employed, including catalytic hexafluorosilicic acid,[60] or more commonly tetrabutylammonium fluoride (TBAF),[61,62] CsF,[63] and various nucleophilic bases.[64] The main disadvantage of TBAF-facilitated desilylations is the subsequent difficulty in the purification of the desired products, free of ionic side products.[65] The low solubility of CsF causes slow reaction progress and often requires additional phase transfer catalysts (e.g. polyethylene glycols) to guarantee a sufficient fluoride concentration in the organic solvent reaction medium.[63] In addition, both base mediated and fluoride deprotection strategies can lead to complications in the subsequent Sonogashira reactions with coupling partners bearing substituents that are sensitive to nucleophiles, including SAc.
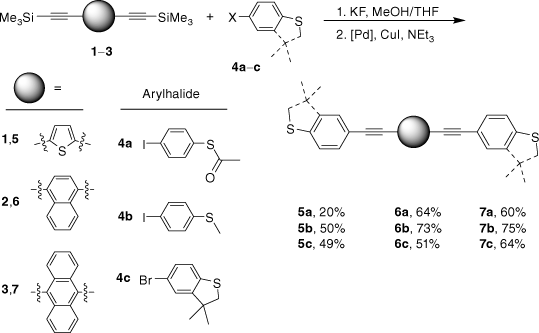
In order to access a series of OAE compounds with systematically varied arylene cores (1,4-naphthalene, 9,10-anthracene and 2,5-thiophene) and sulfur-based anchor groups we have employed modified reaction conditions that permit facile two-step, one-pot synthetic strategy. The reaction conditions are tolerated by the SAc functionality, and are also suitable for other sulfur-based anchor groups such as thioanisole and 3,3-dimethyl-2,3-dihydrobenzo[b]thiophene (DMBT). The molecular conductance of each of these compounds was determined in single-molecule junctions using the scanning tunnelling microscope break junction (STM-BJ) method under a uniform set of conditions. This has allowed an exploration of the interplay and influence of anchor groups and the central arylene core on molecular conductance, and further quantification and testing of the parameters and relationships contained within recently proposed quantum circuit laws.[18]
Results and discussion
Synthetic strategy
Given the competitive cyclo-oligomerisation of S-(4-ethynylphenyl) ethanethiolate under Sonogashira conditions, the reaction strategy outlined in Scheme 2 was adopted here. As proof of principle, the reaction of trimethylsilyl-protected ethynylthioanisole with S-(4-iodophenyl) ethanethiolate[29] was initially investigated (Scheme 2). The desilylation of 4-(trimethylsilylethynyl)thioanisole by KF (2.5 equiv.) in a methanol solution was readily achieved within 2 h. To prevent methanolysis of the thioacyl group in the subsequent coupling reaction, all volatiles were removed under reduced pressure. The residue was directly treated with triethyl amine (NEt3), S-(4-iodophenyl) ethanethioate, [PdCl2(PPh3)2], and CuI and allowed to react for 2 h at ambient temperature that gave the desired product in good yield (Scheme 2). The fluoride-based salts are apparently insufficiently soluble under these conditions to cause deprotection of the SAc group, and A was isolated in 63% yield.

|
The two-step, one-pot deprotection/cross-coupling of bis(trimethylsilylethynyl)-arylenes 1–3 in combination with S-(4-iodophenyl) ethanethiolate was subsequently explored under similar conditions, although the initial desilylation reaction of 9,10-bis(trimethylsilylethynyl)anthracene (3) in the mixed-solvent system required an increase of the temperature to 60°C. After allowing the reaction to cool followed by the removal of the polar solvents, the SAc-functionalised compounds were readily formed from the subsequent room-temperature cross-coupling reactions for 2 h producing 5a (20%), 6a (64%) and 7a (60%) (Scheme 3).
|
With the SAc-functionalised OAE compounds in hand, attention was turned to the preparation of similar compounds bearing other S-based anchor groups. Cross-coupling reactions of 4-iodothioanisole with the terminal dialkynes prepared by in situ deprotection gave 5b (50%), 6b (73%) and 7b (75%) under similar conditions and after similar reaction times. However, the rather electron-rich, and hence deactivated, 5-bromo-3,3-dimethyl-2,3-dihydrobenzo[b]thiophene has been shown to more efficiently enter into Sonogashira cross-coupling reactions when the more active palladium catalyst combination of [Pd2(dba)3]/JohnPhos is employed.[66] Using this catalyst system and rather more forcing conditions (50–90°C; 24 h) 5c, 6c and 7c were formed and isolated in 49–64% yield (Scheme 3).
Each of the compounds was characterised by the usual array of IR spectroscopy, NMR spectroscopies (1H, 13C{1H}) and mass spectrometry, with data consistent with that available in the literature in the cases of known compounds (5a,[48] 5b,[67] 6a,[34] 6b,[51] 7a,[36] 7b[36]). The molecular structure of the precursor 2,5-bis(trimethylsilylethynyl)thiophene (1) and the 7c were also confirmed by single-crystal X-ray diffraction (SI).
Molecular conductance measurements
The single-molecule conductance of each of the compounds 5a–c, 6a–c and 7a–c was determined using the scanning tunneling microscope break junction (STM-BJ) method.[68] Although molecular conductance values for some of these compounds have been reported previously (5a,[69] 5b,[67] 6a,[34] 6b,[51] 7a,[34,36,70] 7b,[36,70,71]), to avoid systematic errors in data comparisons all compounds were measured here using the same experimental conditions including technique, solvent, applied voltage bias, and sample concentration. For each compound, 2000 current–distance (I(z)) traces were obtained, with well-developed plateaus in the range log(G/G0) = −3.4 to −4.1 attributed to the formation of the single-molecular junctions, and subsequently analysed by creating all-data point 1-D conductance histograms and overlaid to construct 2-D conductance–distance cloud maps (Fig. 2–4). Each of the conductance histograms featured high conductance peaks associated with the formation of the metallic Au–Au contact between the STM tip and the gold substrate. A sharp feature around 1 G0 (G0 = 2e2/h = 77.5 μS) in each of the conductance histograms is assigned to the last gold–gold atomic contact, together with a peak arising from the molecular junction that reflects the most probable molecular conductance. Molecular conductance values were extracted by fitting the peak to a Gaussian curve (Table 3). The conductance histograms of compounds 6a and 7a also revealed a second, rather less pronounced conductance peak in a lower conductance region (log(Gexp/G0) = −4.8 to −5.0), which is in good agreement with the previous studies.[34]

