

Element 25 – Manganese
Colette Boskovic AA School of Chemistry, University of Melbourne, Victoria, 3010, Australia. Email: c.boskovic@unimelb.edu.au
Australian Journal of Chemistry 72(6) 407-410 https://doi.org/10.1071/CH19107
Submitted: 6 March 2019 Accepted: 6 March 2019 Published: 25 March 2019
This essay in a series being published in the Australian Journal of Chemistry concerns this author’s favourite element, manganese. I was introduced to the chemistry of manganese when undertaking postdoctoral work with George Christou at Indiana University. My first reaction involved the disproportionation of manganese(vii) and manganese(ii) to give a trinuclear manganese(iii) complex. This one reaction encompasses several of the most appealing aspects of inorganic chemistry – the redox chemistry of metals that exist in multiple oxidation states, the change in a metal ion’s properties upon changing the number of valence electrons, the beautifully symmetric structures of polynuclear metal complexes and, of course, the colours!
There is evidence of the most common manganese-containing mineral, pyrolusite (MnO2), being used in pigments in early Stone Age cave paintings. The same mineral has been utilised to control the colour in glass-making since ancient Egyptian times. Interestingly this use comprises action as both a decolourising agent, through oxidation of other species, and as a colourant. In 2019, over 20 million tonnes of manganese will be used industrially worldwide, with an estimated 85–90 % employed in steel manufacturing. Other industrial applications include incorporation into other alloys, particularly with aluminium and copper, and the employment of manganese dioxide as a cathode material in zinc–carbon and alkaline batteries. In biology, manganese is an essential trace element for all known living organisms.[1] Manganese(ii) ions are integral to many enzyme cofactors and manganese plays the role of redox centre in several enzymes, including mitochondrial superoxide dismutase. However, perhaps the most significant role of manganese in life on Earth is in the oxygen-evolving complex (OEC) or water-oxidation complex (WOC) of Photosystem II. Again, it is the redox properties of manganese that allow the polynuclear manganese-containing active site to oxidise water to dioxygen in the photosynthetic reaction in cyanobacteria, algae, and higher plants.
Historically, the name manganese arises from the region of Magnesia in central Greece, where several brown/black-coloured minerals were found, including pyrolusite. The region also gave its name to magnesium and the iron ore magnetite. In the 1700s, the ore pyrolusite was used by alchemists in Europe for bleaching glass and by the later 1700s there was a view that this mineral contained an as yet unknown metal.[2] In 1774, Swedish chemist Carl Wilhelm Scheele described the formation of the new element chlorine from the reaction of pyrolusite with hydrochloric acid. In the same year, his fellow Swede, Johan Gottlieb Gahn, succeeded in producing a sample of impure manganese metal by reducing the manganese(iv) in the mineral by heating powdered pyrolusite with charcoal in oil. At the time of Mendeleev’s Periodic Table in 1869, manganese was properly placed in Group 7, although its heavier congeners, technetium and rhenium, were yet to be discovered.
Manganese constitutes roughly 0.1 % of the Earth’s crust, making it the 12th most abundant element and the 3rd most abundant transition metal (after iron and titanium). Over 300 different ores are known, containing manganese in the +2, +3, and +4 oxidation states. Some of the commercially most important ores are pyrolusite, rhodochrosite (MnCO3), hausmannite (Mn3O4), braunite (Mn7SiO12), and psilomelane ((Ba,H2O)2Mn5O10). Manganese generally comprises 25–45 wt-% of these ores. Four countries dominate the world manganese market and possess over 90 % of proven reserves: South Africa, Australia, China, and Gabon. In Australia, the economically demonstrated manganese resources are in Western Australia and the Northern Territory. Manganese is also found in the form of manganese nodules on the deep-ocean seabed,[3] including the Cape Leeuwin field off Western Australia.[4] These manganese and iron oxide mineral deposits are formed on, or just below, the sediment-covered sea surface by accretion of oxide layers around a nucleus. Nodules can exhibit various shapes, but commonly range in size from 1 to 12 cm in diameter. Notable are cone- or pyramid-shaped nodules, which appear to have been seeded by shark teeth. The nodules typically have three to six times more manganese than iron and manganese concentrations can be greater than 30 wt-%. Such high potential ore grades have sparked an incredible amount of interest in mining manganese nodules, although their extraction has yet to prove economically viable and the related environmental considerations are significant.
