

Conjugation Approaches for Peptide-Mediated Delivery of Oligonucleotides Therapeutics
Nitin A. Patil A
A
A Biomedicine Discovery Institute, Monash University, Clayton, Vic. 3800, Australia. Email: nitin.patil@monash.edu

Dr Nitin Patil is an Australian National Health and Medical Research Council's Peter Doherty Fellow at Monash University. He leads the chemistry team in the laboratory of Antimicrobial System Pharmacology. The overarching aim of his research is to address challenges of drug-resistant pathogenic bacteria by developing peptide and oligonucleotide-based therapeutics. |
Australian Journal of Chemistry 75(2) 24-33 https://doi.org/10.1071/CH21131
Submitted: 31 May 2021 Accepted: 12 August 2021 Published: 7 September 2021
Journal Compilation © CSIRO 2022 Open Access CC BY
Abstract
Oligonucleotide-based agents are versatile biomolecules that modulate gene expression. The last decade has seen the emergence of oligonucleotide-based tools for biochemical investigations. Importantly, several oligonucleotide-based drugs and vaccines are currently used for various therapeutic applications ranging from anti-inflammatory and anti-viral agents to those used in cardiovascular, ophthalmic, and neuro-muscular disorders. Despite a broad range of applications, achieving efficient oligonucleotide delivery remains a major limitation. A possible solution is to conjugate cell-penetrating peptides with oligonucleotides. This review provides an overview of chemical strategies used to synthesise peptide–oligonucleotide conjugates. The merits and liabilities of these strategies are discussed in the context of synthetic efficiency, and bio-reversible and -irreversible linkages.
Keywords: conjugation, oligonucleotide, peptide, peptide nucleic acids, drug delivery, thiol–maleimide conjugation, click chemistry, 2-cyanoisoicotinamide (CINA) linkage.
Introduction
Oligonucleotides are typically short oligomers (12–24 monomers) of natural or chemically modified nucleotide analogues that bind and modulate the function of their cognate mRNA or DNA sequence to produce a desired pharmacological effect.[1] Over the last decade, several oligonucleotide-based tools for therapeutics, diagnostics, and biochemical investigations have emerged.[2] The increasing impact of oligonucleotides on overall healthcare is demonstrated by the fact that over last two decades, 10 oligonucleotides have received approval from the United States Food and Drug Administration (USFDA). Additionally, over 80 oligonucleotide candidates are currently in clinical trials for various diagnostic and therapeutic applications, including those with anti-inflammatory and anti-viral activities and for cardiovascular, ophthalmic, and neuromuscular disorders.[2–4]
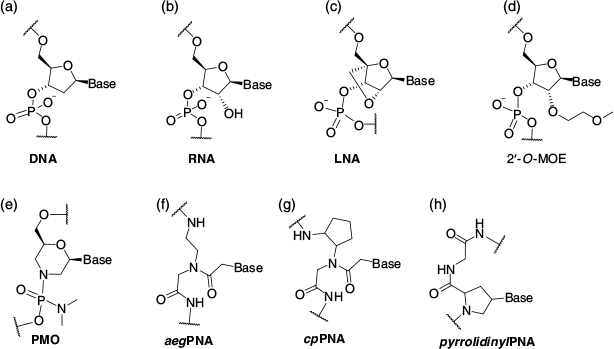
The phosphodiester linkages in the native oligonucleotides, namely DNA (Fig. 1a) and RNA (Fig. 1b), are susceptible to enzymatic degradation in the cellular environment. Therefore, to enhance biological stability and effectiveness, a large number of chemically modified oligonucleotides have been developed including locked nucleic acid (LNA, Fig. 1c),[5] 2′-O-(methoxyethyl) oligonucleotides (2′-O-MOE, Fig. 1d),[6] phosphorodiamidate morpholino oligonucleotides (PMO, Fig. 1e),[7,8] N-(2-aminoethyl)glycyl peptide nucleic acids (aegPNA, Fig. 1f),[9,10] cyclopentyl PNA (cpPNA, Fig. 1g), and pyrrolidinyl peptide nucleic acids (pyrrolidinylPNA, Fig. 1h).[11] More recently, a new generation of oligonucleotides that exhibit improved selectivity towards RNA over DNA have been developed including Nusinersen (belonging to the 2′-O-MOE class)[7] and Eteplirsen[6] (belonging to the PMO class).
|
The current overriding challenge for oligonucleotide therapeutics is to efficiently deliver active oligonucleotides to their target. An enormous amount of work in this area has been undertaken over recent years that has resulted in the development of delivery systems based on lipid nanoparticles,[12–14] polymeric nanoparticles,[15–18] small molecules,[19–21] and peptides.[22–24] Particularly, peptide-based delivery systems have been widely used due to their safety, cell specificity, and efficient membrane permeability.[25–27] Current reviews of the utility of the various peptides developed have focussed primarily on their cell-penetrating ability and target selectivity.[18,28] However, an important aspect of an ideal conjugate is the ability to form bio-reversible and -irreversible linkages between a drug molecule and delivery vehicle to achieve optimum stability and biological activity. Given chemical strategies can be effectively used to obtain conjugates that are either bio-reversible (i.e. release free oligonucleotides within the target cell) or irreversible (i.e. remain bound within the target cell) (Fig. 2),[14,25,29,30] conjugation chemistry has a major role to play in the development of efficient peptide–oligonucleotide conjugates. This review focuses specifically on the various chemical approaches used to form oligonucleotide and peptide conjugates.

