

Crystal Structure of Burgess Inner Salts and their Hydrolyzed Ammonium Sulfaminates*
Anthony J. ArduengoA Department of Chemistry and Biochemistry, The University of Alabama, Tuscaloosa, AL 35487-0336, USA.
B Department of Chemistry, School of Science, Kitasato University, 1-15-1 Kitasato, Minami-ku, Sagamihara, Kanagawa, 252-0373, Japan.
C Corresponding author. Email: aj@ajarduengo.net
Australian Journal of Chemistry 72(11) 867-873 https://doi.org/10.1071/CH19338
Submitted: 21 July 2019 Accepted: 29 July 2019 Published: 23 August 2019
Abstract
The solid-state structures of the Burgess reagent, and its analogous ethyl ester reveal structures indicative of triethylamine solvated sulfonyl imides rather than the more commonly depicted triethylammonium sulfonyl amidate. The existence of a reversibly formed hydrate of Burgess reagent is not supported by present studies, but rather a hydrosylate that does not revert to the Burgess reagent with gentle warming under vacuum was isolated and characterised. Structures of the hydrosylates from both the methyl- and ethyl-amidate esters were determined from X-ray crystallographic analysis and are reported. The crystal structures of the Burgess inner salts exhibit geometries at the sulfur atoms that are intermediate between a planar O2S=NCO2R unit and tetrahedral 4-coordinate sulfur centres that would be expected from a strong single (dative) bond between the triethylamine nitrogen and sulfur. The hydrolysed ammonium sulfaminates are water soluble intermolecular salts composed of triethylammonium ions, Et3NH+, and N-(alkoxycarbonyl)sulfaminate, O(−)SO2NHCO2R {R = CH3 or C2H5}.
Introduction
The Burgess reagent[1–4] has been utilised by synthetic chemists as a powerful and flexible reagent to dehydrate secondary and tertiary alcohols,[5] amides,[6] and nitroalkanes,[7] and form amines[2] (via carbamates) from primary alcohols. The reagent has found further utility in sulfonations,[1] heterocycle formation,[8,9] and most recently oxidation reactions.[10] In spite of these diverse applications, no solid state structure of the Burgess reagent (either as its commercially available methyl carbamate ester 1, nor its ethyl ester 2, the ‘inner salt,’ originally prepared by Atkins and Burgess) has been reported. In the years shortly before his passing (June 2018), Burgess related to one of us (AJA) that this popular reagent reversibly forms a hydrate with water that reverts to the parent powerful dehydrating agent with only mild warming under vacuum. Indeed, Atkins’ dissertation contains a claim to exactly this point.[11] However, contrary to Burgess’s recollection shared with AJA, an X-ray structure on this putative hydrate was never completed.[12] Given the strength of the Burgess reagent as a dehydrating agent, even under mild conditions, the reversible formation of a hydrate is a puzzling and interesting possibility. Hence, we decided to explore this aspect of the chemistry of the Burgess reagent in more detail.
Results and Discussion
Burgess reagents, the inner salt methyl (1) and ethyl (2) esters, were prepared by the reaction of the corresponding alkoxycarbonylsulfamoyl chloride 3a,b with 2 equivalents of triethylamine in benzene (Scheme 1).[2]
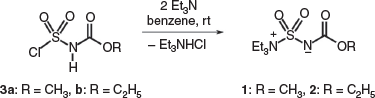
|
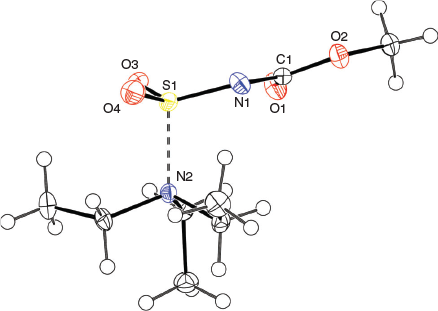
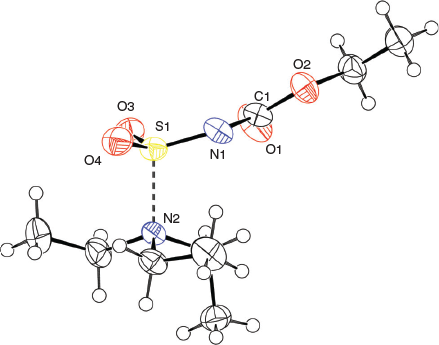
The solid-state structure of the Burgess reagent was determined by single crystal X-ray diffraction. The structures of both the methyl and ethyl carbamate esters were obtained using crystals formed from tetrahydrofuran solutions. The structures of the Burgess reagent reveals a long N(triethylamine)…S(sulfaminate) interatomic distance of 1.90 Å. This S–NEt3 distance is surprisingly long and is intermediate between the dative bond in the trimethylamine–SO2 adduct (2.048 Å)[13] and that found in the trimethylamine–SO3 adduct (1.844 Å).[14] Such a long, weak-dative interaction may also be reflected in reactivity involving dissociation of the triethylamine moiety in the Burgess reagent to reveal a sulfonylimine (O2S=NC(O)OR) that quickly adds an alcohol to give the alkyl-N-carbomethoxysulfamate. This latter alkyl-N-carbomethoxysulfamate subsequently undergoes an internal β-elimination to give an olefin.[5] In addition, one notes that the degree of pyramidalisation of the sulfonylimine moiety is not particularly high when compared with the chlorosulfonyl amide precursor 3b to the Burgess reagent or its hydrosylates 4 and 5 (Table 1).