|

|

|

|
The absolute electrode displacement in the experimental measurements, and hence the length of the molecular junction, was estimated by adding a snapback distance that allows for the rupture of the last Au–Au contact to the experimentally measured displacement Δz* (plateau length).[72,73] The snapback distance is extracted from the current–displacement traces without molecular junctions (Δzcorr = 0.5 nm). The most probable absolute displacements Δz* + Δzcorr are, in most cases, close to the length of the molecule suggesting that, before breaking of molecular junction, the molecule is oriented almost perpendicularly (with a couple of exceptions) in respect to the substrate (Table 3).
The junction formation probability (JFP) was computed by dividing the number of traces containing conductive plateaus by the total number of traces.[42] It was found that JFP for the molecule with the same core follows the trend DMBT = −SMe > −SAc (Table 3). The high JFP of compounds with DMBT and –SMe binding groups can be attributed to the strong interactions of the sulfur moiety with the gold electrodes. Although thioacetate has been widely used as an anchor in molecular electronics research and is known to give rise to Au–S contacts,[47] in our hands, the JFP for thiolate junctions formed from –SAc protecting binding groups was rather low (a matter of a few percent) in the absence of a deprotecting agent that increased to 25–40% upon addition of 1 M THF solution of either tetra-n-butylammonium fluoride (TBAF) (Fig. 2–4) or tetra-n-butylammonium hydroxide (TBAOH; Supplementary Fig. S23).
Quantum circuit law parameters
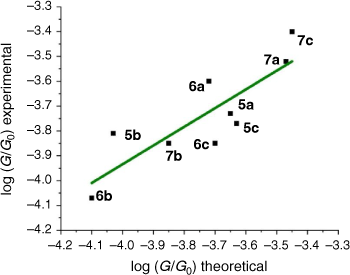
From these experimental data, the relevant anchor group parameters, aX (Table 1), and the circuit rule (Eqn 3) it is possible to calculate, test and refine backbone parameters, bB, for the 2,5-diethynylthiophene (bthioph), 1,4-diethynylnaphthalene (bnaphth) and 9,10-diethynylanthracene (banth) fragments. In the first instance, given the anchor parameters for the thioanisole (aSMe = −1.41) and DMBT (aDMBT = −1.21) moieties,[19] and the experimentally determined molecular conductance of the 2,5-diethynylthiophene compounds 5b (log(G/G0) = −3.81 ± 0.36) and 5c (log(G/G0) = −3.77 ± 0.36) (Table 3) it is possible to estimate the bridge parameter bthioph (−1.17) from the average solutions of Eqn 3.
With values for both bthioph and the thiolate anchor (aS− = −1.22) in hand, from Eqn 3 the logarithm of conductance of the thiolate contacted diethynylthiophene junction derived from 5a can also be estimated as follows.
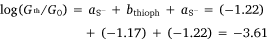
This value is in excellent agreement with the experimentally determined value (−3.73 ± 0.40) of the junction derived from 5a, giving confidence in the significance of the circuit rule.
Alternatively, averaging the values of bthioph obtained from each of the three experimental conductance values from 5a, 5b and 5c gives bthioph = −1.18, a value obviously similar to the two-point estimate given above. Unsurprisingly, using the established anchor parameters (Table 1) and this globally averaged value of the bridge parameter, estimates of molecular conductance (log(Gth/G0)) are also found to be in excellent agreement with the experimental values (log(Gexp/G0)) (Table 3).
In the same manner, a two-point average estimate of bnaphth (−1.34) and banth (−1.00) have been calculated from 6b and 6c, and 7b and 7c respectively, and used to predict the most probable conductance of the structures with thiolate anchor groups:
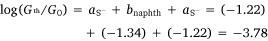
and
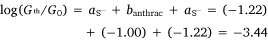
These values are in excellent agreement with the experimental conductance of 6a and 7a (−3.60 ± 0.25 and −3.52 ± 0.49, respectively). Again, globally averaged values of the bridge parameter bnaphth and banth based on experimental conductance values log(Gexp/G0) of 6a, 6b, 6c and 7a, 7b, 7c, have been determined, and summarised in Table 2. Estimates of molecular conductance (log(Gth/G0)) are found to be in excellent agreement with the experimental values (Table 3).
Structure–property relationships
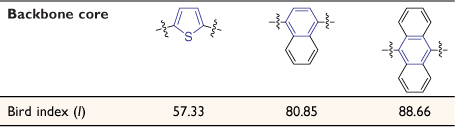
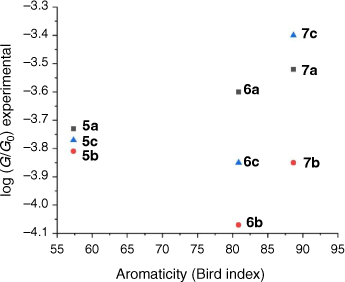
In addition, we have studied the relationship between the aromaticity of the backbone and conductance of single-molecule junctions.[74] Aromaticity of the substituted ring of each core has been estimated by the Bird index.[75] For this, the bond orders (N) were calculated from the bond length (R) from the crystal structure data of compounds, using the Gordy relationship[76] (Eqn 4)

where, for C–C bond a = 6.20 and b = 1.71, and for C–S bond a = 11.9 and b = 2.59.
The coefficient of variation for the bond orders of a heterocycle is estimated by the equation (Eqn 5)
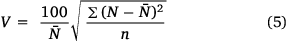
where n is the number of bonds.
Finally, the aromaticity index (I) can be calculated using the coefficient of variation for the bond orders (Eqn 6)
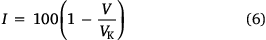
where, for a five-membered ring heterocycle, the coefficient of variation for the bond orders in a non-delocalized Kekule form of the cyclic structure with alternating single and double bonds VK = 35 (thiophen backbones), and for a six-membered ring heterocycle VK = 33.3 (naphthalene and anthracene backbones).
The aromaticity of each compound has been estimated from the metric data contained within the available crystal structures of 2,5-bis((trimethylsilyl)ethynyl)thiophene, 1,4-bis((trimethylsilyl)ethynyl)naphthalene and 9,10-bis((trimethylsilyl)ethynyl)anthracene. The results of these calculations of the Bird index reveal that the aromaticity of the alkynyl substituted ring follows the trend thiophene < naphthalene < anthracene (Table 4). However, there is no strong correlation between the aromaticity index and conductance of molecules 5a,b–7a,b (Fig. 5). Not surprisingly, for a range of molecules that vary in both backbone and anchor group, a single parameter such as the aromaticity of the backbone moiety cannot be the key tool for prediction, comparison, and tuning of molecular conductance of a molecular junction. Structure–property relationships can, however, be rationalised from the molecular circuit rule where the overall molecular conductance of a specific compound is obtained from partitioning the junction into a series of weakly coupled scattering regions (i.e. the molecular backbone and the anchor groups) (Fig. 6).
|
|
|
Conclusions
The in situ desilylation of trimethylsilyl-protect bis(alkynyl)arylenes and subsequent coupling with the appropriate aryl halides offer a convenient method to synthesis the wide variety of oligo(arylene–ethynylene) (OAE) type molecular wires. The molecular conductance values of a range of these molecular wires (5a,b–7a,b) with different arylene ethynylene backbones have been measured using the STM-BJ method. Off-resonance tunnelling through this family of molecules allows the junction to be treated as a series of weakly coupled scattering moieties (anchors and a molecular bridge) in line with the concepts expressed in recently proposed quantum circuit laws. Experimentally determined molecular conductance values have been used to extract the molecular bridge parameters, bB, for 2,5-diethynylthiophene, 1,4-diethynylnaphthalene, and 9,10-diethynylanthracene moieties. Predictions of molecular conductance based on these parameters are in excellent agreement with available experimental results. Whilst there was no simple relationship between molecular conductance and the degree of aromaticity in the arylene fragments, the junction may be partitioned into anchor and backbone regions according to molecular circuit laws. By parameterising these regions with numerical terms (aX, bB) that capture the molecular variables influencing the electrical properties of a junction (e.g. energy level alignment, molecular length, electrode–molecule coupling, etc.), the quantum circuit rules can be used as predictive tools for molecular conductance.
Experimental section
General conditions
All reactions were performed with the use of standard Schlenk line techniques, under an atmosphere of N2. Methanol was dried over activated Mg turnings and distilled under N2 before use. Tetrahydrofuran was dried on an INERT™ column-based solvent purification system and stored under N2. Triethylamine (NEt3) was dried and distilled over KOH under N2 before use. No special precautions were taken to exclude air or moisture during work-up. The compounds S-(4-iodophenyl) ethanethioate,[41] 5-bromo-3,3-dimethyl-2,3-dihydrobenzo[b]thiophene,[66] 4-(trimethylsilylethynyl)thioanisole,[66] 2,5-bis((trimethylsilyl)ethynyl)thiophene,[77] 1,4-bis((trimethylsilyl)ethynyl)naphthalene,[53] 9,10-bis((trimethylsilyl)ethynyl)anthracene,[53] [PdCl2(PPh3)2],[78] [Pd2(dba)3][79] and were prepared by literature methods. All other materials were obtained from commercial suppliers and used as received. NMR spectra were recorded in deuterated solvent solutions on Bruker Avance 400 MHz (1H, 400.1 MHz; 13C, 100.6 MHz) or 500 MHz (1H, 500.1 MHz; 13C, 126 MHz) spectrometers using CDCl3 as the solvent and referenced to internal solvent references (1H, 13C{1H}). Infrared spectra were recorded on an Agilent Technologies Cary 630 spectrometer fitted with an ATR attachment. High-resolution mass spectra were recorded using a Waters LCT Premier XE mass spectrometer using electrospray ionisation (ESI) or atmospheric pressure chemical ionisation (APCI) with Leucine Enkephalin as reference.
Crystallography
Data were collected using an XtaLAB Synergy single source HyPix diffractometer operating at T = 102(2) K. Data were measured using Cu-Kα radiation. The diffraction pattern was indexed and the total number of runs and images was based on the strategy calculation from the program CrysAlisPro 1.171.41.103a. Data reduction, scaling and absorption corrections were performed using CrysAlisPro (Rigaku, V1.171.41.103a, 2021).
The crystals were kept at a steady T = 100 K during data collection. The structures were solved with the ShelXT 2018 solution program using dual methods,[80] and by using Olex2 as the graphical interface.[81] The models were refined with ShelXL using full matrix least squares minimisation on F2.[82] All crystallographic data have been deposited with the CCDC (2107811, 2107812) and can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/, or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK (fax +441223336033; email deposit@ccdc.cam.ac.uk). Crystal and refinement details are given in Supplementary Table S2.
S-(4-((4-(Methylthio)phenyl)ethynyl)phenyl) ethanethioate, A