|
Amide Linkage
The most explored strategy for conjugation of a peptide with an oligonucleotide is through an amide linkage. Importantly, this was the first conjugation strategy investigated to deliver PNA-based oligonucleotides.[31–33] As for peptides, PNAs utilise standard solid-phase peptide synthesis (SPPS) protocols that allow a facile amide linkage to be formed between the peptide and PNA. Therefore, a small delivery peptide sequence (≤15 amino acids) can be synthesised on the same solid support after completion of the PNA sequence (Fig. 3a).[34,35] Winkler and co-workers developed an on-resin phase transfer reaction to form an amide link between peptides and non-PNA oligonucleotides (Fig. 3b).[36] This advanced SPPS strategy employs a benzyl-protected 2-amino-2′-deoxyuridinenucleotide to provide a site for peptide attachment. The orthogonal removal of the benzyl group is achieved by phase-transfer hydrogenation using poly-vinylpyrrolidone (PVP)-stabilised palladium nanoparticles, thereby enabling on-resin conjugation of the peptide through an amide bond.[37,38] Adventitiously, this strategy allows conjugation of a peptide at any position of an oligonucleotide sequence and has been successfully employed to obtain peptide conjugates of many non-PNA oligonucleotides, including PMO.[36–38] On-resin strategies are suitable only for small peptides (<15 amino acids). If larger or more structurally complex peptides are to be used, amide conjugation can be performed in solution using standard coupling conditions (Fig. 3c).[39,40]

|
While peptide–oligonucleotide conjugation via an amide linkage is beneficial in developing bio-irreversible peptide-based delivery of oligonucleotides, formation of the amide bond by in situ activation through succinyl ester or 1-hydroxybenzotriazole (HOBt) esters requires the use of short (≤15 amino acids) and protected peptides. Moreover, protected peptides often form aggregates and are insoluble in aqueous solution. Therefore, formation of an amide bond when using a protected peptide presents several challenges.[41] An alternative approach involves the conjugation of unprotected peptides to oligonucleotides using native chemical ligation (NCL).[42–49] NCL utilises a chemoselective S–N acyl transfer to form an amide linkage.[50] Initially, an unprotected peptide with an activated C-terminal thioester forms a trans-thioester with the N-terminal thiol of the ligating oligonucleotide.[41] The free N-terminal amine then attacks the carbonyl group of the trans-thioester intermediate to form an amide bond. Given this elegant strategy is selective for the N-terminal thiol and employs an unprotected peptide in aqueous buffers, it provides significant advantages over protected peptide conjugation.
Joyce and co-workers employed a template-directed NCL to accelerate conjugation between the delivery peptide and an oligonucleotide (Fig. 4a).[42] In this approach, the activated delivery peptide thioester forms an intermediate trans-thioester with the thiol of PNA 1. The intermediate trans-thioester then binds to the complementary DNA (cDNA) template, allowing access to the free amine of PNA 2 with subsequent rapid amide bond formation. Further enhancement of the NCL strategy to form peptide–oligonucleotides conjugates without cDNA was successfully achieved using cysteine-functionalised oligonucleotides (Fig. 4b).[46,48] With this approach, the 5′-ends of DNA oligonucleotides were modified with a cysteine residue, which allows efficient S–N acyl transfer owing to the close proximity of both a thiol and an amino residue. Additionally, an S-benzyl succinyl moiety at the site of ligation was used to generate N-terminal peptide thioesters. At pH 7, the N-terminal peptide thioesters and 5′-cysteinyl oligonucleotides are then ligated in the presence of tris(2-carboxyethyl)phosphine (TCEP) and thiophenol to form the peptide–oligonucleotide conjugate.[48] A similar approach led to the introduction of a new generation γ-modified PNA with a 5′-end 1,2-aminothiol handle to facilitate NCL of peptide-PNA (Fig. 4c).[45,51] Recently, Sayers et al. described an ‘Se-mediated ligation’ (selenocysteine–seleonoester) approach for the synthesis of peptide-PNA hybrids that is faster than NCL (cysteine–thioester) and allows the rapid detection of microRNA (miRNA).[47]

|
Disulfide Linkage
Disulfide bonds are unique covalent bonds that are stable for mass spectroscopic (MS) characterisation, storage, and handling, but are cleaved in the reductive intracellular environments of the cytoplasm or endosomes. Therefore, disulfide conjugates have been employed to form sufficiently stable but reversible linkages that are cleaved after successful cellular uptake.[52] Disulfide-linked conjugation can be performed through the introduction of thiol-linkers or cysteine residues into the peptide or oligonucleotide sequence during the chemical synthesis process.[53–56] The introduction of a free thiol group in non-PNA oligonucleotides can be performed using O-2-cyanoethyl-N,N-diisopropyl phosphoramidite-mediated coupling of 6-mercaptohexanol at the 5′-end of the oligonucleotides.[57–59] Another advantage of disulfide conjugates is that disulfide bond formation can be achieved by reacting two free thiol groups in aqueous conditions at physiological pH (Fig. 5a). In order to accelerate disulfide formation, in situ activation using 2,2′-dipyridyldithiol (DPDS) can be employed.[60] However, the major drawback of this strategy is the formation of homodimeric by-products. To minimise homodimer formation, one of the participating thiol groups must be pre-protected by a S-pyridylthiol N-terminal group (Fig. 5b).[56]