|
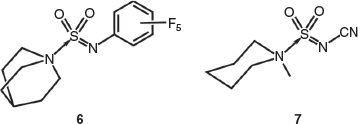
Overall, the structures of the Burgess reagents around the sulfur centre resemble the only two other tertiary amine→sulfonylimine complexes currently recorded in the Cambridge Crystallographic Database, N-(((pentafluorophenyl)imino)sulfonyl)-quinuclidine (6)[15] and N-((cyanamido)sulfonyl)-1-methylpiperidine (7) (Chart 1).[16] The structures of 1, 2, and 3b are depicted in Figs 1, 2, and 3.
|
|
|
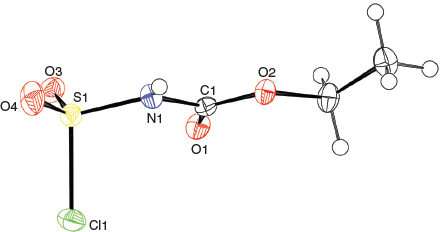
|
The geometry of the Burgess reagent is well modelled at the G3MP2 level.[17] Selected bond distances from experiment and theory are presented in Table 2. Heats of reaction in the gas phase from the amine and sulfonylimine (the negative of the S–N bond dissociation energy (BDE)) were calculated as ΔH(298 K) = −33.5 kcal mol−1 for the NEt3 adduct and −34.2 kcal mol−1 when triethylamine is replaced with NMe3. The corresponding gas phase heats of formation are ΔHf(298 K) = −161.8 kcal mol−1 for the Burgess reagent and −145.2 kcal mol−1 for the NMe3 derivative. Interestingly, the Burgess reagent (inner salt) compounds 1 and 2, with their long, weak, dative amine→sulfonylimine interaction, appear somewhat similar to dative amine→borane interactions.[18,19] The corresponding G3MP2 B-N BDE at 298 K in H3BNMe3 is 38.0 kcal mol−1 which is in agreement with experiment and the value of the B–N BDE for H3BNEt3 is 35.9 kcal mol−1, both of which are similar to the S–N BDEs given above.

|
Burgess Reagent Reaction with Water
The ‘hydrate’ of the Burgess inner salt that was described in Atkins’ Ph.D. thesis was reported to crystallise from moist benzene.[11] Indeed, when water vapour is allowed to diffuse into a dry benzene solution of 1 or 2, crystals form (respectfully 4 and 5). In the case of the water adduct 5, the melting point (89–90°C) and 1H NMR spectra agree well with the material reported by Atkins. There is one notable difference in the proton NMR spectra in that the broad 2-hydrogen resonance reported at δ8.38 by Atkins appears as two resonances of equal intensity at δ7.49 and 9.14 in the 600 MHz spectrum reported herein. These resonances likely correspond to the two acidic (exchangeable) NH protons in 5 that appear as an averaged resonance in the 60 MHz spectrum recorded by Atkins. The infrared spectra are more difficult to compare because a limited selection of peaks (without relative intensities) were reported by Atkins. Nonetheless, there is agreement between some of the absorption frequencies reported for the two samples. Based on the experimental conditions and the physical and spectroscopic properties, it appears likely that the material 5 reported herein and ‘hydrate’ reported by Atkins are one and the same. A very similar reaction takes place with the methyl carbamate ester of the Burgess reagent 1 and produces the adduct 4 that has very similar spectroscopic properties to 5 and a slightly lower melting point. Burgess’s inner salts 1 and 2 are only slowly soluble in water, while compounds 4 and 5 dissolve quickly in water. On the other hand, 1 and 2 are easily soluble in benzene, and 4 and 5 are not. Gentle warming of 4 and 5 in vacuum did not transform them back into 1 and 2 as reported by Atkins, and more aggressive heating eventually led to extensive charring and decomposition of the materials.
