

Singly Occupied Molecular Orbital−Highest Occupied Molecular Orbital (SOMO−HOMO) Conversion
Ryo Murata A , Zhe Wang A and Manabu Abe A B
A and Manabu Abe A B
A Department of Chemistry, Graduate School of Advanced Science and Engineering, Hiroshima University, 1-3-1 Kagamiyama, Higashi-Hiroshima 739-8526, Hiroshima, Japan.
B Corresponding author. Email: mabe@hiroshima-u.ac.jp

Ryo Murata received his bachelor’s degree in 2021 from the University of Hiroshima. He is currently a master’s student at the Basic Chemistry Program, Graduate School of Advanced Science and Engineering, Hiroshima University, under the supervision of Professor Manabu Abe. His current research focuses on SOMO-HOMO conversion in triplet carbenes. |

Zhe Wang received his bachelor’s degree (B.Eng.) from the China University of Petroleum (East China) in 2017. He then joined the research group of Professor Manabu Abe at Hiroshima University. He received his master’s degree (M.Sc.) from the Hiroshima University in 2019 under the supervision of Professor Dr Manabu Abe. He is currently a Ph.D. student at Hiroshima University. His research focuses on the kinetic stabilisation of singlet 2,2-dimethoxycyclopentane-1,3-diyl diradicals. |

Manabu Abe was born in Osaka, Japan. He received his Ph.D. in 1995 from the Kyoto Institute of Technology (KIT) with Professor Akira Oku. In 1995, Manabu became a faculty staff member at Osaka University (HANDAI, Professor Masatomo Nojima’s group). From 1997 to 1998, he was an Alexander-von-Humboldt fellow with Professor Dr Waldemar Adam at the Universität Würzburg in Germany. Manabu was also a visiting researcher at the LMU München (Professor Dr Herbert Mayr) in 2007. He moved to Hiroshima and became a full-time professor of Organic Chemistry at the Department of Chemistry, Graduate School of Science, Hiroshima University (HIRODAI) in 2007. His research focuses on reactive intermediates chemistry, especially on diradicals, organic photochemistry, and unusual molecules. |
Australian Journal of Chemistry 74(12) 827-837 https://doi.org/10.1071/CH21186
Submitted: 3 August 2021 Accepted: 5 October 2021 Published: 11 November 2021
Journal Compilation © CSIRO 2021 Open Access CC BY-NC-ND
Abstract
Singly occupied molecular orbital−highest occupied molecular orbital (SOMO−HOMO) conversion (inversion), SHC, is a phenomenon in which the SOMO is lower in energy than the doubly occupied molecular orbitals (DOMO, HOMO). A non-Aufbau electronic structure leads to unique properties such as a switch in bond dissociation energy and the generation of high-spin species on one-electron oxidation. In addition, the pronounced photostability of these species has been reported recently for application in organic light-emitting devices. In this review article, we summarise the chemistry of SOMO−HOMO converted (inverted) species reported to date.
Keywords: SOMO–HOMO conversion, SOMO–HOMO inversion, high spin species, open shell species, radicals, carbenes, quantum chemical calculation, DFT calculation.
Introduction
Frontier molecular orbital theory[1] is useful for predicting and understanding the reactivity and other properties of molecules. The energy and orbital phase matching between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) is the key to analysing the reactivities of molecules. According to the Aufbau principle, electrons fill atomic orbitals with the lowest available energy levels before occupying the higher levels. Based on this, in closed-shell species, HOMO corresponds to the doubly occupied molecular orbital (DOMO), whereas in open-shell compounds such as radicals, it generally corresponds to the singly occupied molecular orbital (SOMO). Therefore, in general, the chemical behaviour of open-shell species can be predicted and analysed based on the energy level of the SOMO and its orbital phase.
However, several open-shell species, such as metal complexes, distonic radical anions, and stabilised radicals, do not follow the Aufbau rule. Specifically, the SOMO−HOMO can convert the electronic configuration such that the SOMO exhibits a lower energy than the DOMO. Such SOMO−HOMO-converted (inverted) species have garnered scientific interest because of their unique characteristics, such as the switch in bond dissociation energy and the generation of high-spin species in the oxidation state. In organic molecules, this phenomenon was first reported in 1993 by Sugimoto et al.[2] for an aminoxyl radical containing a tetrathiafulvalene (TTF) moiety. However, no specific term was given to this phenomenon. Later, in 2008, for the first time, Nishihara et al. used the term ‘SOMO−HOMO conversion’ (SHC)[3] to describe the phenomenon. Although the technical term is often used, this terminology has not been completely established. Recently, two terms, SHC and SOMO−HOMO inversion (SHI),[4] have been principally used in the literature. Throughout this review, we have used the term SHC.