|
Thioether, Thiol–Maleimide, and Thiol–Aryl Linkages
Reactivity of the thiol group is not limited to the formation of disulfide bridges. Given the susceptibility of the disulfide to reducing conditions may be undesirable, especially when the oligonucleotide target is located intracellularly, the thioether linkage is a widely used strategy where non-reducible or non-reversible conjugates are required for oligonucleotide delivery.[61,62] This strategy employs thiol-mediated nucleophilic substitution of haloacetamides to link the peptides and oligonucleotides (Fig. 6).[63] One of the conjugating partners contains either a free thiol or S-pyridylthiol-protected thiol, while the counterpart possess a haloacetyl handle. Thioether formation can be performed in either an organic solvent or aqueous buffer. The reaction kinetics of thioether formation are slow, and undesired disulfide formation may result in homodimeric products.[64] The formation of a disulfide homodimer can be avoided by employing S-pyridylthiol-protected thiol peptides or oligonucleotides. However, the S-pyridylthiol-protected peptide approach requires additional steps to obtain the conjugating precursors. The haloacetyl peptides or oligonucleotides are susceptible to hydrolysis and require careful handling during the conjugate synthesis.[64]

|
The thiol–maleimide linkage is an advanced strategy that results in the relatively rapid formation of a thioether linkage.[65] A maleimide moiety can be easily introduced into the oligonucleotide or peptide sequences using succinyl spacers.[49,66] The maleimide moiety acts as a Michael acceptor and rapidly reacts with a free thiol group to form the thiol–maleimide linkage.[57,67] Given this linkage can be formed in an aqueous buffer without a catalyst, the thiol–maleimide linkage has become the most commonly used linkage with which to synthesise peptide–oligonucleotide conjugates (Fig. 7).[63,68–70]

|
Nucleophilicity of the thiol group also plays an important role in the formation of thiol–maleimide conjugates. Highly reactive thiols can often form disulfide bonds that limit the availability of the free thiol to form thiol–maleimide linkages. Moreover, competing nucleophiles may also undergo a Michael addition reaction with the maleimide moiety to form an undesired covalently linked conjugate. Importantly, the formation of a thiol–maleimide linkage introduces an additional stereocentre that results in diastereoisomeric products.[71] Despite faster reaction kinetics, compatibility with aqueous conditions, and improved stability compared with disulfide linkages, the thiol–maleimide linkage is still susceptible to in vivo reduction that results in the premature release of oligonucleotides from delivery peptides.[64,69] This can be overcome through the use of a retro-Michael ring-opening of the thiol–maleimide linkage.[72,73]
Recently, Jbara et al. employed a thioaryl ether linkage to conjugate a DNA oligonucleotide with a cysteine-containing peptide.[74] This strategy utilises an elegant bifunctional conjugation handle with N-hydroxysuccinimide (NHS) and palladium(ii)-oxidative addition complex (OAC) moieties for site-selective conjugation (Fig. 8), allowing faster kinetics compared with thiol–maleimide linkages while producing conjugates that are significantly more stable.

|
Cyclohexene Linkage
Seelig et al. reported a peptide oligonucleotide conjugation that uses a Diels–Alder reaction between a diene and a dienophile.[75] This reaction employs modified peptides with a dienophile handle to form a cyclohexene linkage with 5′-modified oligonucleotides.[76] However, the use of 1,3-diene only allows modification at the 5′-end of oligonucleotides, limiting its applicability. Subsequently, diene-modified nucleotides were employed to enable the formation of a Diels–Alder cyclohexene linkage at positions other than the non-terminal position within oligonucleotides (Fig. 9).[77,78] While the Diels–Alder cycloaddition reaction can be performed in aqueous conditions and allows the straightforward synthesis of large peptide–oligonucleotide conjugates, it nevertheless exhibits slow reaction kinetics and utilises a maleimide moiety as the dienophile, which may lead to undesired products.

|
Oxime Linkage
Oxime linkages utilise highly reactive functional groups, namely aldehydes, ketones, and hydroxylamines. Dumy and co-workers were the first to explore the oxime conjugation strategy for chemoselectively linking peptides with oligonucleotides.[79–81] An oxime linkage can be formed by treating an aldehyde-containing oligonucleotide with an aminooxy functionalised peptide. Specially, an aldehyde function can be generated at both the 3′- and 5′-ends of oligonucleotides, which allows the bifunctionalisation of oligonucleotides.[80,81] However, the initial strategy developed by Dumy and co-workers involved the generation of aldehyde oligonucleotides through the glyoxylic moiety (Fig. 10a) and is not compatible with basic pH conditions. Subsequently, further development of oxime linkage employed a relatively stable keto-oxime linkage to obtain peptide–oligonucleotide conjugates under basic pH conditions (Fig. 10b).[82,83] However, the highly reactive amino-oxy functional group requires careful handling of oligonucleotides or peptides, thereby limiting the broad applicability of oxime linkage to form peptide–oligonucleotide conjugates.

|
Hydrazone Linkage
Ollivier et al. employed reactive aldehyde oligonucleotides with N-terminal hydrazide peptides to form hydrazone linkages (Fig. 11).[84] The N-terminal glyoxylyl handle was introduced using (+)-diacetyl-l-tartaric anhydride-mediated acetylation of a DNA oligonucleotide. The non-thiol-based hydrazone ligation chemistry is complementary to the use of cysteine-containing peptides.[84] However, glyoxylyl and hydrazide groups are unstable during the synthesis and purification processes. Therefore, despite orthogonality, the hydrazone linkage has limited applications in peptide–oligonucleotide conjugations.