|
A solution of 4-(trimethylsilylethynyl)thioanisole (200 mg, 0.907 mmol) and KF (264 mg, 4.54 mmol) in dry, degassed MeOH (10 mL) was stirred at room temperature for 2 h. After this time, the solvent was removed under reduced pressure and dry, degassed NEt3 (20 mL), S-(4-iodophenyl) ethanethioate (252 mg, 0.907 mmol), [PdCl2(PPh3)2] (14 mg, 0.02 mmol), and CuI (5 mg, 0.02 mmol) were added to the residue. The reaction mixture was stirred at room temperature for further 20 h. After this time, the solvent was removed in vacuo, and the residue purified by preparative thin layer chromatography on silica eluting with CH2Cl2:hexane (1:5) to give the product as an off-white solid. Yield was 0.135 g (63%). IR (solid state, ATR)/cm−1: ν(C≡C) 2116. 1H NMR (CDCl3, 500 MHz, δ/ppm): 7.54 (d, J = 8.4 Hz, 2H, H9), 7.44 (d, J = 8.5 Hz, 2H, H4), 7.39 (d, J = 8.4 Hz, 2H, H10), 7.21 (d, J = 8.5 Hz, 2H, H3), 2.50 (s, 3H, H1), 2.43 (s, 3H, H13). 13C{1H} NMR (CDCl3, 126 MHz, δ/ppm): 193.63 (C12), 139.92 (C2), 134.36 (C10), 132.24 (C9), 132.09 (C4), 128.06 (C8), 125.99 (C3), 124.75 (C11), 119.28 (C5), 91.07 (C6), 88.88 (C7), 30.42 (C13), 15.49 (C1). Acc-MS(ESI+): m/z 298.0426 [M]+ calculated for C17H14OS2 298.0486.
|
Synthesis of S,S′-((thiophene-2,5-diylbis(ethyne-2,1-diyl))bis(4,1-phenylene))diethanethioate, 5a
|
|
A solution of 2,5-bis((trimethylsilyl)ethynyl)thiophene (100 mg, 0.361 mmol) and KF (53 mg, 0.903 mmol) in dry, degassed MeOH (15 mL) was stirred at room temperature for 3 h. After this time, the solvent was removed under reduced pressure and the residue immediately treated with dry, degassed NEt3 (15 mL), S-(4-iodophenyl) ethanethioate (211 mg, 0.758 mmol), [PdCl2(PPh3)2] (13 mg, 0.02 mmol), and CuI (4 mg, 0.02 mmol). The reaction mixture was stirred at room temperature for further 2 h, after which time the solvent was removed in vacuo, and the residue purified by column chromatography on silica eluting with CH2Cl2:hexane (1:9) to give the product as a yellow solid. Yield was 0.30 g (20%). IR (solid state, ATR)/cm−1: ν(C≡C) 2113. 1H NMR (CDCl3, 500 MHz, δ/ppm): 7.54 (d, J = 8.5 Hz, 2H, H5), 7.40 (d, J = 8.5 Hz, 2H, H4), 7.18 (s, 1H, H10), 2.44 (s, 3H, H1). 13C{1H} NMR (CDCl3, 126 MHz, δ/ppm): 193.45 (C2), 134.40 (C4), 132.42 (C10), 132.15 (C5), 128.77 (C6), 124.80 (C9), 123.91 (C3), 93.68 (C7), 84.01 (C8), 30.47 (C1). Acc-MS(ESI+): m/z 432.0312 [M]+ calculated for C24H16O2S3 432.0312.
|
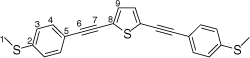
Synthesis of 2,5-bis((4-(methylthio)phenyl)ethynyl)thiophene, 5b

|
A solution of 2,5-bis((trimethylsilyl)ethynyl)thiophene (100 mg, 0.361 mmol) and KF (53 mg, 0.903 mmol) in dry, degassed MeOH (15 mL), was stirred at room temperature for 3 h. After this time solvent was removed under reduced pressure and the residue immediately treated with dry, degassed NEt3 (15 mL), 4-iodothioanisole (190 mg, 0.0.758 mmol), [PdCl2(PPh3)2] (14 mg, 0.02 mmol), and CuI (4 mg, 0.02 mmol). The reaction mixture stirred at room temperature for further 2 h. The solvent was then removed in vacuo, and the residue purified by column chromatography on silica eluting with CH2Cl2:hexane (1:5) to give the product as a yellow solid. Yield was 0.70 g (47%). IR (solid state, ATR)/cm−1: ν(C≡C) 2122. 1H NMR (CDCl3, 500 MHz, δ/ppm): 7.42 (d, J = 8.5 Hz, 4H, H4), 7.26 (s, 1H, H9), 7.21 (d, J = 8.5 Hz, 2H, H3), 2.50 (s, 3H, H1).13C{1H} NMR (CDCl3, 126 MHz, δ/ppm): 140.16 (C2), 131.87 (C4 & C9), 125.94 (C3), 124.77 (C8), 118.93 (C5), 94.11 (C6), 82.57 (C7), 15.44 (C1). Acc-MS(ESI+): m/z 376.0411 [M]+ calculated for C22H16S3 376.0414.
|
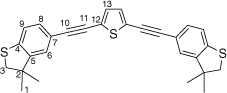
Synthesis of 2,5-bis((3,3-dimethyl-2,3-dihydrobenzo[b]thiophen-5-yl)ethynyl)thiophene, 5c