Molecules exhibiting SHC phenomena reported to date are summarised in Fig. 1. The chemistry is introduced in the following sections: (1) aminoxyl radicals in spin-polarised donors; (2) organometallic complexes; (3) distonic radical anions; (4) biologically important radicals; (5) helicene-based radicals; (6) chiral bicarbazole-based radicals; (7) triarylmethyl radicals; (8) other radicals; and (9) carbenes and diradicals. Although a review article on SHC was published by Corminboeuf, Coote, and their co-workers in 2015,[5] herein, we summarise recent progress of studies on SHC molecules and their chemistry, including those reported after 2015.

|
Aminoxyl Radicals with a Spin-Polarised Donor
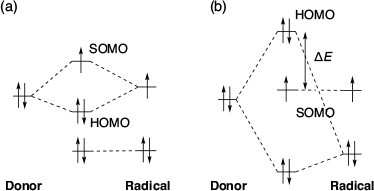
There are several reports on SHC in aminoxyl radicals with spin-polarised donor moieties. Sugimoto et al. in 1993 reported the first ever SHC molecule i.e. the 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) radical derivative with a spin-polarised donor TTF moiety (Fig. 2a).[2] Based on electron paramagnetic resonance (EPR) measurements, they observed that one-electron oxidation of the molecule occurred at the TTF moiety, not at the TEMPO radical moiety, which generated a singlet diradical. Similar oxidation behaviour was observed in some TTF-based nitronyl nitroxides (Fig. 2b).[6–15] In those radicals, triplet diradicals were formed on one-electron oxidation. Sugawara et al. explained the cause of the unusual electronic configurations using the perturbational molecular orbital (PMO) method (Fig. 3).[9] When considering the union of the donor and radical units, two types of interaction patterns, (a) and (b), are possible. In type (a) interaction, the SOMO is destabilised and the HOMO is stabilised by the interaction between the donor’s HOMO and the radical’s SOMO, resulting in a normal configuration. In type (b) interaction, the donor’s HOMO is destabilised by interacting with the radical’s HOMO owing to the mismatch between the donor’s HOMO and the radical’s SOMO. In the type (b) interactions, SHC can be achieved if the energy gap between HOMO and SOMO (ΔE) is lower than the on-site Coulombic repulsion of the SOMO. These aminoxyl radicals with spin-polarised donors such as TTF can be expected to be applied to organic magnetic materials owing to their SHC electronic structures and their ability to generate a high spin state by one-electron oxidation. Particularly, it is expected they will be applied to organic magnetic conductors.[16–19]

|
|
Organometallic Complexes
In 2008, Nishihara et al. reported SHC in a dithiolate complex (tempodt)Pt (Fig. 4a).[3] Tempodt itself does not show SHC, whereas on coordination with the Pt ion containing 4,4′-di-tert-butyl-2,2′-bipyridyl (PttBubpy)2+, it produces SHC. The interactions between the HOMO of tempodt, originating from the π-conjugated dithiolate moiety, and the HOMO of (PttBubpy)2+ increased the HOMO energy of the resulting complex ((tempodt)Pt). This high-lying HOMO caused an unusual electronic configuration in this species. The one-electron oxidation of (tempodt)Pt caused the removal of an electron from the HOMO and gave a corresponding dimer product. Generation of the dimer product was suggested by monitoring changes in the UV-vis spectra and mass spectrometry (MS) measurements. The electron spin resonance (ESR) measurements of the dimer product showed a triplet signal. These results suggested the removal of one electron from the HOMO of (tempodt)Pt and its SOMO−HOMO-converted electronic configuration. Density functional theory (DFT) calculations at the UMPW1PW91/6-31G(d) level for C, H, N, S, and O, and LanL2DZ(Hay–Wadt ECP) for Pt/PCM (CH2Cl2) (PCM = polarisable continuum model) supported SHC in these species. Additionally, Nishihara et al. reported SHC in another dithiolate molecule in 2010 (Fig. 4b).[20] One-electron oxidation of this molecule resulted in a unique cyclisation reaction. When one-electron oxidation was conducted, electrons were removed from their HOMO in the π-conjugated dithiolene moiety. As the generated π radical was close to the nitroxide radical, a unique intramolecular cyclisation was observed, as shown in Fig. 4b. SHC in these species was also supported by DFT calculations at the UB3LYP/6-31G+(d) for C, H, N, S, and O, LanL2DZ for Ni, and MVB60 for Au level of theory.