|
Urea and Carbonyl Linkage
Fujii and co-workers developed a peptide–oligonucleotide conjugation process involving sequential fragment condensation on a solid support to establish urea or a carbonyl linkages.[85,86] In this approach, an α,ω-diisocyanatoalkane or a carbonyl diimidazole (CDI) is initially allowed to react with a resin-bound oligonucleotide containing a free amino group. Subsequently, the protected peptide fragment containing a free amino group reacts with the carbonyl imidazole or isocyanatoalkane moiety present on resin-bound oligonucleotide (Fig. 12).[87] Urea and carbonyl linkages require organic solvents and are only compatible with protected peptides and oligonucleotides. However, the resulting oligonucleotide–peptide conjugate is non-reversible and stable to intracellular enzymatic degradation.[87]

|
Triazole Linkage
The elegant biorthogonal alkyne and azide cycloaddition reaction (click reaction) has been a key strategy for conjugating peptides to oligonucleotides (Fig. 13).[88] The selective nature of ‘click conjugation’ results in a stable, non-bioreversible 1,2,3-triazole link with high yields. Importantly, alkyne and azide residues are commercially available and can be easily introduced during solid-phase synthesis of peptides and oligonucleotides. Notably, the copper-free click conjugation reaction utilises aqueous buffers at room temperature.[89–92] Additionally, the mild reaction conditions and orthogonality of the copper(i)-catalysed azide–alkyne cycloaddition (CuAAC) reaction are extremely useful in the synthesis of peptide–oligonucleotide conjugates (Fig. 13).[88] Triazole linkage has been widely used in non-bioreversible peptide–oligonucleotide conjugates.

|
Thiazolidine and Cyanobenzothiazole Linkage
Thiazolidine and cyanobenzothiazole linkages are highly selective for 1,2-aminothiol moieties such as N-terminal cysteine residues (Fig. 14). Thiazolidine conjugation utilises an aldehyde and cysteine residue to form peptide–oligonucleotide conjugates (Fig. 14a).[79,93] However, the utility of the thiazolidine linkage to synthesise versatile peptide–oligonucleotide conjugates is limited by the cross-reactivity of aldehydes with other nucleophiles, and instability at basic pH. We recently developed a cysteine–cyanobenzothiazole (Cys-CBT) conjugation strategy that is highly selective for N-terminal cysteine residues.[64] The cysteine–cyanobenzothiazole linkage employs a luciferin-derived 2-cyanobenzothiazole residue that reacts explicitly with an N-terminal cysteine residue (Fig. 14b). This elegant reaction involves fast reaction kinetics, is compatible with both organic and aqueous buffers, and has high selectivity towards N-terminal cysteine residues.