|
A solution of 2,5-bis((trimethylsilyl)ethynyl)thiophene (100 mg, 0.361 mmol) and KF (53 mg, 0.903 mmol) in dry, degassed MeOH (15 mL) was stirred at room temperature for 2 h. After this time, the solvent was removed under reduced pressure and the residue immediately treated with dry and degassed NEt3 (15 mL), 5-bromo-3,3-dimethyl-2,3-dihydrobenzo[b]thiophene (182 mg, 0.758 mmol), [Pd2(dba)3] (9 mg, 0.009 mmol), JohnPhos (6 mg, 0.02 mmol) and CuI (4 mg, 0.02 mmol). The reaction mixture was allowed to react for 24 h at 50°C temperature, and then cooled t room temperature before the solvent was removed in vacuo. The residue was purified by column chromatography on silica eluting with hexane to give the product as a yellow solid. Yield was 0.80 g (49%). IR (solid state, ATR)/cm−1: ν(C≡C) 2131. 1H NMR (CDCl3, 500 MHz, δ/ppm): 7.28 (dd, J = 8.0, 1.4 Hz, 2H, H8), 7.18 (d, J = 1.4 Hz, 2H, H6), 7.16 (d, J = 8.0 Hz, 2H, H9), 7.12 (s, 2H, H13), 3.20 (s, 4H, H3), 1.39 (s, 6H, H1). 13C{1H} NMR (CDCl3, 126 MHz, δ/ppm): 148.50 (C5), 142.36 (C4), 131.71 (C13), 130.88 (C8), 125.84 (C6), 124.76 (C12) 122.50 (C9), 118.56 (C7), 94.62 (C10), 81.95 (C11), 47.46 (C3), 47.42 (C2), 27.50 (C1). Acc-MS(ESI+): m/z 456.1042 [M]+ calculated for C28H24S3 456.1040.
|
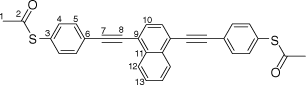
Synthesis of S,S′-((naphthalene-1,4-diylbis(ethyne-2,1-diyl))bis(4,1-phenylene))diethanethioate, 6a

|
A solution of 1,4-bis((trimethylsilyl)ethynyl)naphthalene (100 mg, 0.312 mmol) and KF (46 mg, 0.78 mmol) in a solution of dry, degassed MeOH (10 mL) and THF (2 mL), was stirred at room temperature for 3 h. After this time the reaction mixture was allowed to cool to room temperature. The solvent was removed under reduced pressure and the residue immediately treated with dry and degassed NEt3 (10 mL) and THF (2 mL), S-(4-iodophenyl) ethanethioate (183 mg, 0.655 mmol), [Pd(PPh3)2Cl2] (11 mg, 0.02 mmol), and CuI (3 mg, 0.02 mmol). The reaction mixture was allowed to stir at room temperature for further 2 h, before the solvent was removed in vacuo, and the residue purified by column chromatography on silica eluting with CH2Cl2:hexane (1:6) followed by CH2Cl2:hexane (1:1) to give the product as a yellow solid. Yield was 0.95 g (64%). IR (solid state, ATR)/cm−1: ν(C≡C) 2108. 1H NMR (CDCl3, 400 MHz, δ/ppm): 8.45 (dd, J = 6.4, 3.3 Hz, 2H, H12), 7.75 (s, 2H, H10), 7.71–7.63 (m, 6H, H13 & H5), 7.46 (d, J = 8.4 Hz, 4H, H4), 2.46 (s, 6H, H1). 13C{1H} NMR (CDCl3, 100 MHz, δ/ppm): 193.52 (C2), 134.49 (C4), 133.17 (C11), 132.39 (C5), 130.02 (C10), 128.64 (C6), 127.59 (C13), 126.73 (C12), 124.52 (C3), 121.60 (C9), 95.45 (C8), 89.27 (C7), 30.48 (C1). Acc-MS(ESI+): m/z 477.0981 [M + H]+ calculated for C30H21S2O2 477.0981.
|
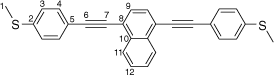
Synthesis of 1,4-bis((4-(methylthio)phenyl)ethynyl)naphthalene, 6b

|
A solution of 1,4-bis((trimethylsilyl)ethynyl)naphthalene (100 mg, 0.312 mmol) and KF (46 g, 0.78 mmol) in a solution of dry, degassed MeOH (10 mL) and THF (2 mL), was stirred at room temperature for 3 h. After this time the reaction mixture was allowed to cool, the solvent was removed under reduced pressure and the residue treated with dry and degassed NEt3 (10 mL) and THF (2 mL), 4-iodothioanisole (164 mg, 0.0.655 mmol), [Pd(PPh3)2Cl2] (11 mg, 0.02 mmol) and CuI (3 mg, 0.02 mmol). After that, the reaction mixture was allowed to stirred at room temperature for further 2 h. The solvent removed in vacuo, and the residue purified by column chromatography on silica eluting with hexane followed by CH2Cl2:hexane (1:8) to give the product as a yellow solid. Yield was 0.96 g (73%). IR (solid state, ATR)/cm−1: ν(C≡C) 2114. 1H NMR (CDCl3, 500 MHz, δ/ppm): 8.45 (dd, J = 6.4, 3.3 Hz, 2H, H11), 7.72 (s, 2H, H9), 7.65 (dd, J = 6.4, 3.3 Hz, 2H, H12), 7.56 (d, J = 8.5 Hz, 4H, H4), 7.26 (d, J = 8.5 Hz, 2H, H3), 2.53 (s, 6H, H1). 13C{1H} NMR (CDCl3, 126 MHz, δ/ppm): 139.98 (C2), 133.17 (C10), 132.09 (C4), 129.77 (C9), 127.37 (C12), 126.78 (C11), 126.08 (C3), 121.61 (C8), 119.58 (C5), 96.01 (C7), 87.84 (C6) 15.54 (C1). Acc-MS(ESI+): m/z 420.1005 [M]+ calculated for C28H20S2 420.1006.
|
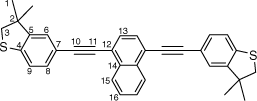
Synthesis of 1,4-bis((3,3-dimethyl-2,3-dihydrobenzo[b]thiophen-5-yl)ethynyl)naphthalene, 6c