|
Distonic Radical Anions
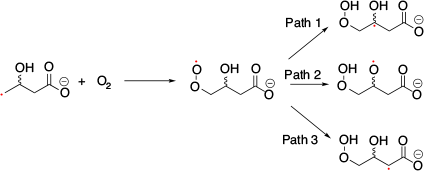
SHC in distonic radical anions was first discovered by Coote et al. in 2013.[21] They observed that distonic radical anions, COO––(CH2)n–COO• and COO––TEMPO-containing aminoxyl radicals exhibited SHC electronic structures on calculation (Fig. 5a). They confirmed SHC in these species with ab initio (i.e. UHF, MP2, and MCSCF) and DFT calculations (i.e. M06-2X). Protonation of these SHC species results in the restoration of normal electronic configuration, which is one of the unique characteristics of these species. Protonation can switch the SHC species into non-SHC species. Additionally, they computed the bond dissociation energy of the distonic radical anions and their protonated species using CH3• as a reference to evaluate the stability of the radical using G3(MP2)-RAD(+), G3(MP2,CC)(+), and G4(MP2)-6X methods. They observed that the bond dissociation energy of the SHC radical anions was smaller than that of the corresponding protonated radical with a non-SHC phenomenon, which suggested the enhancement of radical stability caused by SHC. Another computational study reported by Coote et al. using DFT calculations (i.e. M06-2X/6-31+G(d)) suggested that this unique stability originated from the interactions between the anion and the radical moiety.[22] Additionally, they reported the loss of this stabilisation effect in polar solvents such as water. SHC in distonic radical anions can stabilise the radical by the interaction between anions and radicals, which indicates enhancement of the acidity of the conjugate acid. Coote and group experimentally confirmed the difference in acidity caused by SHC by measuring the gas-phase acidity.[21] Additionally, they suggested that SHC in distonic radical anions can occur in various substrates (Fig. 5b). To confirm the stabilisation of the radical induced by SHC, the change in the bond dissociation energy induced by protonation was investigated. In 2014, Lucarini et al. investigated the difference in bond dissociation energy between SHC and non-SHC species in MeCN and DMSO using the EPR radical equilibration technique (Fig. 6).[23] They obtained the equilibrium constant (Keq) and Gibbs free energy (ΔG) values, but there was no significant difference between the SOMO−HOMO-converted radical and the non-converted radical at 298 K. This result was consistent with the computational study reported by the Coote group, in which the loss of the bond dissociation energy change was induced by SHC in a polar solvent.[22] Additionally, they determined the enthalpy change ΔH (= SHC − non-SHC) and the entropy change ΔS (= SHC − non-SHC) from the experimentally obtained ΔG value in the temperature range of 293−358 K. The values obtained (ΔH = −3.1 kcal mol−1 (1 kcal mol−1 = 4.186 kJ mol−1) and ΔS = 9.13 cal mol−1 K−1) suggested that entropic factors suppressed the ΔG switch. The reactivity of peroxide radicals with SHC configurations was reported by Silva and co-workers in 2020.[24] They studied the reaction of •CH2CH(OH)CH2C(O)O– with O2 using ion-trap MS in the gas phase and suggested a possible reaction mechanism (Fig. 7). Computational studies were conducted to compare the reactivity of the distonic radical anion and neutral radical anion at the M06-2X/aug-cc-pVTZ level of theory. Path 1 was found to have the lowest energy barrier in the distonic radical anion, while Path 2 had the lowest energy barrier in the neutral radical. In the case of the distonic radical anion, Path 1 exhibited a charge-driven reaction originating from the existence of the carboxylate, while in the case of the neutral radical anion, Path 1 exhibited a radical-driven reaction. SHC was related to this difference in the reaction between the SHC peroxide radical and neutral radicals.

|

|
|
Biologically Important Radicals
In 2014, Sevilla et al. reported SHC in one-electron oxidised A−T, G−C base pairs (A•+−T, G•+−C) using theoretical calculations (Fig. 8).[4] The proton-transferred (PT) products of A•+−T and G•+−C did not exhibit SHC at the Uωb97x, UB3LYP or UM06-2x/6-31++G(d) level of theory. Further oxidation of these species was investigated by calculating each species and ionisation potential. Their calculations suggested that the doubly oxidised triplet of A•+−T•+ was preferred for A−T, while the doubly oxidised singlet of G(−H)+−(H+)C was preferred for G−C. This difference in the response of A−T and G−C towards two-electron oxidation suggested that SHC is involved in biological reactions. Additionally, in 2018, they suggested that SHC can occur in several biologically important radicals.[25] They confirmed the occurrence of SHC in 20 diatomic molecules such as O2•– and in the radicals generated from 1-methylcytosine, 2-thiothymine, 2-thiocytosine, 2-selenothymine, phenylselenyl, 8-bromopurine, and 6-azapyrimidine by DFT calculations at the UB3LYP-PCM/6-31++G(d,p) level of theory.