|
Conclusion and Outlook
While chemical efficiency is an important aspect of peptide–oligonucleotide conjugation, the site of release and stability to enzymatic degradation must be considered when determining an appropriate linkage strategy. The above-mentioned peptide–oligonucleotide linkages each have their own advantages and disadvantages that must be considered depending on the particular circumstances. Whereas the amide linkage approach provides significant enzymatic stability, it often results in low yields and is limited to small peptides (<15 amino acids). Stepwise peptide–oligonucleotide conjugation through SPPS provides high yields and often requires one purification step, although it is limited to short peptide–oligonucleotide conjugates and requires the use of organic solvents. Disulfide, triazole, thiol–maleimide, and Cys-CBT conjugation can be performed in aqueous conditions that are often advantageous for conjugate unprotected or complex peptides with oligonucleotides. Importantly, these conjugation strategies can also be employed and optimised to form bioreversible (e.g. disulfide and ester) and non-bioreversible (e.g. thioether, alkyne-azide, and Cys-CBT) linkages. The intracellular redox-cleavable disulfide linkages are advantageous for the release of the oligonucleotides and therefore achieving the desired biological effect.[64,94] Recently, other linkages such as cysteine–cyanoisonicotinic acid (CINA) conjugation have been successfully employed for peptide–protein or peptide–small molecule conjugation.[95] While the use of CINA conjugation to obtain peptide–oligonucleotide conjugates is yet to be explored, the increasing demand for and versatile applications of oligonucleotides will no doubt ensure further exploration of CINA conjugation in the near future.
Data Availability Statement
Data sharing is not applicable as no new data were generated or analysed during this study.
Conflicts of Interest
The authors declare no conflicts of interest.
Declaration of Funding
Dr Patil was supported by the Australian National Health and Medical Research Council’s Early Career Fellowship (Grant ID: APP1158171).
Acknowledgements
The author thanks Professor Jian Li, Dr Julien Tailhades, and Dr Phillip Bergen for the critical view of the manuscript.
References
[1] C. F. Bennett, E. E. Swayze, Annu. Rev. Pharmacol. Toxicol. 2010, 50, 259.| Crossref | GoogleScholarGoogle Scholar | 20055705PubMed |
[2] D. P. Potaczek, H. Garn, S. D. Unger, H. Renz, J. Allergy Clin. Immunol. 2016, 137, 1334.
| Crossref | GoogleScholarGoogle Scholar | 27155029PubMed |
[3] T. C. Roberts, R. Langer, M. J. A. Wood, Nat. Rev. Drug Discov. 2020, 19, 673.
| Crossref | GoogleScholarGoogle Scholar | 32782413PubMed |
[4] R. Gambari, Expert Opin. Ther. Pat. 2014, 24, 267.
| Crossref | GoogleScholarGoogle Scholar | 24405414PubMed |
[5] A. A. Koshkin, P. Nielsen, M. Meldgaard, V. K. Rajwanshi, S. K. Singh, J. Wengel, J. Am. Chem. Soc. 1998, 120, 13252.
| Crossref | GoogleScholarGoogle Scholar |
[6] R. S. Finkel, C. A. Chiriboga, J. Vajsar, J. W. Day, J. Montes, D. C. De Vivo, M. Yamashita, F. Rigo, G. Hung, E. Schneider, D. A. Norris, S. Xia, C. F. Bennett, K. M. Bishop, Lancet 2016, 388, 3017.
| Crossref | GoogleScholarGoogle Scholar | 27939059PubMed |
[7] J. J. Dowling, Nat. Rev. Neurol. 2016, 12, 675.
| Crossref | GoogleScholarGoogle Scholar | 27857122PubMed |
[8] J. Summerton, D. Weller, Antisense Nucleic Acid Drug Dev. 1997, 7, 187.
| Crossref | GoogleScholarGoogle Scholar | 9212909PubMed |
[9] A. Porcheddu, G. Giacomelli, Curr. Med. Chem. 2005, 12, 2561.
| Crossref | GoogleScholarGoogle Scholar | 16248816PubMed |
[10] C. Sharma, S. K. Awasthi, Chem. Biol. Drug Des. 2017, 89, 16.
| 27490868PubMed |
[11] T. H. S. Tan, D. T. Hickman, J. Morral, I. G. Beadham, J. Micklefield, Chem. Commun. 2004, 516.
| Crossref | GoogleScholarGoogle Scholar |
[12] L. Huang, Y. Liu, Annu. Rev. Biomed. Eng. 2011, 13, 507.
| Crossref | GoogleScholarGoogle Scholar | 21639780PubMed |
[13] H. Yin, R. L. Kanasty, A. A. Eltoukhy, A. J. Vegas, J. R. Dorkin, D. G. Anderson, Nat. Rev. Genet. 2014, 15, 541.
| Crossref | GoogleScholarGoogle Scholar | 25022906PubMed |