|
A solution of 1,4-bis((trimethylsilyl)ethynyl)naphthalene (100 mg, 0.312 mmol) and KF (46 mg, 0.78 mmol) in a solution of dry, degassed MeOH (10 mL) and THF (2 mL) was stirred at room temperature for 2 h. After this time, the solvent was removed under reduced pressure and the residue immediately treated with dry, degassed NEt3 (10 mL) and THF (2 mL), 5-bromo-3,3-dimethyl-2,3-dihydrobenzo[b]thiophene (166 mg, 0.5 mmol), [Pd2(dba)3] (8 mg, 0.008 mmol), JohnPhos (6 mg, 0.02 mmol) and CuI (4 mg, 0.02 mmol). The reaction mixture then heated for further 24 h at 50°C temperature. After this time, the solution was cooled, the solvent was removed in vacuo, and the residue purified by column chromatography on silica eluting with hexane: CH2Cl2 (8:1) to give the product as a yellow solid. Yield was 0.80 g (51%). IR (solid state, ATR)/cm−1: ν(C≡C) 2109. 1H NMR (CDCl3, 500 MHz, δ/ppm): 8.46 (dd, J = 6.4, 3.3 Hz, 2H, H15), 7.71 (s, 2H, H13), 7.65 (dd, J = 6.4, 3.3 Hz, 2H, H16), 7.42 (dd, J = 8.0, 1.5 Hz, 2H, H8), 7.30 (d, J = 1.5 Hz, 2H, H6), 7.22 (d, J = 8.0 Hz, 2H, H9), 3.23 (s, 4H, H3), 1.43 (s, 12H, H1). 13C{1H} NMR (CDCl3, 126 MHz, δ/ppm): 148.55 (C5), 142.19 (C4), 133.18 (C14), 131.13 (C8), 129.72 (C13), 127.31 (C16), 126.83 (C15), 125.98 (C6), 122.57 (C9), 121.63 (C12), 119.20 (C7), 96.54 (C10), 87.20 (C11), 47.51 (C3), 47.47 (C2), 27.55 (C1). Acc-MS(ESI+): m/z 500.1637 [M]+ calculated for C34H28S2 500.1632.
|
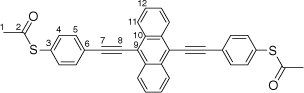
Synthesis of S,S′-((anthracene-9,10-diylbis(ethyne-2,1-diyl))bis(4,1-phenylene))diethanethioate, 7a

|
A solution of 9,10-bis((trimethylsilyl)ethynyl)anthracene (100 mg, 0.269 mmol) and KF (40 mg, 0.673 mmol) in a solution of dry, degassed MeOH (10 mL) and THF (5 mL), was stirred at 60°C temperature for 3 h. After this time the reaction mixture was allowed to cool, and the solvent was removed under reduced pressure. The residue was treated with dry, degassed NEt3 (10 mL) and THF (2 mL), S-(4-iodophenyl) ethanethioate (157 mg, 0.565 mmol), [PdCl2(PPh3)2] (10 mg, 0.013 mmol) and CuI (3 mg, 0.013 mmol), and the mixture allowed to stirred at room temperature for further 2 h. After this time, the solvent removed in vacuo, and the residue purified by column chromatography on silica eluting with CH2Cl2:hexane (1:8) followed by CH2Cl2:hexane (1:3) to give the product as a yellow solid. Yield was 0.85 g (60%). IR (solid state, ATR)/cm−1: ν(C≡C) 2113. 1H NMR (CDCl3, 500 MHz, δ/ppm): 8.67 (dd, J = 6.6, 3.3 Hz, 4H, H11), 7.81 (d, J = 8.4 Hz, 4H, H5), 7.66 (dd, J = 6.6, 3.3 Hz, 4H, H12), 7.51 (d, J = 8.4 Hz, 4H, H4), 2.48 (s, 6H, H1). 13C{1H} NMR (CDCl3, 126 MHz, δ/ppm): 193.54 (C2), 134.60 (C3), 132.38 (C5), 132.31 (C10), 128.77 (C6), 127.33 (C11), 127.19 (C12), 124.70 (C4), 118.54 (C9), 101.86 (C7), 88.27 (C8), 30.50 (C1). Acc-MS(ESI+): m/z 526.1069 [M]+ calculated for C34H22O2S2 526.1061.
|
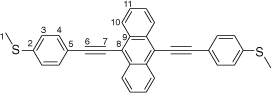
Synthesis of 9,10-bis((4-(methylthio)phenyl)ethynyl)anthracene, 7b

|
A solution of 9,10-bis((trimethylsilyl)ethynyl)anthracene (100 mg, 0.269 mmol) and KF (40 mg, 0.673 mmol) in dry, degassed MeOH (10 mL) and THF (2 mL), was stirred at 60°C temperature for 2 h. After this time the reaction mixture was allowed to cool, and the solvent was removed under reduced pressure. The residue was immediately treated with dry, degassed NEt3 (10 mL) and THF (2 mL), 4-iodothioanisole (142 mg, 0.565 mmol), [PdCl2(PPh3)2] (10 mg, 0.013 mmol), and CuI (3 mg, 0.013 mmol). The reaction mixture was allowed to stirred at room temperature for further 2 h, before the solvent was removed in vacuo, and the residue purified by column chromatography on silica eluting with hexane followed by CH2Cl2:hexane (1:8) to give the product as a yellow solid. Yield was 0.95 g (73%). IR (solid state, ATR)/cm−1: ν(C≡C) 2113. 1H NMR (CDCl3, 500 MHz, δ/ppm): δ 8.68 (dd, J = 6.6, 3.3 Hz, 4H, H10), 7.69 (d, J = 8.4 Hz, 4H, H4), 7.64 (dd, J = 6.6, 3.3 Hz, 4H, H11), 7.32 (d, J = 8.4 Hz, 2H, H3), 2.56 (s, 6H, H1). 13C{1H} NMR (CDCl3, 126 MHz, δ/ppm): 140.15 (C2), 132.20 (C4), 132.08 (C9), 127.41 (C10), 126.95 (C11), 126.17 (C3), 119.81 (C5), 118.58 (C8), 102.49 (C6), 86.87 (C7), 15.58 (C1). Acc-MS(ESI+): m/z 470.1157 [M]+ calculated for C32H22S2 470.1163.
|
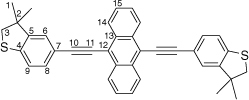
Synthesis of 9,10-bis((3,3-dimethyl-2,3-dihydrobenzo[b]thiophen-5-yl)ethynyl)anthracene, 7c