|
Helicene-Based Radicals
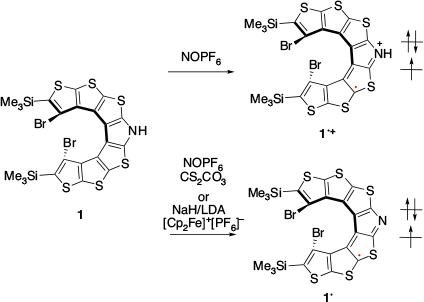
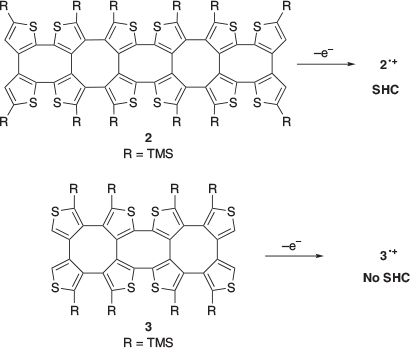
In 2016, Rajca and co-workers reported the first example of SHC in helicene-based radicals.[26] Radical cation 1•+ was prepared by the one-electron oxidation of 1 (Fig. 9). Neutral radical 1• was prepared by proton transfer of 1•+ with a weak base or by the one-electron oxidation of 1–. SHC in both the radicals (1•+ and 1•) was suggested by DFT calculations at the UB3LYP/6-31G(d,p)/PCM(CH2Cl2)+ZPVE level of theory. Additionally, correlation between the cyclic voltammetry (CV) results for 1•+ and the computed difference between the SOMO and HOMO supported SHC. In 2021, SHC in thiophene-based double helices was reported by Rajca et al. (Fig. 10).[27] DFT calculations suggested the presence of SHC in the radical cation 2•+ and a normal electronic configuration in the radical cation 3•+ at the UωB97xD/6-31G(d,p)/PCM (water or DCM) level of theory. 2•+ and 3•+ were experimentally generated, and their half-lives were estimated by EPR measurements. The half-life of 2•+ was nearly 5 min at room temperature, while that of 3•+ was of the order of hours. The radical cation 2•+ exhibited a lower stability than 3•+. Additionally, 2•+ exhibited the presence of SHC whereas 3•+ did not. This difference in stability was explained by the difference in spin density. The spin density of 2•+ was localised on the terminal helicene moiety, whereas that of 3•+ was localised on the central helicene moiety. In other words, the unpaired electron of 3•+ was more sterically protected and showed a longer half-life than that of the SHC radical cation 2•+.
|
|
Chiral Bicarbazole-Based Radicals
SHC in chiral bicarbazole-based radicals was reported by Favereau, Autschbach, and their co-workers in 2020 (Fig. 11).[28] DFT calculations at the PBE0/def2-SV(P)/PCM (CH2Cl2/MeCN) level of theory suggested the occurrence of SHC in the radical cations 4•+ and 5•+. CV measurements on compound 4 showed the first full reversible oxidation. The stability in the first oxidation was unique for non-protected carbazole radicals, and it was derived from the SHC electronic structure. The half-life of the radical cation 4•+ was determined to be 3 h in CH2Cl2 under air. In contrast, compound 5 exhibited the first irreversible oxidation peak. Radical cation 5•+ formed complex oligomers immediately, although it exhibited the presence of SHC. The difference in stability between SOMO−HOMO-converted 4•+ and 5•+ was related to the electronic coupling between the radical centre and the second carbazole fragment. Because the dihedral angle between the two carbazole units of 4•+ was larger than that of 5•+, the interaction between the radical unit and the second carbazole unit was stronger for 5•+ than for 4•+. An attempt to synthesise stable chiral diradicals was made by utilising the unprecedented persistence of 4•+ with SHC. Although 42•2+ generated by further oxidation of 4•+ was highly unstable, by using sterically protected 6•+, a relatively more stable diradical 62•2+ was successfully generated. This 62•2+ diradical showed near-infrared electronic circular dichroism intensity up to 1100 nm and almost degenerate singlet−triplet ground states with weak antiferromagnetic interactions.

|
Triarylmethylradicals
In 2019, Kusamoto, Nishihara, and co-workers reported SHC in TPA-R• and its protonated compound (TPA-[RH]•+) (Fig. 12).[29] The generation of a high-spin state was confirmed in these species. By CV measurements, oxidation peaks of TPA-R• and TPA-[RH]•+ were obtained at 0.38 and 0.44 V (versus ferrocenium/ferrocene (Fc+/Fc)), respectively. This value was closer to the oxidation peak of the precursor of TPA-R• (0.53 V versus Fc+/Fc) compared with that of (3,5-dichloro-4-pyridyl)bis(2,4,6-trichlorophenyl)methyl radical (PyBTM) (0.98 V versus Fc+/Fc). This result suggested that one-electron oxidation occurred from HOMO, not from SOMO, owing to the SOMO−HOMO-converted electronic configuration. SHC in this compound was also supported by DFT calculation at the UB3LYP/6-31G(d) level of theory. In 2019, Li, Bredas, Friend, and their co-workers reported the donor–acceptor radical (D−A•) type SHC material PTM-3NCz and its application to organic optoelectronic devices (OLEDs) (Fig. 12).[30] SHC in PTM-3NCz was supported by DFT calculations at the ωB97XD/6-31+G(d,p) level of theory, CV, and ultraviolet photoelectron spectroscopy (UPS) measurements. The fluorescence intensity half-life for PTM-3NCz was determined to be 9.13 × 106 s in cyclohexane, which was much greater than the half-life of the parent PTM, i.e. 1.02 × 102 s, in the same solvent. The reason for this significantly high photostability was unclear. However, the charge-transfer (CT) nature of the excited state or its SOMO−HOMO-converted electronic configuration were considered possible reasons. PTM-3NCz showed 54 % of the maximum fluorescence quantum yield, and its device showed near-infrared (NIR) emission at 700 nm with a maximum external quantum efficiency of 5.3 %. This suggested the possible application of SHC materials in OLEDs. However, a further experimental attempt to apply SHC species to OLEDs has not been reported yet. As a computational study, the D−A• type SHC species consisting of a substituted triarylamine donor and a perchlorotriphenylmethyl acceptor was suggested at the SRSH LC-ωhPBE/6-31G(d,p) level of theory.[31] In future, many studies will be required to elucidate the relationship between photostability and electronic configuration.

