

Carbon Dioxide Activation by a Palladium Terminal Imido Complex
Stephen J. Goodner A , Annette Grünwald A , Frank W. Heinemann A and Dominik Munz A BA Friedrich-Alexander-Universität Erlangen-Nürnberg, Department of Chemistry and Pharmacy, Institute for General and Inorganic Chemistry, Egerlandstraße 1, 91058 Erlangen, Germany.
B Corresponding author. Email: dominik.munz@fau.de
Australian Journal of Chemistry 72(11) 900-903 https://doi.org/10.1071/CH19323
Submitted: 15 July 2019 Accepted: 09 August 2019 Published: 12 September 2019
Abstract
We recently reported the first example of a palladium(ii) terminal imido complex. We proposed that this complex features exceptional high nucleophilicity at the nitrogen atom and a peculiar zwitterionic electronic structure with an anti-bonding highest-occupied molecular orbital (HOMO). This complex swiftly activated moderately acidic CH, OH, and NH bonds and also reacted with dihydrogen. However, unambiguous nucleophilic reactivity with substrates not featuring a hydrogen atom could not be observed. Herein, we now show that this nucleophilic complex also reacts with CO2 to give a ring-strained four-membered palladium(ii) carbamate complex. Remarkably, the same product is obtained in the reaction of the related bisamido complex, albeit at a slower reaction rate. Density functional theory calculations indicate that the addition of CO2 does not proceed via initial 1,2-addition across the Pd–N bond, but instead through nucleophilic attack by the imido (amido respectively) nitrogen atom.
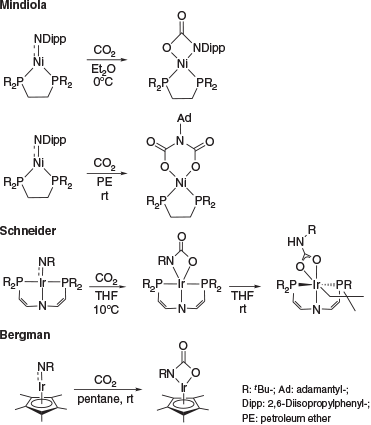
Multiple-bonded late transition metal complexes are an intriguing class of coordination compounds commonly regarded to be too unstable to be isolable.[1–6] This is due to the population of antibonding orbitals with increasing d-electron count, which typically renders these complexes excessively reactive.[7,8] Accordingly, only a handful of late transition metal complexes with a formal double bond to a nitrogen atom (i.e. terminal imido or nitrene complexes) are known.[1–6,9] Congeners with d8 configuration are extremely rare and only nickel imido complexes, which all feature considerable open-shell character, have been reported.[9–12] Whereas the reactivity of early transition metal imido complexes is comparatively well understood,[13–19] the reactivity of late transition metal congeners remains largely unexplored. To our knowledge, the only exceptions are a study by D. Mindiola (Scheme 1) on Hillhouse’s seminal d8 nickel terminal imido complex[20] and R. Bergman’s[21] as well as S. Schneider’s[22] investigations of d6-configured iridium complexes. Notably, the nickel complex was shown to react with either one equivalent of CO2 or two equivalents depending on the substituent at the imido group (aromatic versus aliphatic). Schneider’s iridium complex with a PNP-pincer ligand rearranged at room temperature to give a carbamyldiide ligand after intramolecular CH bond activation.
|
Following our computational predictions,[23,24] we reported recently the first example for a terminal imido complex of palladium (1).[25] This d8-configured nitrene complex is surprisingly stable owing to the strong ligand field, which is exerted by the ancillary cyclic (alkyl)(amino)carbene (CAAC) ligand.[26] We showed that it activates C–H, O–H, N–H, and H–H bonds even at room temperature. However, all these reactions feature hydrogen atoms in the substrate. We hence sought to explore the reactivity with other electrophiles. The study of the coordination chemistry of carbon dioxide, although dating back to the 70s,[27] is witnessing a renaissance in recent years.[28] This is because of carbon dioxide’s capacity as a ubiquitous and hence affordable C1 source in organic synthesis. Consequently, it shows potential for the synthesis of polycarbonates[29] and carbamates[22] as well as reduction chemistry for energy storage.[30] Herein, we report on the reactivity of the terminal sulfonimido complex 1 as well as the related bisamido complex 3 with carbon dioxide.
Treating a solution of 1 in pyridine with carbon dioxide led to immediate decolorization of the strongly yellow-colored solution to faint yellow (Scheme 2). The 1H NMR spectroscopic analysis confirmed quantitative and clean conversion and the 13C NMR spectrum provided indication of a carboxylated adduct with a signal at 165.3 ppm. In perfect agreement, a band in the infrared spectrum (νCO 1677 cm−1) corroborated the introduction of a carbonyl group.