[14] K. Lu, Q.-P. Duan, L. Ma, D.-X. Zhao, Bioconjug. Chem. 2010, 21, 187.
| Crossref | GoogleScholarGoogle Scholar | 19856957PubMed |
[15] M. E. Davis, J. E. Zuckerman, C. H. J. Choi, D. Seligson, A. Tolcher, C. A. Alabi, Y. Yen, J. D. Heidel, A. Ribas, Nature 2010, 464, 1067.
| Crossref | GoogleScholarGoogle Scholar | 20305636PubMed |
[16] O. M. Merkel, T. Kissel, J. Control. Release 2014, 190, 415.
| Crossref | GoogleScholarGoogle Scholar | 24933599PubMed |
[17] D. W. Pack, A. S. Hoffman, S. Pun, P. S. Stayton, Nat. Rev. Drug Discov. 2005, 4, 581.
| Crossref | GoogleScholarGoogle Scholar | 16052241PubMed |
[18] T. Lehto, E. Wagner, Nanomedicine 2014, 9, 2843.
| Crossref | GoogleScholarGoogle Scholar | 25535686PubMed |
[19] K. Phelps, A. Morris, P. A. Beal, ACS Chem. Biol. 2012, 7, 100.
| Crossref | GoogleScholarGoogle Scholar | 22148740PubMed |
[20] R. L. Juliano, X. Ming, O. Nakagawa, Acc. Chem. Res. 2012, 45, 1067.
| Crossref | GoogleScholarGoogle Scholar | 22353142PubMed |
[21] J. Winkler, Ther. Deliv. 2013, 4, 791.
| Crossref | GoogleScholarGoogle Scholar | 23883124PubMed |
[22] T. Coursindel, P. Järver, M. J. Gait, Nucleic Acid Ther. 2012, 22, 71.
| Crossref | GoogleScholarGoogle Scholar | 22409235PubMed |
[23] H. M. Moulton, J. D. Moulton, Biomembranes 2010, 1798, 2296.
| Crossref | GoogleScholarGoogle Scholar |
[24] U. Lächelt, E. Wagner, Chem. Rev. 2015, 115, 11043.
| Crossref | GoogleScholarGoogle Scholar | 25872804PubMed |
[25] R. L. Juliano, Nucleic Acids Res. 2016, 44, 6518.
| Crossref | GoogleScholarGoogle Scholar | 27084936PubMed |
[26] R. Juliano, M. R. Alam, V. Dixit, H. Kang, Nucleic Acids Res. 2008, 36, 4158.
| Crossref | GoogleScholarGoogle Scholar | 18558618PubMed |
[27] S. Benizri, A. Gissot, A. Martin, B. Vialet, M. W. Grinstaff, P. Barthélémy, Bioconjug. Chem. 2019, 30, 366.
| Crossref | GoogleScholarGoogle Scholar | 30608140PubMed |
[28] T. Lehto, K. Ezzat, M. J. A. Wood, S. El Andaloussi, Adv. Drug Deliv. Rev. 2016, 106, 172.
| Crossref | GoogleScholarGoogle Scholar | 27349594PubMed |
[29] F. Muttach, A. Rentmeister, Methods 2016, 107, 3.
| Crossref | GoogleScholarGoogle Scholar | 26884260PubMed |
[30] P. L. Patel, N. K. Rana, M. R. Patel, S. D. Kozuch, D. Sabatino, ChemMedChem 2016, 11, 252.
| Crossref | GoogleScholarGoogle Scholar | 26663095PubMed |
[31] J. Haralambidis, M. Chai, G. W. Tregear, Nucleic Acids Res. 1987, 15, 4857.
| Crossref | GoogleScholarGoogle Scholar | 3110740PubMed |
[32] P. Nielsen, M. Egholm, R. Berg, O. Buchardt, Science 1991, 254, 1497.
| Crossref | GoogleScholarGoogle Scholar | 1962210PubMed |
[33] P. Sazani, F. Gemignani, S.-H. Kang, M. A. Maier, M. Manoharan, M. Persmark, D. Bortner, R. Kole, Nat. Biotechnol. 2002, 20, 1228.
| Crossref | GoogleScholarGoogle Scholar | 12426578PubMed |
[34] J. Tailhades, H. Takizawa, M. J. Gait, D. A. Wellings, J. D. Wade, Y. Aoki, F. Shabanpoor, Front Chem. 2017, 5, 81.
| Crossref | GoogleScholarGoogle Scholar | 29094037PubMed |
[35] T. Soudah, S. Khawaled, R. I. Aqeilan, E. Yavin, ACS Omega 2019, 4, 13954.
| Crossref | GoogleScholarGoogle Scholar | 31497713PubMed |
[36] J. Winkler, E. Urban, D. Losert, V. Wacheck, H. Pehamberger, C. R. Noe, Nucleic Acids Res. 2004, 32, 710.
| Crossref | GoogleScholarGoogle Scholar | 14757835PubMed |
[37] J. Winkler, E. Urban, C. R. Noe, Bioconjug. Chem. 2005, 16, 1038.
| Crossref | GoogleScholarGoogle Scholar | 16029048PubMed |
[38] J. Winkler, M. Gilbert, A. Kocourková, M. Stessl, C. R. Noe, ChemMedChem 2008, 3, 102.
| Crossref | GoogleScholarGoogle Scholar | 17979170PubMed |
[39] K. Shinbara, W. Liu, R. H. P. van Neer, T. Katoh, H. Suga, Front Chem. 2020, 8, 447.
| Crossref | GoogleScholarGoogle Scholar | 32626683PubMed |
[40] M. Kye, Z. Zhang, Y.-b. Lim, Pept. Sci. 2021, 113, e24193.
| Crossref | GoogleScholarGoogle Scholar |
[41] P. Dawson, T. Muir, I. Clark-Lewis, S. Kent, Science 1994, 266, 776.
| Crossref | GoogleScholarGoogle Scholar | 7973629PubMed |
[42] R. K. Bruick, P. E. Dawson, S. B. H. Kent, N. Usman, G. F. Joyce, Chem. Biol. 1996, 3, 49.
| Crossref | GoogleScholarGoogle Scholar | 8807828PubMed |
[43] O. Zavoiura, U. Resch-Genger, O. Seitz, Bioconjug. Chem. 2018, 29, 1690.