|
A solution of 9,10-bis((trimethylsilyl)ethynyl)anthracene (100 mg, 0.269 mmol), KF (40 mg, 0.673 mmol) in dry, degassed MeOH (10 mL) and THF (2 mL) was stirred at 60°C temperature for 2 h. After this time, the solvent was removed under reduced pressure and the residue immediately treated with dry and degassed NEt3 (10 mL) and THF (2 mL), 5-bromo-3,3-dimethyl-2,3-dihydrobenzo[b]thiophene (137 mg, 0.565 mmol), [Pd2(dba)3] (7 mg, 0.007 mmol), JohnPhos (6 mg, 0.02 mmol) and CuI (4 mg, 0.02 mmol). The mixture was allowed to react for further 24 h at reflux temperature. After this time, the solvent was removed in vacuo, and the residue purified by column chromatography on silica eluting with hexane followed by CH2Cl2:hexane (1:9) to give the product as a yellow solid. Yield was 0.95 g (64%). IR (solid state, ATR)/cm−1: ν(C≡C) 2112. 1H NMR (CDCl3, 500 MHz, δ/ppm): 8.69 (dd, J = 6.6, 3.3 Hz, 4H, H14), 7.65 (dd, J = 6.6, 3.3 Hz, 6H, H15), 7.54 (dd, J = 8.0, 1.5 Hz, 2H, H8), 7.41 (d, J = 1.5 Hz, 2H, H6), 7.28 (d, J = 8.0 Hz, 2H, H9), 3.26 (s, 6H, H3), 1.48 (s, 12H, H1). 13C{1H} NMR (CDCl3, 126 MHz, δ/ppm): 148.66 (C5) 142.39 (C4), 132.19 (C13), 131.20 (C8), 127.46 (C14), 126.88 (C15), 125.89 (C6), 122.67 (C9), 119.44 (C7), 118.60 (C12), 103.04 (C10), 86.20 (C11), 47.56 (C3), 47.53 (C2), 27.59 (C1). Acc-MS(ESI+): m/z 550.1786 [M]+ calculated for C38H30S2 550.1789.
|
Single-molecule conductance characterisation
The general methods and principles of the single-molecule conductance measurements by STM-BJ have been reported in detail elsewhere by others.[83,84] In brief, for the STM-BJ experiment, the gold-on-glass substrates (ArrandeeTM) were cleaned by immersion in a freshly prepared piranha solution (1 part H2O2 (33%) in 3 parts H2SO4 (98%)). After cleaning, the substrates were rinsed with deionised water and dried under a nitrogen stream. A freshly cut gold wire (99.99% purity, 0.25 mm diameter) was used as the STM tip. Analyte solutions (1 mM) were prepared in 1,3,5-trimethylbenzene (TMB). For compounds 5a, 6a, and 7a in 1 mM TMB solution, a few drops of a 1 M solution of tetra-n-butylammonium fluoride (TBAF) in tetrahydrofuran were added as a deprotecting agent to facilitate the removal of the acyl groups. The substrate surface was examined by STM imaging in the analyte solution before starting the conductance–distance measurements. Molecular junctions were formed and broken repeatedly between the sharp tip and the gold substrate by moving the tip into and away from the substrate at the rate of 5 nm s−1 (Vbias = −0.1 V). In the case where no molecule(s) were trapped between electrodes while withdrawing the tip, the current–distance trace demonstrated only exponential decay. In contrast, if target molecule(s) bridged the gap between electrodes, conductive plateau-like features were observed. Typically, 2000 individual traces were recorded for each compound.
Data availability
The data that support this study are available in the article and accompanying online supplementary material.
Conflicts of interest
The authors declare no conflicts of interest.
Declaration of funding
This work has been supported by the Australian Research Council through project (DP190100073; DP190100074) and infrastructure (LE150100148) funding. S. A. M. holds an ARC Future Fellowship (FT2001000243). M. N. thanks the Forrest Research Foundation for a Research Scholarship.
Supplementary material
Crystal structure and refinement details as well as plots of 1H and 13C{1H} NMR spectra are available in supplementary material online.
Acknowledgements
Further support for research carried out by the team from the UK Engineering and Physical Sciences Research Council is gratefully acknowledged.
References
[1] S Marques-Gonzalez, PJ Low, Aust J Chem 2016, 69, 244.| Crossref | GoogleScholarGoogle Scholar |
[2] SV Aradhya, L Venkataraman, Nat Nanotechnol 2013, 8, 399.
| Crossref | GoogleScholarGoogle Scholar | 23736215PubMed |
[3] K Wang, BQ Xu, Top Curr Chem 2017, 375, 17.
| Crossref | GoogleScholarGoogle Scholar |
[4] Y Komoto, S Fujii, M Iwane, M Kiguchi, J Mater Chem C 2016, 4, 8842.
| Crossref | GoogleScholarGoogle Scholar |
[5] JC Lacroix, Curr Opin Electrochem 2018, 7, 153.
| Crossref | GoogleScholarGoogle Scholar |
[6] A Vilan, D Aswal, D Cahen, Chem Rev 2017, 117, 4248.
| Crossref | GoogleScholarGoogle Scholar | 28177226PubMed |
[7] E Gorenskaia, KL Turner, S Martin, P Cea, PJ Low, Nanoscale 2021, 13, 9055.
| Crossref | GoogleScholarGoogle Scholar | 34042128PubMed |
[8] L Herrer, S Martin, P Cea, Appl Sci 2020, 10, 6064.
| Crossref | GoogleScholarGoogle Scholar |
[9] G Puebla-Hellmann, K Venkatesan, M Mayor, E Lortscher, Nature 2018, 559, 232.
| Crossref | GoogleScholarGoogle Scholar | 29995866PubMed |
[10] SK Saxena, UM Tefashe, M Supur, RL McCreery, ACS Sensors 2021, 6, 513.
| Crossref | GoogleScholarGoogle Scholar | 33315386PubMed |
[11] AJ Bergren, L Zeer-Wanklyn, M Semple, N Pekas, B Szeto, RL McCreery, J Phys Condens Mat 2016, 28, 094011.
| Crossref | GoogleScholarGoogle Scholar |
[12] D Xiang, XL Wang, CC Jia, T Lee, XF Guo, Chem Rev 2016, 116, 4318.
| Crossref | GoogleScholarGoogle Scholar | 26979510PubMed |
[13] Lambert CJ. Quantum transport in nanostructures and molecules. In: An introduction to molecular electronics [Online]. IOP Publishing; 2021.
| Crossref |
[14] CJ Lambert, Chem Soc Rev 2015, 44, 875.