|
Other Radicals
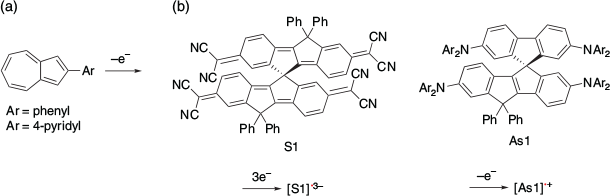
Several other SHC compounds have been reported. For example, SHC in one-electron-oxidised azulene derivatives was suggested using DFT calculations by Mazaki et al. in 2019 (Fig. 13).[32] Another example was reported by Casado, Nakamura, and their co-workers in 2021.[33] They observed that the radical trianion [S1]•3– and the radical cation [As1]•+ exhibited SHC. SHC in these species was also supported by DFT calculations at the UB3LYP/6-31G(d,p) level of theory. Autschbach et al. conducted a computational study of some SHC radicals reported in the past and suggested the design of SHC at the PCM-PBE0/def2-SV(P) level of theory.[34] They calculated the closed-shell compounds formed by adding one electron to the SHC radicals and compared them with the original radicals. They proposed that SHC was likely to occur when the repulsion between the HOMO and other MOs in the parent closed shell compound was weak. This suggested that a small overlap between the SOMO and HOMO orbitals of the radicals is favoured for SHC, which is consistent with previously reported examples. They also suggested that the stabilisation caused by the loss of coulomb repulsion by removing one of the electrons from the HOMO of the parent closed shell species is the key to SHC.
|
Carbenes and Diradicals
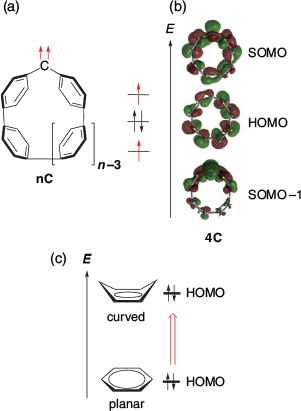
In 2021, SHC in triplet carbenes were reported by Abe, Antol, and their co-workers.[35] The SHC in nC, triplet carbenes embedded in cycloparaphenylene (CPP), and simple triplet diaryl carbenes was confirmed computationally (Figs 14 and 15) at the UB3LYP/6-31G(d) level of theory. CPP is known to have a higher HOMO level with a smaller ring size due to the increase in quinoidal character (Fig. 14c).[36] By calculating triplet carbenes bearing various sizes of CPP with the symmetry constraint method, it was suggested that the high-lying HOMO originating from the CPP moiety contributed significantly to SHC in nC. In fact, while SHC was observed clearly in 4C, 11C, which had a relatively larger ring size and lower HOMO level, showed a normal electronic configuration.
|
|
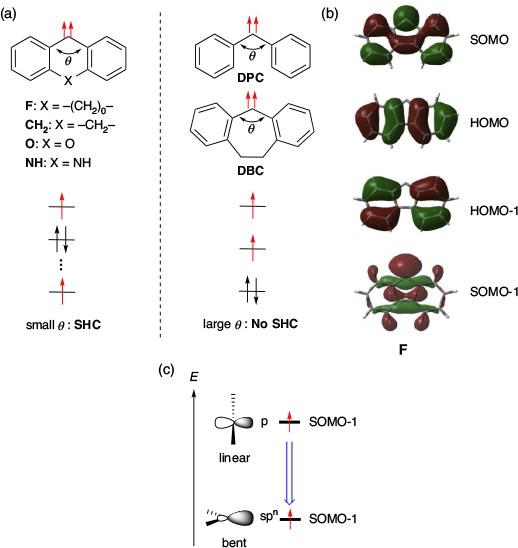
They also investigated the causes of SHC in triplet diaryl carbenes by comparing several carbenes (Fig. 15a, b). Consequently, it was suggested that the increase in s character induced by the small carbene angle was the key cause of SHC (Fig. 15c). For example, the SOMO−1 energy of F, which has a small carbene angle θ (θ = 112°), was very low (−7.16 eV) and exhibited SHC. For the same reason, CH2 (θ = 128°, SOMO−1 = −6.61 eV), O (θ = 125°, SOMO−1 = −6.92 eV), and NH (θ = 127°, SOMO−1 = −6.66 eV) also exhibited SHC. In contrast, diphenyl carbene, DPC (θ = 142°, SOMO−1 = −5.74 eV) and dibenzocycloheptadienylidene, DBC (θ = 142°, SOMO−1 = −6.02 eV) with a large carbene angle, did not show SHC. Thus, the small carbene angle was one of the main factors causing SHC in these diaryl carbenes.