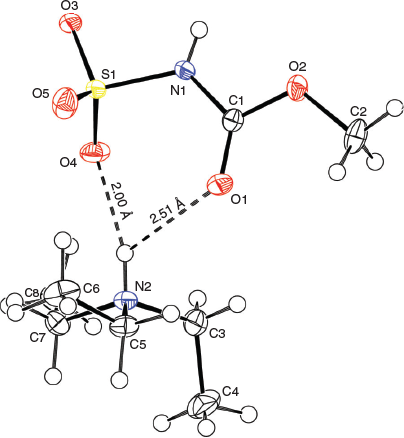
X-Ray crystallographic structure analyses reveal the structures of 4 and 5 to be hydrolysates, not simple hydrates. Thus, the overall reactions are irreversible hydrolyses (Scheme 2). Crystals of hydrolysed ammonium methoxy- and ethoxycarbonyl-sulfaminates may be obtained from benzene containing 1 % water or by allowing water vapour to slowly diffuse into benzene solutions of the Burgess reagents. The solid-state structures of salts 4 and 5 consist of hydrogen-bonded triethylammonium cations and alkoxycarbonylsulfaminate anion pairs. These ammonium salt structures are depicted in Figs 4 and 5.
|
|
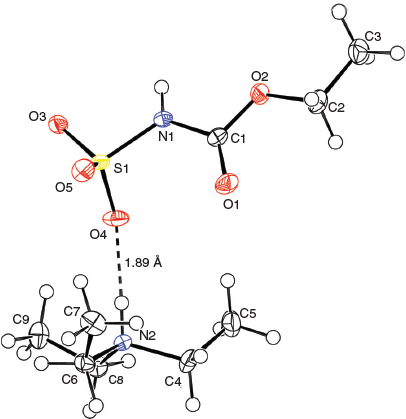
|
Although the spectroscopic characteristics described herein for 4 and 5, and those reported by Atkins for the putative ‘hydrate’ appear consistent, there is one aspect of the report of reversible formation of a hydrate by Atkins that remains bothersome and that is the gentle dehydration of Atkins’ hydrate to reform the Burgess inner salt. Based on the X-ray structures of 4 and 5 there is no doubt that these materials are hydrosylates – not simple hydrates. As Atkins points out in his Ph.D. thesis, his putative hydrate ‘dehydrated under conditions milder than would be expected for the dehydration of’ the ammonium salt 5. It remains possible that the structural and spectroscopic characterisation of the isolated material are different from the ‘hydrate’ as it is initially formed. Subsequent dissolution in chloroform for characterisation and purification might have led to the observed decomposition and the formation of the hydrosylate. Such a later-stage hydrolysis during characterisation and purification could obscure the presence of a very marginally stable hydrate. This could be the case if the BDE of the S–N dative bound triethylamine is only slightly higher than the BDE of the hydrogen bond(s) that bind the water to the nucleophilic sites of the Burgess reagent in a possible hydrate. This energetic ordering seems likely based on the theoretical modelling studies described above. Work is currently in progress to find even milder conditions to generate a potential hydrate of the Burgess reagent that would require no further manipulation to obtain samples suitable for spectroscopic and structural characterisation.
Conclusion
The solid-state structures of methyl- and ethyl-carbamate esters of the Burgess reagent exhibit weak dative bonding geometries between the triethylamine and sulfonylimine units comprising them. These geometric features include long N(triethylamine)–S(sulfonyl) interactions and a sulfur coordination sphere that is intermediate between tetrahedral and planar. These dative N→S interactions are reproduced at the G3MP2 level of theory and the calculated N→S bond dissociation energies are consistent with the chemistry of the Burgess reagent towards nucleophiles. Furthermore, the dative N→S interactions in the Burgess reagent are reminiscent of dative N→B interactions in amine boranes.
If a true ‘hydrate’ of the Burgess reagent exists its stability is so marginal that even simple sample manipulations at room temperature lead to its transformation to hydrosylates. The spectroscopic and physical data reported previously for this putative hydrate actually correspond to a hydrosylate (triethylammonium N-(alkoxycarbonyl)sulfaminate). As expected, these triethylammonium N-(alkoxycarbonyl)sulfaminate do not easily revert to the initial Burgess reagents.