| Crossref | GoogleScholarGoogle Scholar | 25255961PubMed |
[15] J Ferrer, CJ Lambert, VM Garcia-Suarez, DZ Manrique, D Visontai, L Oroszlany, R Rodriguez-Ferradas, I Grace, SWD Bailey, K Gillemot, H Sadeghi, LA Algharagholy, New J Phys 2014, 16, 093029.
| Crossref | GoogleScholarGoogle Scholar |
[16] M Naher, DC Milan, OA Al-Owaedi, IJ Planje, S Bock, J Hurtado-Gallego, P Bastante, ZM Abd Dawood, L Rincon-Garcia, G Rubio-Bollinger, SJ Higgins, N Agrait, CJ Lambert, RJ Nichols, PJ Low, J Am Chem Soc 2021, 143, 3817.
| Crossref | GoogleScholarGoogle Scholar | 33606524PubMed |
[17] A Aggarwal, V Kaliginedi, PK Maiti, Nano Lett 2021, 21, 8532.
| Crossref | GoogleScholarGoogle Scholar | 34622657PubMed |
[18] DZ Manrique, Q Al-Galiby, WJ Hong, CJ Lambert, Nano Lett 2016, 16, 1308.
| Crossref | GoogleScholarGoogle Scholar | 26784577PubMed |
[19] E Gorenskaia, M Naher, L Daukiya, SA Moggach, DC Milan, A Vezzoli, CJ Lambert, RJ Nichols, T Becker, PJ Low, Aust J Chem 2021, 74, 806.
| Crossref | GoogleScholarGoogle Scholar |
[20] N Xin, JX Guan, CG Zhou, XJN Chen, CH Gu, Y Li, MA Ratner, A Nitzan, JF Stoddart, XF Guo, Nat Rev Phys 2019, 1, 211.
| Crossref | GoogleScholarGoogle Scholar |
[21] I Kaur, XT Zhao, MR Bryce, PA Schauer, PJ Low, R Kataky, ChemPhysChem 2013, 14, 431.
| Crossref | GoogleScholarGoogle Scholar | 23316022PubMed |
[22] H Hakkinen, Nat Chem 2012, 4, 443.
| Crossref | GoogleScholarGoogle Scholar | 22614378PubMed |
[23] YH Jang, WA Goddard, J Phys Chem C 2010, 114, 4646.
| Crossref | GoogleScholarGoogle Scholar |
[24] T Park, H Kang, S Seong, S Han, YJ Son, E Ito, T Hayashi, M Hara, J Noh, J Phys Chem C 2019, 123, 9096.
| Crossref | GoogleScholarGoogle Scholar |
[25] MH Schoenfisch, JE Pemberton, J Am Chem Soc 1998, 120, 4502.
| Crossref | GoogleScholarGoogle Scholar |
[26] JM Tour, AM Rawlett, M Kozaki, YX Yao, RC Jagessar, SM Dirk, DW Price, MA Reed, CW Zhou, J Chen, WY Wang, I Campbell, Chem Eur J 2001, 7, 5118.
| Crossref | GoogleScholarGoogle Scholar | 11775685PubMed |
[27] LA Bumm, JJ Arnold, MT Cygan, TD Dunbar, TP Burgin, L Jones, DL Allara, JM Tour, PS Weiss, Science 1996, 271, 1705.
| Crossref | GoogleScholarGoogle Scholar |
[28] R Frisenda, D Stefani, HSJ van der Zant, Acc Chem Res 2018, 51, 1359.
| Crossref | GoogleScholarGoogle Scholar | 29862817PubMed |
[29] LJ O’Driscoll, MR Bryce, Nanoscale 2021, 13, 10668.
| Crossref | GoogleScholarGoogle Scholar | 34110337PubMed |
[30] XT Zhao, CC Huang, M Gulcur, AS Batsanov, M Baghernejad, WJ Hong, MR Bryce, T Wandlowski, Chem Mater 2013, 25, 4340.
| Crossref | GoogleScholarGoogle Scholar |
[31] CS Wang, AS Batsanov, MR Bryce, J Org Chem 2006, 71, 108.
| Crossref | GoogleScholarGoogle Scholar |
[32] Q Lu, K Liu, HM Zhang, ZB Du, XH Wang, FS Wang, ACS Nano 2009, 3, 3861.
| Crossref | GoogleScholarGoogle Scholar | 19916506PubMed |
[33] R Kitouni, P Selvanathan, O Galangau, L Norel, B Ouarda, S Rigaut, Synth Commun 2018, 48, 1052.
| Crossref | GoogleScholarGoogle Scholar |
[34] V Kaliginedi, P Moreno-Garcia, H Valkenier, WJ Hong, VM Garcia-Suarez, P Buiter, JLH Otten, JC Hummelen, CJ Lambert, T Wandlowski, J Am Chem Soc 2012, 134, 5262.
| Crossref | GoogleScholarGoogle Scholar | 22352944PubMed |
[35] H Valkenier, EH Huisman, PA van Hal, DM de Leeuw, RC Chiechi, JC Hummelen, J Am Chem Soc 2011, 133, 4930.
| Crossref | GoogleScholarGoogle Scholar | 21384876PubMed |
[36] XT Wang, TLR Bennett, A Ismael, LA Wilkinson, J Hamill, AJP White, IM Grace, OV Kolosov, T Albrecht, BJ Robinson, NJ Long, LF Cohen, CJ Lambert, J Am Chem Soc 2020, 142, 8555.
| Crossref | GoogleScholarGoogle Scholar |
[37] A Yamada, QG Feng, Q Zhou, A Hoskins, KM Lewis, BD Dunietz, J Phys Chem C 2017, 121, 10298.
| Crossref | GoogleScholarGoogle Scholar |
[38] R Chinchilla, C Najera, Chem Rev 2007, 107, 874.
| Crossref | GoogleScholarGoogle Scholar | 17305399PubMed |
[39] R Chinchilla, C Najera, Chem Soc Rev 2011, 40, 5084.
| Crossref | GoogleScholarGoogle Scholar | 21655588PubMed |
[40] MM Huq, MR Rahman, M Naher, MMR Khan, MK Masud, GMG Hossain, NY Zhu, YH Lo, M Younus, WY Wong, J Inorg, Organomet Polym 2016, 26, 1243.
| Crossref | GoogleScholarGoogle Scholar |
[41] ZF Shi, LJ Wang, H Wang, XP Cao, HL Zhang, Org Lett 2007, 9, 595.
| Crossref | GoogleScholarGoogle Scholar | 17253702PubMed |
[42] P Moreno-Garcia, M Gulcur, DZ Manrique, T Pope, WJ Hong, V Kaliginedi, CC Huang, AS Batsanov, MR Bryce, C Lambert, T Wandlowski, J Am Chem Soc 2013, 135, 12228.
| Crossref | GoogleScholarGoogle Scholar | 23875671PubMed |
[43] N Weibel, A Blaszczyk, C von Haenisch, M Mayor, I Pobelov, T Wandlowski, F Chen, NJ Tao, Eur J Org Chem 2008, 136.
| Crossref | GoogleScholarGoogle Scholar |
[44] CR Parker, E Leary, R Frisenda, ZM Wei, KS Jennum, E Glibstrup, PB Abrahamsen, M Santella, MA Christensen, EA Della Pia, T Li, MT Gonzalez, XB Jiang, TJ Morsing, G Rubio-Bollinger, BW Laursen, K Norgaard, H van der Zant, N Agrait, MB Nielsen, J Am Chem Soc 2014, 136, 16497.
| Crossref | GoogleScholarGoogle Scholar | 25375316PubMed |
[45] K Liu, XH Wang, FS Wang, ACS Nano 2008, 2, 2315.
| Crossref | GoogleScholarGoogle Scholar | 19206398PubMed |
[46] MS Inkpen, AJP White, T Albrecht, NJ Long, Chem Commun 2013, 49, 5663.
| Crossref | GoogleScholarGoogle Scholar |