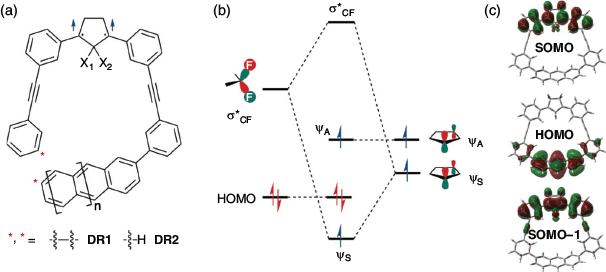
Recently, the Abe group reported computational studies on SHC in cyclopentane-1,3-diyl diradicals DR1F1 (X1 = X2 = F, n = 1) and DR1HF1 (X1 = H, X2 = F, n = 1) at the UB3LYP, UωB97xD or UM06–2X/6-31G(d) level of theory.[37] Moreover, the dihydro-substituted DR1H1 (X1 = X2 = H, n = 1), naphthyl-substituted DR1H0 (X1 = X2 = H, n = 0), and DR1F0 (X1 = X2 = F, n = 0) showed similar electronic configurations, which indicated that the stabilisation of SOMO–1 (ψS) via a bond interaction[38] caused by the fluoro substituents at the C2 position of the cyclopentane unit and the anthracyl moiety with a high HOMO energy level were the key causes for SHC (Fig. 16). The HOMO is mainly located at the anthracyl unit, without interaction with the cyclopentane unit (SOMO domain), which made it possible to modify the SOMO−HOMO order by molecular design. The one-electron oxidised cation DR1F1+ was an SHC radical species whose singly occupied SOMO was lower-lying than the doubly occupied HOMO. In contrast, the dication DR1F12+ was found to be a triplet diradical following the Aufbau principle.
|
The non-macrocyclic diradical DR2F1 (X1 = X2 = F, n = 1) also exhibited SHC in its triplet state. Although the HOMO energy was 0.05 eV lower than that of DR1F1, it was still 0.29 eV higher than that of SOMO–1 at the UB3LYP/6-31G(d) level of theory. The higher HOMO energy in DR1F1 was caused by the slightly bent anthracyl structure in the macrocyclic compound.
The meta-linkage effect on SHC was confirmed by the computational results for DR3a,b (Fig. 17). In DR3a, the linkage pattern (meta or para) of the anthracyl unit with the phenyl unit did not affect the SHC. Additionally, the 2-(3-anthracylphenyl)ethynyl unit in DR3b, present at the para position of the 1,3-diphenyl unit, did not exhibit SHC owing to destabilisation of SOMO–1 by π-conjugation.

|
Conclusion
In this review article, open-shell compounds with SHC configurations and their chemistry were summarised to demonstrate the importance of SHC molecules in the disciplines of material science and life science. Computational and experimental studies revealed the rational molecular design of SHC species: (1) SOMO and HOMO should not overlap each other; (2) the SOMO should localise; (3) the SOMO energy should be highly stabilised by the effect of heteroatoms and hyperconjugation; (4) the HOMO energy should be highly destabilised by strong electron-donating substituents and curved π-conjugations; (5) the energy gap between the HOMO and SOMO in SHC molecules should be lower than the on-site Coulombic repulsion of the SOMO. There are many cases where SHC is supported by both experimental results and DFT calculations.[3,20,26,28–30] Also, some researchers have studied the functional dependency of SHC in DFT calculations and they reported that the unusual electronic configuration is maintained with various functions.[4,37] Thus, DFT is considered to give reliable results about SHC. Further studies on SHC species may provide opportunities for their application in the new field of materials science, such as magnetic materials and OLEDs.
Data Availability Statement
Data sharing is not applicable as no new data were generated or analysed during this study.
Conflicts of Interest
Manabu Abe is an Associate Editor of the Australian Journal of Chemistry but was blinded from the peer-review process for this paper.
Declaration of Funding
This work was supported by JSPS KAKENHI grant nos 17H03022 (MA), 20K21197 (MA), and 21H01921 (MA). Additionally, it was supported by the International Collaborative Research Program of Institute for Chemical Research, Kyoto University (grant 2020–43).