Experimental
General
1H and 13C{1H} NMR spectra were recorded on a Bruker Advance-II 600 (600 MHz for 1H and 151 MHz for 13C) spectrometer at room temperature with the inner standards of TMS (δH 0.00), acetone-d6 (δH 2.06), CDCl3 (δC 77.0), and acetone-d6 (δC 30.6). Protons and carbons of products were assigned by DEPT135 and 2D NMR techniques (HSQC, HMBC, and HHCOSY). Splitting types of protons are designated as s, d, t, and q, for singlet, doublet, triplet, and quartet signals respectively. Melting points were measured using a Yanaco Micro Melting Point apparatus. IR Spectroscopy was conducted using a FT/IR-4200 of JASCO equipped with MicromATRvision of Czitek for measurement of the solid state and two NaCl plates for measurement in CHCl3 solution. Relative intensities of IR absorption were defined by w, m, s, and vs, to identify weak, medium, strong, and very strong signals respectively. X-Ray crystallographic analysis was performed by SMART APEX II of Bruker AXS Co. Electrospray ionisation–mass spectrometry (ESI-MS) was conducted on a Thermo Fisher Exactive Plus Orbitrap Mass Spectrometer. Reagents and solvents for synthesis were purchased from Tokyo Chemical Industry Co., LTD, Kanto Chemical Co., INC., Wako Pure Chemical Industries, Ltd, Sigma–Aldrich Co. LLC., Cambridge Isotope Laboratories. Benzene, MeOH, EtOH, and triethylamine were used after distillation over CaH2. Dried THF and Et2O were used from the solvent cans of Kanto without any further purification.
Synthesis of Ethyl(chlorosulfonyl)carbamate 3b
Ethyl(chlorosulfonyl)carbamate 3b was prepared using a modification (substituting ethanol for methanol) of the Organic Syntheses procedure published by Burgess et al.[2] Compound 3b was obtained as colourless crystals (mp 45.5–47°C), yield 87 %. δH (600 MHz, CH3CN–acetone-d6) 1.13 (3H, t, JHH 7.2, OCH2CH3), 4.13 (2H, q, JHH 7.2, OCH2CH3). δC (151 MHz, CH3CN–acetone-d6) 14.84 (OCH2CH3), 65.70 (OCH2CH3), 150.7 (C=O).
Synthesis of Methyl(carboxysulfamoyl)triethylammonium Hydroxide 1
Methyl(carboxysulfamoyl)triethylammonium hydroxide 1 was prepared using the Organic Syntheses procedure published by Burgess et al.[2] Compound 1 was obtained as colourless crystals (mp 67.2–69.5°C, lit[2] mp 71–72°C), yield 63 %. δH (600 MHz, CDCl3) 1.41 (9H, t, JHH 7.2, NCH2CH3), 3.47 (6H, q, JHH 7.2, NCH2CH3, 3.70 (3H, s, CH3), δC (151 MHz, CDCl3) 9.38 (NCH2CH3), 50.43 (NCH2CH3), 53.26 (OCH3), 158.2 (C=O). νmax (CHCl3)/cm−1 3020 (w), 2951 (w), 1698 (m), 1460 (w), 1438 (w), 1342 (w), 1262 (s), 1222 (w), 1202 (w), 1187 (w), 1110 (w), 1039 (w), 965 (w), 866 (w), 734 (w), 714 (w), 668 (w), 623 (w), 590 (w), 577 (w), 563 (w). νmax (solid)/cm−1 3046 (w), 2996 (w), 2979 (w), 2951 (w), 1719 (w), 1688 (m), 1455 (m), 1441 (m), 1402 (w), 1338 (w), 1326 (m), 1304 (w), 1241 (s), 1219 (m), 1186 (m), 1111 (m), 1092 (m), 1042 (m), 1023 (m), 960 (m), 893 (w), 856 (m), 835 (m), 787 (m), 717 (m), 695 (w), 679 (w), 668 (w), 651 (w), 641 (w), 627 (m), 600 (s), 577 (m).