|
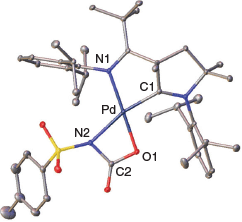
Single crystals suitable for the determination of the solid-state structure were obtained by condensing diethyl ether into a saturated solution of 2 in pyridine (Fig. 1). Indeed, 2 features a cyclic structure with the formation of a cyclic palladacarbamate (metalla-lactam respectively). Compound 2 was stable at room temperature as a solid. In contrast, solutions in pyridine or benzene showed signs of further transformation to unidentified products over the course of days.
|
Indicating a stronger trans-effect of the alkoxo in comparison with the amido ligand, the amido group is positioned trans to the strong-field carbene donor ligand. The moderate elongation of the Pd–N2 bond indicates slightly more single-bond character in 2 (2.115(2) Å) compared with 1 (2.042(2) Å). The C1–Pd bond shows a slightly increased length from 1 (1.922(2) Å) to 2 (1.934(2) Å). We attribute this to weaker π-backbonding from the palladium metal to the CAAC carbon, which is in line with reduced multiple-bond character of the amido functionality relative to the sulfonimido group. The N2–Pd–O1 angle is remarkably low (64.48(6)°), whereas the N2–C2–O1 angle is much larger (108.2(2)°) than for an idealized rectangle with 90°. We conclude that the cyclic palladacarbamate exhibits considerably cyclic ring strain. This ring strain seems to be a common pattern in agreement with the previously reported metallalactams by Mindiola and Schneider. Mindiola’s nickel complex has an N–Ni–O angle that is also low (69.24(6)°), whereas the opposite N–C–O angle is large (107.2(2)°).[20] Schneider’s iridium complex shows similar strain of 63.0(1)° and 106.50(3)° respectively.[22]
In order to compare the reactivity of the imido complex 1 with that of the related bisamido complex 3, we reacted 3 as well with carbon dioxide (Scheme 3). This reaction also proceeded quantitatively, albeit slowly at room temperature. Following the reaction by 1H NMR spectroscopic analysis revealed quantitative conversion of 3 and a mixture of compounds (including 2) after 1 h. After a prolonged reaction time (48 h), stoichiometric formation of 2 with concomitant release of para-toluenesulfonamide (TosNH2) was obtained.

|
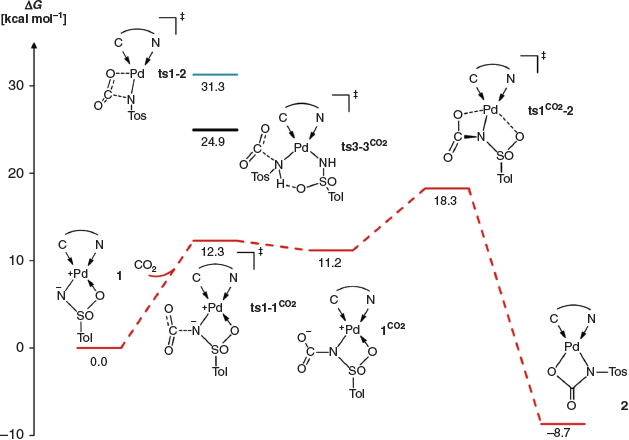
The activation of carbon dioxide by 1 could proceed by essentially two different mechanisms. First, the C=O bond could add via a 1,2-addition across the vicinal zwitterion (i.e. formal double bond) of the sulfonimido ligand. Second, carbon dioxide activation could proceed via direct nucleophilic addition of the imido nitrogen atom to the carbon dioxide molecule to give a carbamate. The latter mechanism is expected for the amido complex 3. In order to elucidate the mechanism, we therefore performed density functional theory (DFT) calculations (at the PBE0-D3BJ/def2-TZVPP(SMD)//PBE0-D3BJ/def2-SVP level of theory; Scheme 4).
|
The calculations surprisingly predicted that the 1,2-addition pathway is too high in energy (1,2ts1–1CO2, Gibbs free energy of activation ΔG‡ +31.3 kcal mol−1 [1 kcal mol−1 = 4.186 kJ mol−1]) for a reaction proceeding at room temperature. In contrast, nucleophilic attack of the imido nitrogen atom, which carries a negative partial charge, should be a very facile process (ts1–1CO2, ΔG‡ +12.3 kcal mol−1). A further energetically low-lying transition state (ts1CO2−2, ΔG‡ +18.3 kcal mol−1), which corresponds to substitution of the neutral κO coordinate sulfonyl ligand by the carboxylate group, then connects the endergonic intermediate 1CO2 (ΔG +11.2 kcal mol−1) with the final product 2. This rearrangement determines the overall reaction rate (rate-determining transition state). For the bisamido complex, the nucleophilic addition of the amido ligand onto carbon dioxide was estimated to be feasible, though slow at room temperature (ts3–3CO2, ΔG +24.9 kcal mol−1). This is consistent with the experiment (cf. Scheme 3), where 2 formed only slowly over the course of 2 days with the initial formation of a mixture.