| Crossref | GoogleScholarGoogle Scholar | 29694033PubMed |
[44] M. Lovrinovic, R. Seidel, R. Wacker, H. Schroeder, O. Seitz, M. Engelhard, R. S. Goody, C. M. Niemeyer, Chem. Commun. 2003, 822.
| Crossref | GoogleScholarGoogle Scholar |
[45] S. Ficht, A. Mattes, O. Seitz, J. Am. Chem. Soc. 2004, 126, 9970.
| Crossref | GoogleScholarGoogle Scholar | 15303871PubMed |
[46] M. McPherson, M. C. Wright, P. A. Lohse, Synlett 1999, 978.
| Crossref | GoogleScholarGoogle Scholar |
[47] J. Sayers, R. J. Payne, N. Winssinger, Chem. Sci. 2018, 9, 896.
| Crossref | GoogleScholarGoogle Scholar | 29629156PubMed |
[48] D. A. Stetsenko, M. J. Gait, J. Org. Chem. 2000, 65, 4900.
| Crossref | GoogleScholarGoogle Scholar | 10956469PubMed |
[49] M. Dastpeyman, J. A. Karas, A. Amin, B. J. Turner, F. Shabanpoor, Front Chem. 2021, 9, 627329.
| Crossref | GoogleScholarGoogle Scholar | 33738276PubMed |
[50] J. Tailhades, N. A. Patil, M. A. Hossain, J. D. Wade, J. Pept. Sci. 2015, 21, 139.
| Crossref | GoogleScholarGoogle Scholar | 25641053PubMed |
[51] S. Ficht, C. Dose, O. Seitz, ChemBioChem 2005, 6, 2098.
| Crossref | GoogleScholarGoogle Scholar | 16208732PubMed |
[52] G. Saito, J. A. Swanson, K.-D. Lee, Adv. Drug Deliv. Rev. 2003, 55, 199.
| Crossref | GoogleScholarGoogle Scholar | 12564977PubMed |
[53] J. Y. Legendre, A. Trzeciak, B. Bohrmann, U. Deuschle, E. Kitas, A. Supersaxo, Bioconjug. Chem. 1997, 8, 57.
| Crossref | GoogleScholarGoogle Scholar | 9026036PubMed |
[54] E. Wagner, C. Plank, K. Zatloukal, M. Cotten, M. L. Birnstiel, Proc. Natl. Acad. Sci. USA 1992, 89, 7934.
| Crossref | GoogleScholarGoogle Scholar | 1518816PubMed |
[55] D. R. Corey, D. Munoz-Medellin, A. Huang, Bioconjug. Chem. 1995, 6, 93.
| Crossref | GoogleScholarGoogle Scholar | 7711111PubMed |
[56] F. M. Uckun, S. Qazi, I. Dibirdik, D. E. Myers, Integr. Biol. 2013, 5, 122.
| Crossref | GoogleScholarGoogle Scholar |
[57] R. Eritja, A. Pons, M. Escarcellar, E. Giralt, F. Albericio, Tetrahedron 1991, 47, 4113.
| Crossref | GoogleScholarGoogle Scholar |
[58] J.-P. Bongartz, A.-M. Aubertain, P. G. Milhaud, B. Lebleu, Nucleic Acids Res. 1994, 22, 4681.
| Crossref | GoogleScholarGoogle Scholar | 7984418PubMed |
[59] J. J. Turner, D. Williams, D. Owen, M. J. Gait, Curr. Protoc. Nucleic Acid Chem. 2006, 24, 4.28.1.
| Crossref | GoogleScholarGoogle Scholar |
[60] N. A. Patil, J. A. Karas, J. D. Wade, M. A. Hossain, J. Tailhades, Chem. – Eur. J. 2019, 25, 8599.
| Crossref | GoogleScholarGoogle Scholar | 30924212PubMed |
[61] K. Arar, A.-M. Aubertin, A.-C. Roche, M. Monsigny, R. Mayer, Bioconjug. Chem. 1995, 6, 573.
| Crossref | GoogleScholarGoogle Scholar | 8974456PubMed |
[62] S. Abes, G. D. Ivanova, R. Abes, A. A. Arzumanov, D. Williams, D. Owen, B. Lebleu, M. J. Gait, in Macromolecular Drug Delivery: Methods and Protocols (Ed. M. Belting) 2009, pp. 85–99 (Humana Press: Totowa, NJ).
[63] H. Yin, A. F. Saleh, C. Betts, P. Camelliti, Y. Seow, S. Ashraf, A. Arzumanov, S. Hammond, T. Merritt, M. J. Gait, M. J. A. Wood, Mol. Ther. 2011, 19, 1295.
| Crossref | GoogleScholarGoogle Scholar | 21505427PubMed |
[64] N. A. Patil, J. A. Karas, B. J. Turner, F. Shabanpoor, Bioconjug. Chem. 2019, 30, 793.
| Crossref | GoogleScholarGoogle Scholar | 30645945PubMed |
[65] X. Ming, M. R. Alam, M. Fisher, Y. Yan, X. Chen, R. L. Juliano, Nucleic Acids Res. 2010, 38, 6567.
| Crossref | GoogleScholarGoogle Scholar | 20551131PubMed |
[66] M. Dastpeyman, R. Sharifi, A. Amin, J. A. Karas, B. Cuic, Y. Pan, J. A. Nicolazzo, B. J. Turner, F. Shabanpoor, Int. J. Pharm. 2021, 599, 120398.
| Crossref | GoogleScholarGoogle Scholar | 33640427PubMed |
[67] N. J. Ede, G. W. Tregear, J. Haralambidis, Bioconjug. Chem. 1994, 5, 373.
| Crossref | GoogleScholarGoogle Scholar | 7948105PubMed |
[68] M. R. Alam, X. Ming, M. Fisher, J. G. Lackey, K. G. Rajeev, M. Manoharan, R. L. Juliano, Bioconjug. Chem. 2011, 22, 1673.
| Crossref | GoogleScholarGoogle Scholar | 21755983PubMed |
[69] A. D. Baldwin, K. L. Kiick, Bioconjug. Chem. 2011, 22, 1946.
| Crossref | GoogleScholarGoogle Scholar | 21863904PubMed |
[70] Y. Kim, S. O. Ho, N. R. Gassman, Y. Korlann, E. V. Landorf, F. R. Collart, S. Weiss, Bioconjug. Chem. 2008, 19, 786.