Synthesis of Ethyl(carboxysulfamoyl)triethylammonium Hydroxide 2
Ethyl(carboxysulfamoyl)triethylammonium hydroxide 2 was prepared using the Organic Syntheses procedure published by Burgess et al.[2] Compound 2 was obtained as colourless crystals (mp 81–82°C, lit[1,11] mp 66–69°C), yield 43 %. δH (600 MHz, CDCl3) 1.285 (3H, t, JHH 7.2, OCH2CH3), 1.41 (9H, t, JHH 7.2, NCH2CH3), 3.48 (6H, q, JHH 7.2, NCH2CH3), 4.13 (2H, q, JHH 7.2, OCH2CH3), δC (151 MHz, CDCl3) 9.34 (NCH2CH3), 14.29 (OCH2CH3), 50.34 (NCH2CH3), 62.06 (OCH2CH3), 157.6 (C = O). νmax (CHCl3)/cm−1 3023 (m), 2987 (m), 2948 (w), 2900 (w), 1687 (vs), 1461 (m), 1391 (m), 1367 (m), 1339 (vs), 1258 (vs), 1206 (s), 1170 (m), 1105 (vs), 1022 (m), 1041 (s), 1010 (w), 902 (w), 806 (w), 687 (w), 655 (w), 619 (m), 567 (w), 544 (w), 483 (m), 466 (m). νmax (solid)/cm−1 2990 (w), 2945 (w), 2909 (w), 1718 (w), 1686 (s), 1470 (m), 1449 (m), 1398 (m), 1366 (m), 1325 (s), 1232 (vs), 1207 (s), 1181 (m), 1163 (m), 1095 (s), 1041 (m), 1019 (s), 899 (m), 866 (s), 812 (m), 792 (s), 739 (m), 718 (m), 599 (s), 576 (vs), 547 (s), 496 (m), 470 (w), 454 (w), 445 (w), 431 (m), 412 (w), 402 (s).
Synthesis of Triethylammonium Methylcarboxysulfaminate 4
Burgess reagent methyl derivative 1 (199.9 mg, 0.839 mmol) in a 10 mL sample tube was dissolved in benzene (5 mL) with gentle warming. The tube containing the solution of the Burgess reagent was placed upright in a 100 mL round-bottom flask equipped with a three-way stopcock. Water (50 μL, 2.8 mmol) was added to the bottom of the flask (outside the tube containing the Burgess reagent solution) via a 0.5 mL syringe. The round-bottom flask was stoppered and stored at room temperature under N2 for 21 h. The solid that crystallised from benzene in the presence of water was collected by filtration. The residue in the sample tube was rinsed with CHCl3 (10 mL × 2) to collect all products. The solutions were transferred to another 100 mL round-bottom flask by a Pasteur pipette and evaporated under vacuum at 30°C producing an oily residue that was stored at room temperature under N2. After 1 day these oily residues solidified. The combined solids were washed with hexane and then dried under vacuum to provide the hydrolysed Burgess reagent methyl derivative 4 (183.6 mg, 0.719 mmol) as colourless crystals in 86 % yield, mp 87–89°C. δH (600 MHz, CDCl3) 1.36 (9H, t, JHH 6.6, NCH2CH3), 3.22 (6H, q, JHH 6.6, NCH2CH3, 3.68 (3H, s, CH3), 7.31 (1H, br s, SO2NHCO), 9.14 (1H, br s, NHCH2CH3), δC (151 MHz, CDCl3) 8.47 (NCH2CH3), 46.41 (NCH2CH3), 52.26 (OCH3), 153.7 (C=O). νmax (CHCl3)/cm−1 3422 (w), 3015 (m), 2954 (w), 1731 (m), 1457 (m), 1409 (m), 1336 (w), 1272 (m), 1216 (m), 1041 (m), 949 (w), 836 (w), 763 (vs), 756 (vs), 666 (m), 621 (m), 599 (w), 578 (w), 566 (w). νmax (solid)/cm−1 3219 (w), 3051 (w), 3015 (w), 3000 (w), 2888 (w), 2760 (w), 1718 (m), 1467 (m), 1402 (m), 1360 (w), 1310 (w), 1266 (m), 1236 (m), 1203 (s), 1086 (m), 1065 (m), 1039 (s), 1013 (m), 898 (w), 831 (m), 782 (w), 730 (w), 701 (w), 685 (w), 675 (m), 662 (m), 624 (s), 586 (w), 569 (m), 555 (w). m/z (ESI, CH3CN, +ve) 102.1281, calcd for C6H16N: 102.1277. m/z (ESI, CH3CN, −ve) 153.9806, calcd for C2H4NO5S: 153.9805.