References
[1] K. Fukui, T. Yonezawa, H. Shingu, J. Chem. Phys. 1952, 20, 722.| Crossref | GoogleScholarGoogle Scholar |
[2] T. Sugimoto, S. Yamaga, M. Nakai, K. Ohmori, M. Tsujii, H. Nakatsuji, H. Fujita, J. Yamauchi, Chem. Lett. 1993, 22, 1361.
| Crossref | GoogleScholarGoogle Scholar |
[3] T. Kusamoto, S. Kume, H. Nishihara, J. Am. Chem. Soc. 2008, 130, 13844.
| Crossref | GoogleScholarGoogle Scholar | 18821762PubMed |
[4] A. Kumar, M. D. Sevilla, J. Phys. Chem. B 2014, 118, 5453.
| Crossref | GoogleScholarGoogle Scholar | 24798145PubMed |
[5] G. Gryn’ova, M. L. Coote, C. Corminboeuf, Wiley Interdiscip. Rev. Comput. Mol. Sci. 2015, 5, 440.
| Crossref | GoogleScholarGoogle Scholar | 27774112PubMed |
[6] R. Kumai, M. M. Matsushita, A. Izuoka, T. Sugawara, J. Am. Chem. Soc. 1994, 116, 4523.
| Crossref | GoogleScholarGoogle Scholar |
[7] R. Kumai, H. Sakurai, A. Izuoka, T. Sugawara, Mol. Cryst. Liq. Cryst. Sci. Technol. Sect. A 1996, 279, 133.
| Crossref | GoogleScholarGoogle Scholar |
[8] J. Nakazaki, M. M. Matsushita, A. Izuoka, T. Sugawara, Tetrahedron Lett. 1999, 40, 5027.
| Crossref | GoogleScholarGoogle Scholar |
[9] H. Sakurai, A. Izuoka, T. Sugawara, J. Am. Chem. Soc. 2000, 122, 9723.
| Crossref | GoogleScholarGoogle Scholar |
[10] J. Nakazaki, Y. Ishikawa, A. Izuoka, T. Sugawara, Y. Kawada, Chem. Phys. Lett. 2000, 319, 385.
| Crossref | GoogleScholarGoogle Scholar |
[11] A. Izuoka, M. Hiraishi, T. Abe, T. Sugawara, K. Sato, T. Takui, J. Am. Chem. Soc. 2000, 122, 3234.
| Crossref | GoogleScholarGoogle Scholar |
[12] J. Nakazaki, I. G. Chung, M. M. Matsushita, T. Sugawara, R. Watanabe, A. Izuoka, Y. Kawada, J. Mater. Chem. 2003, 13, 1011.
| Crossref | GoogleScholarGoogle Scholar |
[13] S. Hiraoka, T. Okamoto, M. Kozaki, D. Shiomi, K. Sato, T. Takui, K. Okada, J. Am. Chem. Soc. 2004, 126, 58.
| Crossref | GoogleScholarGoogle Scholar | 14709058PubMed |
[14] M. M. Matsushita, H. Kawakami, Y. Kawada, T. Sugawara, Chem. Lett. 2007, 36, 110.
| Crossref | GoogleScholarGoogle Scholar |
[15] H. Komatsu, R. Mogi, M. M. Matsushita, T. Miyagi, Y. Kawada, T. Sugawara, Polyhedron 2009, 28, 1996.
| Crossref | GoogleScholarGoogle Scholar |
[16] M. M. Matsushita, H. Kawakami, T. Sugawara, M. Ogata, Phys. Rev. B Condens. Matter Mater. Phys. 2008, 77, 1.
| Crossref | GoogleScholarGoogle Scholar |
[17] T. Sugawara, M. M. Matsushita, J. Mater. Chem. 2009, 19, 1738.
| Crossref | GoogleScholarGoogle Scholar |
[18] H. Komatsu, M. M. Matsushita, S. Yamamura, Y. Sugawara, K. Suzuki, T. Sugawara, J. Am. Chem. Soc. 2010, 132, 4528.
| Crossref | GoogleScholarGoogle Scholar | 20222711PubMed |
[19] T. Sugawara, H. Komatsu, K. Suzuki, Chem. Soc. Rev. 2011, 40, 3105.
| Crossref | GoogleScholarGoogle Scholar | 21409235PubMed |
[20] T. Kusamoto, S. Kume, H. Nishihara, Angew. Chem. Int. Ed. 2010, 49, 529.
| Crossref | GoogleScholarGoogle Scholar |
[21] G. Gryn’ova, D. L. Marshall, S. J. Blanksby, M. L. Coote, Nat. Chem. 2013, 5, 474.
| Crossref | GoogleScholarGoogle Scholar | 23695628PubMed |
[22] G. Gryn’ova, M. L. Coote, J. Am. Chem. Soc. 2013, 135, 15392.
| Crossref | GoogleScholarGoogle Scholar | 24090128PubMed |
[23] P. Franchi, E. Mezzina, M. Lucarini, J. Am. Chem. Soc. 2014, 136, 1250.