| Crossref | GoogleScholarGoogle Scholar | 18275130PubMed |
[71] L. P. T. Hong, J. A. Scoble, L. Doughty, G. Coia, C. C. Williams, Aust. J. Chem. 2011, 64, 779.
| Crossref | GoogleScholarGoogle Scholar |
[72] H. Wu, P. J. LeValley, T. Luo, A. M. Kloxin, K. L. Kiick, Bioconjug. Chem. 2018, 29, 3595.
| Crossref | GoogleScholarGoogle Scholar | 30285419PubMed |
[73] S. D. Fontaine, R. Reid, L. Robinson, G. W. Ashley, D. V. Santi, Bioconjug. Chem. 2015, 26, 145.
| Crossref | GoogleScholarGoogle Scholar | 25494821PubMed |
[74] M. Jbara, J. Rodriguez, H. H. Dhanjee, A. Loas, S. L. Buchwald, B. L. Pentelute, Angew. Chem. Int. Ed. Engl. 2021, 60, 12109.
| Crossref | GoogleScholarGoogle Scholar | 33730425PubMed |
[75] B. Seelig, A. Jäschke, Tetrahedron Lett. 1997, 38, 7729.
| Crossref | GoogleScholarGoogle Scholar |
[76] K. W. Hill, J. Taunton-Rigby, J. D. Carter, E. Kropp, K. Vagle, W. Pieken, D. P. C. McGee, G. M. Husar, M. Leuck, D. J. Anziano, D. P. Sebesta, J. Org. Chem. 2001, 66, 5352.
| Crossref | GoogleScholarGoogle Scholar | 11485455PubMed |
[77] V. Borsenberger, S. Howorka, Nucleic Acids Res. 2009, 37, 1477.
| Crossref | GoogleScholarGoogle Scholar | 19139071PubMed |
[78] V. Marchán, S. Ortega, D. Pulido, E. Pedroso, A. Grandas, Nucleic Acids Res. 2006, 34, e24.
| Crossref | GoogleScholarGoogle Scholar | 16478710PubMed |
[79] D. Forget, D. Boturyn, E. Defrancq, J. Lhomme, P. Dumy, Chem. – Eur. J. 2001, 7, 3976.
| Crossref | GoogleScholarGoogle Scholar | 11596939PubMed |
[80] Y. Singh, O. P. Edupuganti, M. Villien, É. Defrancq, P. Dumy, C. R. Chim. 2005, 8, 789.
| Crossref | GoogleScholarGoogle Scholar |
[81] O. P. Edupuganti, O. Renaudet, E. Defrancq, P. Dumy, Bioorg. Med. Chem. Lett. 2004, 14, 2839.
| Crossref | GoogleScholarGoogle Scholar | 15125943PubMed |
[82] S. Ghosh, E. Defrancq, J. H. Lhomme, P. Dumy, S. Bhattacharya, Bioconjug. Chem. 2004, 15, 520.
| Crossref | GoogleScholarGoogle Scholar | 15149179PubMed |
[83] T. Hamma, P. S. Miller, Bioconjug. Chem. 2003, 14, 320.
| Crossref | GoogleScholarGoogle Scholar | 12643742PubMed |
[84] N. Ollivier, C. Olivier, C. Gouyette, T. Huynh-Dinh, H. Gras-Masse, O. Melnyk, Tetrahedron Lett. 2002, 43, 997.
| Crossref | GoogleScholarGoogle Scholar |
[85] Y. Anno, T. Kubo, R. Ueki, M. Yano, K. Sasaki, H. Ohba, M. Fujii, Nucleosides Nucleotides Nucleic Acids 2003, 22, 1451.
| Crossref | GoogleScholarGoogle Scholar | 14565440PubMed |
[86] T. Kubo, M. Morikawa, H. Ohba, M. Fujii, Org. Lett. 2003, 5, 2623.
| Crossref | GoogleScholarGoogle Scholar | 12868874PubMed |
[87] T. Kubo, Z. Zhelev, B. Rumiana, H. Ohba, K. Doi, M. Fujii, Org. Biomol. Chem. 2005, 3, 3257.
| Crossref | GoogleScholarGoogle Scholar | 16132084PubMed |
[88] B. B. Kasten, X. Ma, H. Liu, T. R. Hayes, C. L. Barnes, S. Qi, K. Cheng, S. C. Bottorff, W. S. Slocumb, J. Wang, Z. Cheng, P. D. Benny, Bioconjug. Chem. 2014, 25, 579.
| Crossref | GoogleScholarGoogle Scholar | 24568284PubMed |
[89] C. S. McKay, M. G. Finn, Chem. Biol. 2014, 21, 1075.
| Crossref | GoogleScholarGoogle Scholar | 25237856PubMed |
[90] A. H. El-Sagheer, T. Brown, Chem. Soc. Rev. 2010, 39, 1388.
| Crossref | GoogleScholarGoogle Scholar | 20309492PubMed |
[91] J. Willibald, J. Harder, K. Sparrer, K.-K. Conzelmann, T. Carell, J. Am. Chem. Soc. 2012, 134, 12330.
| Crossref | GoogleScholarGoogle Scholar | 22812910PubMed |
[92] D. C. Dieterich, J. J. L. Hodas, G. Gouzer, I. Y. Shadrin, J. T. Ngo, A. Triller, D. A. Tirrell, E. M. Schuman, Nat. Neurosci. 2010, 13, 897.
| Crossref | GoogleScholarGoogle Scholar | 20543841PubMed |
[93] T. S. Zatsepin, D. A. Stetsenko, A. A. Arzumanov, E. A. Romanova, M. J. Gait, T. S. Oretskaya, Bioconjug. Chem. 2002, 13, 822.
| Crossref | GoogleScholarGoogle Scholar | 12121138PubMed |
[94] K. Braun, P. Peschke, R. Pipkorn, S. Lampel, M. Wachsmuth, W. Waldeck, E. Friedrich, J. Debus, J. Mol. Biol. 2002, 318, 237.
| Crossref | GoogleScholarGoogle Scholar | 12051833PubMed |
[95] N. A. Patil, J.-P. Quek, B. Schroeder, R. Morewood, J. Rademann, D. Luo, C. Nitsche, ACS Med. Chem. Lett. 2021, 12, 732.
| Crossref | GoogleScholarGoogle Scholar | 34055219PubMed |