Synthesis of Triethylammonium Ethylcarboxysulfaminate 5
Burgess reagent ethyl derivative 2 (203.2 mg, 0.805 mmol) in a 10 mL sample tube was dissolved in benzene (5 mL) with gentle warming. The tube containing the solution of the Burgess reagent was placed upright in a 100 mL round-bottom flask equipped with a three-way stopcock. Water (50 μL, 2.8 mmol) was added to the bottom of the flask (outside the tube containing the Burgess reagent solution) via a 0.5 mL syringe. The round-bottom flask was stoppered and stored at room temperature under N2 for 21 h. The solid that crystallised from benzene in the presence of water was collected by filtration. The residue in the sample tube was rinsed with CHCl3 (10 mL × 2) to collect all products. The solutions were transferred to another 100 mL round-bottom flask by a Pasteur pipette and evaporated under vacuum at 30°C producing an oily residue that was stored at room temperature under N2. After 1 day these oily residues solidified. The combined solids were washed with hexane and then dried under vacuum to provide the hydrolysed Burgess reagent ethyl derivative 5 (107.5 mg, 0.398 mmol) in 49 % yield as colourless crystals (mp 89–90°C). δH (600 MHz, CDCl3) 1.24 (3H, t, JHH 7.2, OCH2CH3), 1.37 (9H, t, JHH 7.2, NHCH2CH3), 3.22 (6H, qd, JHH 7.2, 4.8, NHCH2CH3), 4.11 (2H, q, JHH 7.2, OCH2CH3), 7.09 (1H, br s, SO2NHCO), 9.16 (1H, br s, NHCH2CH3). δH (151 MHz, CDCl3) 8.48 (NHCH2CH3), 14.42 (OCH2CH3), 46.44 (NHCH2CH3), 61.21 (OCH2CH3), 153.3 (C=O). νmax (CHCl3)/cm−1 3418 (w), 3223 (w), 3008 (m), 2805 (w), 2737 (w), 1726 (s), 1471 (m), 1443 (m), 1388 (m), 1356 (w), 1320 (w), 1269 (s), 1237 (s), 1199 (m), 1008 (m), 869 (m), 757 (w), 666 (m), 594 (m), 546 (s), 502 (m), 493 (m). νmax (solid)/cm−1 3250 (w), 3007 (w), 2789 (w), 2710 (w), 1721 (m), 1474 (m), 1442 (m), 1402 (m), 1384 (w), 1358 (w), 1261 (s), 1225 (s), 1200 (s), 1085 (w), 1039 (s), 1007 (m), 907 (w), 811 (m), 781 (m), 749 (m), 667 (m), 617 (s), 573 (m), 542 (m), 511 (w), 499 (w), 485 (m), 472 (w), 463 (w), 441 (m), 430 (w). m/z (ESI, CH3CN, +ve) 102.1281, calcd for C6H16N: 102.1277. m/z (ESI, CH3CN, −ve) 167.9963, calcd for C2H6NO5S: 167.9961.
X-Ray Crystallographic Analyses
Crystal Data for Burgess Reagent 1
C8H18N2O4S (238.30 amu); data were collected on a colourless block obtained by slow evaporation of a THF solution measuring 0.48 × 0.43 × 0.21 mm3. Data collection at −153°C with MoKα radiation (λ 0.71073 Å); monoclinic, P21/n, Z 4; a 6.8066(5), b 12.8754(9), c 13.1397(9) Å, β 93.8074(8)°; μ(Mo) 0.280 mm−1; with index ranges of −6 ≤ h ≤ 9, −17 ≤ k ≤ 13, and −15 ≤ l ≤ 17; 6633 reflections were collected yielding 2741 independent reflections. The structure was solved by direct methods and expanded using Fourier techniques (SHELX 2018). All non-hydrogen atoms were refined anisotropically, while hydrogen atoms were refined isotropically. Data to parameter ratio 2741/140; R1 0.0274, wR2 0.0742 (I > 2σ(I)); R1 0.0314, wR2 0.0768 (all data); GOF 1.053. CCDC deposition number 1941098.
Crystal Data for Burgess Reagent (Ethylcarbamate Ester Analogue) 2
C9H20N2O4S (252.33 amu); data were collected on a colourless block obtained by slow evaporation of a THF solution measuring 0.51 × 0.50 × 0.33 mm3. Data collection at 23°C with MoKα radiation (λ 0.71073 Å); monoclinic, P21, Z 2; a 6.6023(9), b 12.8477(17), c 7.8470(10) Å, β 106.2153(16)°; μ(Mo) 0.256 mm−1; with index ranges of −8 ≤ h ≤ 5, −17 ≤ k ≤ 6, −10 ≤ l ≤ 9; 3796 reflections were collected yielding 2838 independent reflections. The structure was solved by direct methods and expanded using Fourier techniques (SHELX 2018). All non-hydrogen atoms were refined anisotropically, while hydrogen atoms were refined isotropically. Data to parameter ratio 2838/149; R1 0.0295, wR2 0.0691 (I > 2σ(I)); R1 0.0326, wR2 0.0710 (all data); GOF 1.044. The Flack absolute structure parameter was 0.05(3), indicating refinement of the correct configuration for solid state 2. CCDC deposition number 1941099.