| Crossref | GoogleScholarGoogle Scholar | 24410365PubMed |
[24] S. So, B. B. Kirk, U. Wille, A. J. Trevitt, S. J. Blanksby, G. Da Silva, Phys. Chem. Chem. Phys. 2020, 22, 2130.
| Crossref | GoogleScholarGoogle Scholar | 31912066PubMed |
[25] A. Kumar, M. D. Sevilla, J. Phys. Chem. B 2018, 122, 98.
| Crossref | GoogleScholarGoogle Scholar | 29240424PubMed |
[26] Y. Wang, H. Zhang, M. Pink, A. Olankitwanit, S. Rajca, A. Rajca, J. Am. Chem. Soc. 2016, 138, 7298.
| Crossref | GoogleScholarGoogle Scholar | 27219299PubMed |
[27] A. Rajca, C. Shu, H. Zhang, S. Zhang, H. Wang, S. Rajca, Photochem. Photobiol. 2021,
| Crossref | GoogleScholarGoogle Scholar | 34152605PubMed |
[28] S. Kasemthaveechok, L. Abella, M. Jean, M. Cordier, T. Roisnel, N. Vanthuyne, T. Guizouarn, O. Cador, J. Autschbach, J. Crassous, L. Favereau, J. Am. Chem. Soc. 2020, 142, 20409.
| Crossref | GoogleScholarGoogle Scholar |
[29] A. Tanushi, S. Kimura, T. Kusamoto, M. Tominaga, Y. Kitagawa, M. Nakano, H. Nishihara, J. Phys. Chem. C 2019, 123, 4417.
| Crossref | GoogleScholarGoogle Scholar |
[30] H. Guo, Q. Peng, X. Chen, Q. Gu, S. Dong, E. W. Evans, A. J. Gillett, X. Ai, M. Zhang, D. Credgington, V. Coropceanu, R. H. Friend, J. Brédas, F. Li, Nat. Mater. 2019, 18, 977.
| Crossref | GoogleScholarGoogle Scholar | 31332338PubMed |
[31] E. Cho, V. Coropceanu, J. L. Brédas, J. Am. Chem. Soc. 2020, 142, 17782.
| Crossref | GoogleScholarGoogle Scholar | 32997939PubMed |
[32] T. Tsuchiya, Y. Katsuoka, K. Yoza, H. Sato, Y. Mazaki, ChemPlusChem 2019, 84, 1659.
| Crossref | GoogleScholarGoogle Scholar | 31943874PubMed |
[33] S. Medina Rivero, R. Shang, H. Hamada, Q. Yan, H. Tsuji, E. Nakamura, J. Casado, Bull. Chem. Soc. Jpn. 2021, 94, 989.
| Crossref | GoogleScholarGoogle Scholar |
[34] L. Abella, J. Crassous, L. Favereau, J. Autschbach, Chem. Mater. 2021, 33, 3678.
| Crossref | GoogleScholarGoogle Scholar |
[35] R. Murata, Z. Wang, Y. Miyazawa, I. Antol, S. Yamago, M. Abe, Org. Lett. 2021, 23, 4955.
| Crossref | GoogleScholarGoogle Scholar | 34048267PubMed |
[36] T. Iwamoto, Y. Watanabe, Y. Sakamoto, T. Suzuki, S. Yamago, J. Am. Chem. Soc. 2011, 133, 8354.
| Crossref | GoogleScholarGoogle Scholar | 21542589PubMed |
[37] Z. Wang, R. Murata, M. Abe, ACS Omega 2021, 6, 22773.
| Crossref | GoogleScholarGoogle Scholar | 34514248PubMed |
[38] (a) M. Abe, J. Ye, M. Mishima, Chem. Soc. Rev. 2012, 41, 3808.
| Crossref | GoogleScholarGoogle Scholar | 22377794PubMed |
(b) Z. Wang, R. Akisaka, S. Yabumoto, T. Nakagawa, S. Hatano, M. Abe, Chem. Sci. 2021, 12, 613.
| Crossref | GoogleScholarGoogle Scholar |
(c) M. Abe, H. Furunaga, D. Ma, L. Gagliardi, G. J. Bodwell, J. Org. Chem. 2012, 77, 7612.
| Crossref | GoogleScholarGoogle Scholar |
(d) T. Nakamura, A. Takegami, M. Abe, J. Org. Chem. 2010, 75, 1956.
| Crossref | GoogleScholarGoogle Scholar |
(e) M. Abe, S. Kawanami, C. Ishihara, M. Nojima, J. Org. Chem. 2004, 69, 5622.
| Crossref | GoogleScholarGoogle Scholar |
(f) M. Abe, C. Ishihara, A. Tagegami, J. Org. Chem. 2004, 69, 7250.
| Crossref | GoogleScholarGoogle Scholar |
(g) M. Abe, Chem. Rev. 2013, 113, 7011.
| Crossref | GoogleScholarGoogle Scholar |