In conclusion, we report the activation of carbon dioxide by a palladium(ii) terminal sulfonimido complex and its bisamido congener. The reaction with the terminal imido complex proceeded rapidly at room temperature and produced a peculiar four-membered, cyclic palladacarbamate. In contrast, the bisamido complex is less reactive but gave the cyclic palladacarbamate as well. DFT calculations suggest that the activation of carbon dioxide proceeds, surprisingly, via nucleophilic attack of the nitrogen atom and not via a 1,2-addition mechanism. In the largely unexplored sphere of late-stage transition metal terminal imido complexes, this investigation yields a useful introduction to palladium carbamate complex formation. It hence implies scope in the synthetic outlook for carbon dioxide utilization.
Supplementary Material
Synthetic procedures, spectroscopic data, and computational and crystallographic details are available on the Journal’s website.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
D. M. thanks the Fonds der Chemischen Industrie for a Liebig fellowship, the RRZE Erlangen for computational resources and the German–American Fulbright commission (Bundesministerium für Bildung und Forschung BMBF respectively) for financial support. Continuing support by K. Meyer is gratefully acknowledged.
References
[1] K. Ray, F. Heims, F. F. Pfaff, Eur. J. Inorg. Chem. 2013, 3784.| Crossref | GoogleScholarGoogle Scholar |
[2] P. R. Sharp, J. Chem. Soc., Dalton Trans. 2000, 2647.
| Crossref | GoogleScholarGoogle Scholar |
[3] A. R. Eikey, M. M. Abu-Omar, Coord. Chem. Rev. 2003, 243, 83.
| Crossref | GoogleScholarGoogle Scholar |
[4] J. F. Berry, Comments Inorg. Chem. 2009, 30, 28.
| Crossref | GoogleScholarGoogle Scholar |
[5] L. H. Gade, P. Mountford, Coord. Chem. Rev. 2001, 216–217, 65.
| Crossref | GoogleScholarGoogle Scholar |
[6] C. T. Saouma, J. C. Peters, Coord. Chem. Rev. 2011, 255, 920.
| Crossref | GoogleScholarGoogle Scholar | 21625302PubMed |
[7] J. R. Winkler, H. B. Gray, in Molecular Electronic Structures of Transition Metal Complexes I. Structure and Bonding (Eds D. Mingos, P. Day, J. Dahl) 2011, Vol. 142, pp. 17–28 (Springer: Berlin).
[8] J. M. Mayer, Comments Inorg. Chem. 1988, 8, 125.
| Crossref | GoogleScholarGoogle Scholar |
[9] D. J. Mindiola, G. L. Hillhouse, J. Am. Chem. Soc. 2001, 123, 4623.
| Crossref | GoogleScholarGoogle Scholar | 11457258PubMed |
[10] N. D. Harrold, G. L. Hillhouse, Chem. Sci. 2013, 4, 4011.
| Crossref | GoogleScholarGoogle Scholar |
[11] C. A. Laskowski, A. J. M. Miller, G. L. Hillhouse, T. R. Cundari, J. Am. Chem. Soc. 2011, 133, 771.
| Crossref | GoogleScholarGoogle Scholar | 21175213PubMed |
[12] V. M. Iluc, A. J. Miller, J. S. Anderson, M. J. Monreal, M. P. Mehn, G. L. Hillhouse, J. Am. Chem. Soc. 2011, 133, 13055.
| Crossref | GoogleScholarGoogle Scholar | 21797224PubMed |
[13] P. J. Tiong, A. Nova, L. R. Groom, A. D. Schwarz, J. D. Selby, A. D. Schofield, E. Clot, P. Mountford, Organometallics 2011, 30, 1182.
| Crossref | GoogleScholarGoogle Scholar |
[14] A. E. Guiducci, C. L. Boyd, E. Clot, P. Mountford, Dalton Trans. 2009, 5960.