Crystal Data for Ethyl(chlorosulfonyl)carbamate 3b
C3H6NO4ClS (187.60 amu); data were collected on a colourless plate obtained by slow evaporation of a benzene/hexane solution measuring 0.50 × 0.32 × 0.05 mm3. Data collection at −153°C with MoKα radiation (λ 0.71073 Å); monoclinic, P21/c, Z 2; a 8.6823(14), b 9.5159(16), c 9.3527(15) Å, β 99.645(2)°; μ(Mo) 0.734 mm−1; with index ranges of −11 ≤ h ≤ 5, −12 ≤ k ≤ 12, −12 ≤ l ≤ 12; 4334 reflections were collected yielding 1813 independent reflections. The structure was solved by direct methods and expanded using Fourier techniques (SHELX 2018). All non-hydrogen atoms were refined anisotropically, while hydrogen atoms were refined isotropically. Data to parameter ratio 1813/96; R1 0.0305, wR2 0.0659 (I > 2σ(I)); R1 0.0416, wR2 0.0707 (all data); GOF 1.043. CCDC deposition number 1941100.
Crystal Data for Triethylammonium Methylcarboxysulfaminate 4
C8H20N2O5S (256.32 amu); data were collected on a colourless block obtained by slow evaporation of a benzene solution measuring 0.43 × 0.41 × 0.40 mm3. Data collection at −153°C with MoKα radiation (λ 0.71073 Å); monoclinic, P21/n, Z 4; a 8.3942(5), b 15.2853(8), c 9.9254(5) Å, β 90.1786(7)°; μ(Mo) 0.263 mm−1; with index ranges of −10 ≤ h ≤ 11, −19 ≤ k ≤ 17, −10 ≤ l ≤ 13; 7352 reflections were collected yielding 3034 independent reflections. The structure was solved by direct methods and expanded using Fourier techniques (SHELX 2018). All non-hydrogen atoms were refined anisotropically, while hydrogen atoms were refined isotropically. Data to parameter ratio 3034/157; R1 0.0272, wR2 0.0716 (I > 2σ(I)); R1 0.0295, wR2 0.0729 (all data); GOF 1.044. CCDC deposition number 1941101.
Crystal Data for Triethylammonium Ethylcarboxysulfaminate 5
C9H22N2O5S (256.32 amu); data were collected on a colourless block obtained by slow evaporation of a benzene solution measuring 0.53 × 0.33 × 0.23 mm3. Data collection at −153°C with MoKα radiation (λ 0.71073 Å); triclinic, P-1, Z 4; a 8.4796(8), b 8.7778(8), c 10.2367(10) Å, α 106.1971(12)°, β 93.6684(12)°, γ 108.6712(11)°; μ(Mo) = 0.245 mm−1; with index ranges of −11 ≤ h ≤ 9, −9 ≤ k ≤ 11, −11 ≤ l ≤ 13; 3993 reflections were collected yielding 3073 independent reflections. The structure was solved by direct methods and expanded using Fourier techniques (SHELX 2018). All non-hydrogen atoms were refined anisotropically, while hydrogen atoms were refined isotropically. Data to parameter ratio 3073/166; R1 0.0269, wR2 0.0669 (I > 2σ(I)); R1 0.0295, wR2 0.0686 (all data); GOF 1.074. CCDC deposition number 1941102.
Crystallographic Data
Crystallographic data has been deposited with the Cambridge Crystallographic Data Center for 1, 2, 3b, 4, and 5 as: CCDC 1941098, 1941099, 1941100, 1941101, and 1941102 respectively. These latter data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge, CB2 1EZ, UK; Fax: +44 1223 336033; or deposit@ccdc.cam.ac.uk).
Supplementary Material
Final energy-minimised geometries for the structures selected for G2MP2 modelling, X-ray single crystal diffraction data for 1, 2, 3b, 4, and 5 in tabular form, and images of relevant text and structures from George M. Atkins’ Ph.D. thesis are available on the Journal’s website.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
All authors thank The University of Alabama for support of the StanCE project (Technology for a Sustainable Chemical Economy). YU thanks Kitasato University for its support. DAD thanks the Robert Ramsay Chair Fund of the University of Alabama for support. The computational work was supported by the DOE BES Catalysis Program by a subcontract from Pacific Northwest National Laboratory (KC0301050-47319). The authors are especially grateful to George Atkins and Donald VanDerveer for helpful discussions.