[47] F Jiang, DI Trupp, N Algethami, HN Zheng, WX He, A Alqorashi, CX Zhu, C Tang, RH Li, JY Liu, H Sadeghi, J Shi, R Davidson, M Korb, AN Sobolev, M Naher, S Sangtarash, PJ Low, WJ Hong, CJ Lambert, Angew Chem Int Ed 2019, 58, 18987.
| Crossref | GoogleScholarGoogle Scholar |
[48] A Seidler, J Svoboda, V Dekoj, JV Chocholousova, J Vacek, IG Stara, I Stary, Tetrahedron Lett 2013, 54, 2795.
| Crossref | GoogleScholarGoogle Scholar |
[49] SM Wu, MT Gonzalez, R Huber, S Grunder, M Mayor, C Schonenberger, M Calame, Nat Nanotechnol 2008, 3, 569.
| Crossref | GoogleScholarGoogle Scholar |
[50] A Operamolla, A Punzi, GM Farinola, Asian J Org Chem 2017, 6, 120.
| Crossref | GoogleScholarGoogle Scholar |
[51] M Gantenbein, XH Li, S Sangtarash, J Bai, G Olsen, A Alqorashi, WJ Hong, CJ Lambert, MR Bryce, Nanoscale 2019, 11, 20659.
| Crossref | GoogleScholarGoogle Scholar | 31641715PubMed |
[52] N Tanifuji, M Irie, K Matsuda, J Am Chem Soc 2005, 127, 13344.
| Crossref | GoogleScholarGoogle Scholar | 16173768PubMed |
[53] W Fudickar, T Linker, J Am Chem Soc 2012, 134, 15071.
| Crossref | GoogleScholarGoogle Scholar | 22881365PubMed |
[54] S Fraysse, C Coudret, JP Launay, J Am Chem Soc 2003, 125, 5880.
| Crossref | GoogleScholarGoogle Scholar | 12733929PubMed |
[55] TX Neenan, GM Whitesides, J Org Chem 1988, 53, 2489.
| Crossref | GoogleScholarGoogle Scholar |
[56] K Danel, JT Lin, Arkivoc 2002, 1, 12.
| Crossref | GoogleScholarGoogle Scholar |
[57] MA Fabre, J Jaud, JJ Bonvoisin, Inorg Chim Acta 2005, 358, 2384.
| Crossref | GoogleScholarGoogle Scholar |
[58] S Fraysse, C Coudret, JP Launay, Tetrahedron Lett 1998, 39, 7873.
| Crossref | GoogleScholarGoogle Scholar |
[59] A Slodek, M Filapek, E Schab-Balcerzak, M Grucela, S Kotowicz, H Janeczek, K Smolarek, S Mackowski, JG Malecki, A Jedrzejowska, G Szafraniec-Gorol, A Chrobok, B Marcol, S Krompiec, M Matussek, J Org Chem 2016, 2016, 4020.
| Crossref | GoogleScholarGoogle Scholar |
[60] D Lasanyi, A Meszaros, Z Novak, GL Tolnai, J Org Chem 2018, 83, 8281.
| Crossref | GoogleScholarGoogle Scholar | 29842774PubMed |
[61] AM DiLauro, WJ Seo, ST Phillips, J Org Chem 2011, 76, 7352.
| Crossref | GoogleScholarGoogle Scholar | 21846123PubMed |
[62] ZL Zhou, LH Zhao, S Zhang, K Vincent, S Lam, D Henze, Synth Commun 2012, 42, 1622.
| Crossref | GoogleScholarGoogle Scholar |
[63] JS Capani, JE Cochran, J Liang, J Org Chem 2019, 84, 9378.
| Crossref | GoogleScholarGoogle Scholar | 31194913PubMed |
[64] U Halbes, P Pale, Tetrahedron Lett 2002, 43, 2039.
| Crossref | GoogleScholarGoogle Scholar |
[65] Y Kaburagi, Y Kishi, Org Lett 2007, 9, 723.
| Crossref | GoogleScholarGoogle Scholar | 17286380PubMed |
[66] M Naher, S Bock, ZM Langtry, KM O’Malley, AN Sobolev, BW Skelton, M Korb, PJ Low, Organometallics 2020, 39, 4667.
| Crossref | GoogleScholarGoogle Scholar |
[67] D Miguel, LA de Cienfuegos, A Martin-Lasanta, SP Morcillo, LA Zotti, E Leary, M Burkle, Y Asai, R Jurado, DJ Cardenas, G Rubio-Bollinger, N Agrait, JM Cuerva, MT Gonzalez, J Am Chem Soc 2015, 137, 13818.
| Crossref | GoogleScholarGoogle Scholar | 26452050PubMed |
[68] L Wang, L Wang, L Zhang, D Xiang, Top Curr Chem 2017, 375, 61.
| Crossref | GoogleScholarGoogle Scholar |
[69] J Bails, A Daaoub, S Sangtarash, XH Li, YX Tang, Q Zou, H Sadeghi, S Liu, XJ Huang, ZB Tan, JY Liu, Y Yang, J Shi, G Meszaros, WB Chen, C Lambert, WJ Hong, Nat Mater 2019, 18, 364.
[70] A Ismael, XT Wang, TLR Bennett, LA Wilkinson, BJ Robinson, NJ Long, LF Cohen, CJ Lambert, Chem Sci 2020, 11, 6836.
| Crossref | GoogleScholarGoogle Scholar | 33033599PubMed |
[71] M Schmidt, D Wassy, M Hermann, MT Gonzalez, N Agrait, LA Zotti, B Esser, E Leary, Chem Commun 2021, 57, 745.
[72] WJ Hong, DZ Manrique, P Moreno-Garcia, M Gulcur, A Mishchenko, CJ Lambert, MR Bryce, T Wandlowski, J Am Chem Soc 2012, 134, 2292.
| Crossref | GoogleScholarGoogle Scholar |
[73] SY Quek, M Kamenetska, ML Steigerwald, HJ Choi, SG Louie, MS Hybertsen, JB Neaton, L Venkataraman, Nat Nanotechnol 2009, 4, 230.
| 19350032PubMed |
[74] WB Chen, HX Li, JR Widawsky, C Appayee, L Venkataraman, R Breslow, J Am Chem Soc 2014, 136, 918.
| Crossref | GoogleScholarGoogle Scholar |
[75] CW Bird, Tetrahedron 1985, 41, 1409.
| Crossref | GoogleScholarGoogle Scholar |
[76] W Gordy, J Chem Phys 1947, 15, 305.
| Crossref | GoogleScholarGoogle Scholar |
[77] SS Roy, SR Chowdhury, S Mishra, SK Patra, Chem Asian J 2020, 15, 3304.
| Crossref | GoogleScholarGoogle Scholar | 32790947PubMed |
[78] MA Fox, JE Harris, S Heider, V Perez-Gregorio, ME Zakrzewska, JD Farmer, DS Yufit, JAK Howard, PJ Low, J Organomet Chem 2009, 694, 2350.
| Crossref | GoogleScholarGoogle Scholar |
[79] SS Zalesskiy, VP Ananikov, AJ Reay, IJS Fairlamb, Inorg Synth 2018, 37, 183.
[80] GM Sheldrick, Acta Crystallogr A 2015, 71, 3.
| Crossref | GoogleScholarGoogle Scholar |
[81] OV Dolomanov, LJ Bourhis, RJ Gildea, JAK Howard, H Puschmann, J Appl Crystallogr 2009, 42, 339.
[82] A Thorn, GM Sheldrick, Acta Crystallogr A 2008, 64, C221.
| Crossref | GoogleScholarGoogle Scholar |
[83] BQ Xu, NJJ Tao, Science 2003, 301, 1221.
| Crossref | GoogleScholarGoogle Scholar |
[84] L Venkataraman, JE Klare, C Nuckolls, MS Hybertsen, ML Steigerwald, Nature 2006, 442, 904.
| Crossref | GoogleScholarGoogle Scholar | 16929295PubMed |
† This paper is dedicated to Professor Glen Deacon, a true icon of Australian chemistry, in celebration of his 85th Birthday.
# These authors contributed equally.