| Crossref | GoogleScholarGoogle Scholar | 19623397PubMed |
[15] S. R. Dubberley, A. Friedrich, D. A. Willman, P. Mountford, U. Radius, Chem. – Eur. J. 2003, 9, 3634.
| Crossref | GoogleScholarGoogle Scholar | 12898691PubMed |
[16] Z. J. Tonzetich, R. R. Schrock, P. Müller, Organometallics 2006, 25, 4301.
| Crossref | GoogleScholarGoogle Scholar |
[17] C. L. Boyd, E. Clot, A. E. Guiducci, P. Mountford, Organometallics 2005, 24, 2347.
| Crossref | GoogleScholarGoogle Scholar |
[18] B. D. Ward, G. Orde, E. Clot, A. R. Cowley, L. H. Gade, P. Mountford, Organometallics 2005, 24, 2368.
| Crossref | GoogleScholarGoogle Scholar |
[19] B. D. Ward, E. Clot, S. R. Dubberley, L. H. Gade, P. Mountford, Chem. Commun. 2002, 2618.
| Crossref | GoogleScholarGoogle Scholar |
[20] D. J. Mindiola, R. Waterman, V. M. Iluc, T. R. Cundari, G. L. Hillhouse, Inorg. Chem. 2014, 53, 13227.
| Crossref | GoogleScholarGoogle Scholar | 25437507PubMed |
[21] D. S. Glueck, J. Wu, F. J. Hollander, R. G. Bergman, J. Am. Chem. Soc. 1991, 113, 2041.
| Crossref | GoogleScholarGoogle Scholar |
[22] M. Kinauer, M. Diefenbach, H. Bamberger, S. Demeshko, E. J. Reijerse, C. Volkmann, C. Würtele, J. van Slageren, B. de Bruin, M. C. Holthausen, S. Schneider, Chem. Sci. 2018, 9, 4325.
| Crossref | GoogleScholarGoogle Scholar | 29780564PubMed |
[23] D. Munz, Chem. Sci. 2018, 9, 1155.
| Crossref | GoogleScholarGoogle Scholar | 29675160PubMed |
[24] A. Grünwald, D. Munz, J. Organomet. Chem. 2018, 864, 26.
| Crossref | GoogleScholarGoogle Scholar |
[25] A. Grünwald, N. Orth, A. Scheurer, F. W. Heinemann, A. Pöthig, D. Munz, Angew. Chem. Int. Ed. 2018, 57, 16228.
| Crossref | GoogleScholarGoogle Scholar |
[26] (a) For the first report of a CAAC, see: V. Lavallo, Y. Canac, C. Präsang, B. Donnadieu, G. Bertrand, Angew. Chem. Int. Ed. 2005, 44, 5705.[Angew. Chem. 2005, 117, 5851].
| Crossref | GoogleScholarGoogle Scholar |
(b) For a leading review on CAACs, see: M. Melaimi, R. Jazzar, M. Soleilhavoup, G. Bertrand, Angew. Chem. Int. Ed. 2017, 56, 10046.[Angew. Chem. 2017, 129, 10180].
| Crossref | GoogleScholarGoogle Scholar |
(c) For the synthesis of donor-substituted CAACs, see: J. Chu, D. Munz, R. Jazzar, M. Melaimi, G. Bertrand, J. Am. Chem. Soc. 2016, 138, 7884.
| Crossref | GoogleScholarGoogle Scholar |
(d) D. Munz, J. Chu, R. Jazzar, M. Melaimi, G. Bertrand, Angew. Chem. Int. Ed. 2016, 55, 12886.[Angew. Chem. 2016, 128, 13078].
| Crossref | GoogleScholarGoogle Scholar |
(e) For a concise and recent review on the electronic properties of carbene ligands, see: D. Munz, Organometallics 2018, 37, 275.
| Crossref | GoogleScholarGoogle Scholar |
[27] M. Aresta, R. Gobetto, E. Quaranta, I. Tommasi, Inorg. Chem. 1992, 31, 4286.
| Crossref | GoogleScholarGoogle Scholar |
[28] T. Sakakura, J.-C. Choi, H. Yasuda, Chem. Rev. 2007, 107, 2365.
| Crossref | GoogleScholarGoogle Scholar | 17564481PubMed |
[29] S. Liu, X. Wang, Curr. Opin. Green Sustain. Chem. 2017, 3, 61.
| Crossref | GoogleScholarGoogle Scholar |
[30] D. Hildebrandt, D. Glasser, B. Hausberger, B. Patel, B. J. Glasser, Science 2009, 323, 1680.
| Crossref | GoogleScholarGoogle Scholar | 19325103PubMed |