References
[1] G. M. Atkins, E. M. Burgess, J. Am. Chem. Soc. 1968, 90, 4744.| Crossref | GoogleScholarGoogle Scholar |
[2] E. M. Burgess, H. R. Penton, E. A. Taylor, W. M. Williams, Org. Synth. 1977, 56, 40.
| Crossref | GoogleScholarGoogle Scholar |
[3] S. Burckhardt, Synlett 2000, 2000, 559.
| Crossref | GoogleScholarGoogle Scholar |
[4] C. Lamberth, J. Prakt. Chem. 2000, 342, 518.
| Crossref | GoogleScholarGoogle Scholar |
[5] E. M. Burgess, H. R. Penton, E. A. Taylor, J. Org. Chem. 1973, 38, 26.
| Crossref | GoogleScholarGoogle Scholar |
[6] D. A. Claremon, B. T. Phillips, Tetrahedron Lett. 1988, 29, 2155.
| Crossref | GoogleScholarGoogle Scholar |
[7] N. Maugein, A. Wagner, C. Mioskowski, Tetrahedron Lett. 1997, 38, 1547.
| Crossref | GoogleScholarGoogle Scholar |
[8] T. A. Metcalf, R. Simionescu, T. Hudlicky, J. Org. Chem. 2010, 75, 3447.
| Crossref | GoogleScholarGoogle Scholar | 20397656PubMed |
[9] Y. Uchiyama, S. Kimura, T. Kurotaki, Y. Sawamura, Y. Kikuchi, S. Abe, J. W. Runyon, J. S. Dolphin, C. Schinnen, A. J. Arduengo, Phosphorus Sulfur Silicon Relat. Elem. 2019, in press.
[10] P. R. Sultane, C. W. Bielawski, J. Org. Chem. 2017, 82, 1046.
| Crossref | GoogleScholarGoogle Scholar | 28035837PubMed |
[11] M. G. Atkins, Jr, Reactions of N-Sulfonylamines 1968, Ph.D. thesis, Georgia Institute of Technology. From page 42: ‘The inner salt XXXI [2 in this present manuscript], upon treatment with water, gave a monohydrate (XXXII) which reverted to XXXI on gentle heating under reduced pressure. This hydrate was considered to be the hydrogen-bonded structure shown and not the product of hydrolysis, LIII [5 in this present manuscript], because of the following spectral data.’ Images of the relevant pages are included with the Supplementary Material. Available at: https://smartech.gatech.edu/handle/1853/27323 (accessed 18 July 2019)
[12] In a conversation with AJA, Professor Burgess recalled that the X-ray structure of the putative hydrate had [may have] been completed by Dr Donald VanDerveer at Georgia Tech. Subsequent conversations between AJA and Dr. VanDerveer provided no supporting data or recollection that such a single crystal structure determination had been conducted. Neither Atkins’ Ph.D. thesis nor those of other Ph.D. candidates from Professor Burgess’s laboratory working on the chemistry of N-sulfonylamines contain any mention of this Burgess reagent hydrate X-ray structure. See also theses for: Harold R. Penton, Jr (https://smartech.gatech.edu/handle/1853/27534) and Edward A. Taylor (https://smartech.gatech.edu/handle/1853/26969) (accessed 18 July 2019).
[13] J. J. Oh, M. S. LaBarge, J. Matos, J. W. Kampf, K. W. Hillig, R. L. Kuczkowski, J. Am. Chem. Soc. 1991, 113, 4732.
| Crossref | GoogleScholarGoogle Scholar |
[14] A. J. Morris, C. H. L. Kennard, J. R. Hall, G. Smith, A. H. White, Acta Crystallogr. Sect. C 1983, C39, 81.
| Crossref | GoogleScholarGoogle Scholar |
[15] U. Jäger, W. Sundermeyer, H. Pritzkow, Chem. Ber. 1987, 120, 1191.
| Crossref | GoogleScholarGoogle Scholar |
[16] C. M. M. Hendriks, P. Lamers, J. Engel, C. Bolm, Adv. Synth. Catal. 2013, 355, 3363.
| Crossref | GoogleScholarGoogle Scholar |
[17] L. A. Curtiss, P. C. Redfern, K. Raghavachari, V. Rassolov, J. A. Pople, J. Chem. Phys. 1999, 110, 4703.
| Crossref | GoogleScholarGoogle Scholar |
[18] D. J. Grant, M. H. Matus, K. D. Anderson, D. M. Camaioni, S. Neufeldt, C. F. Lane, D. A. Dixon, J. Phys. Chem. A 2009, 113, 6121.
| Crossref | GoogleScholarGoogle Scholar | 19422181PubMed |
[19] R. G. Potter, D. M. Camaioni, M. Vasiliu, D. A. Dixon, Inorg. Chem. 2010, 49, 10512.
| Crossref | GoogleScholarGoogle Scholar | 20932027PubMed |
* In memory of Edward M. Burgess, 1934–2018, scientist, mentor, and friend.