Although first isolated in 1774, pure samples of elemental manganese were not accessible until the 1930s, following the development of a purification method involving electrolysis of manganese sulfate obtained by leaching manganese from ore with sulfuric acid. For incorporation into steel, the manganese ore is mixed with iron ore and carbon, and then reduced either in a blast furnace or in an electric arc furnace. The resulting ferromanganese alloy can have a manganese content of 30 to 80 % and is then employed in steel manufacturing to allow precise control of the amount of manganese that is incorporated.
There are four known allotropes of manganese; the form stable at room temperature is referred to as α-manganese.[5] The structure of the α-form is more complex than that of the other 3d metals, comprising a body centred cubic unit cell with 58 Mn atoms of four distinct types. The magnetic behaviour of manganese also differs from that of its 3d neighbours. At room temperature, α-manganese is paramagnetic, undergoing a transition to a complex non-collinear antiferromagnetic phase at a Néel temperature of 95 K.[6] Manganese metal is silver-grey in colour. It is hard, brittle, and easy to oxidise.
Manganese heads Group 7 in the Periodic Table, marking the end of the early versus late 3d transition elements. It is more electropositive than its neighbours and generally more reactive. Inspection of many of the physical properties of the 3d transition metals reveals that local minima (melting point, boiling point, enthalpy of atomisation) or maxima (metallic radius) occur for manganese. These features arise from the large exchange energy associated with the 4s23d5 valence electronic structure and the resulting non-Aufbau configuration of the metallic band structures, resulting in weaker metallic bonding than for neighbouring metals. The most readily-available states of manganese are +2, +3, +4, +6, and +7, though all oxidation states from −3 to +7 have been observed. This redox versatility arises from the large number of valence d electrons with orbital energies not yet below those of the inert electron core. Many d electrons are available for bonding in manganese, affording very high oxidation states following the loss of d electrons, but also allowing π back bonding to stabilise low oxidation states. It is in fact the accessibility of numerous oxidation states that is key for many of the applications of manganese. The most stable oxidation state for manganese is +2 and in aqueous solution, the chemistry of manganese is dominated by the pink-coloured aqua ion [Mn(OH2)6]2+ in acidic and neutral solution. Divalent manganese ions are labile and often compete with the similar sized Mg2+ ions in biological systems. The other very well known manganese complex ion is the permanganate [MnO4]− oxoanion, incorporating high oxidation state manganese(vii). Available in neutral to alkaline solution, this tetrahedral ion is stabilised by π-donor oxo ligands. It has a characteristic purple colour and is used as an oxidant for purifying drinking water and in redox titrations (permanganometry) for analytical purposes.
By far the largest application for manganese is as an additive to steel to increase hardness, as well as act as an oxygen/sulfur scavenger. Most steels contain 0.15 to 0.8 % manganese and high strength alloys often contain 1 to 1.8 % manganese. At ~1.5 % manganese content, the steel becomes brittle, and this trait increases until ~4 to 5 % manganese content is reached, although further increase in the manganese content will increase both hardness and ductility. Most notably, mangalloy, also called manganese steel or Hadfield steel, is an alloy steel containing an average of around 13 % manganese. Mangalloy was created by Robert Hadfield in 1882 and achieved commercial success as the first alloy steel to exhibit behaviour radically differing from carbon steel. Once in its work hardened state, mangalloy has very high impact strength and abrasion resistance and is employed when resistance to severe mechanical shock and wear is required. As well as increasing the hardness of steel, manganese also plays a critical role as a scavenger. Manganese can scavenge sulfur to reduce undesirable brittleness, forming stable, high-melting manganese sulfide particles. It can also react with oxygen to prevent the formation of bubbles and pin holes and enhance corrosion resistance.
In biology, dioxygen is essential to all higher life forms, making the OEC of Photosystem II one of nature’s most important catalysts. The OEC is an oxo-bridged {Mn4CaO5} coordination cluster comprised of four manganese ions of varying oxidation state and one divalent calcium ion (Fig. 1).[7,8] The mechanism of water oxidation is yet to be fully elucidated, but the overall reaction involves extraction of four electrons and four protons from two molecules of water to generate a dioxygen molecule. The OEC operates in the photocatalytic Kok cycle (Fig. 1), involving progressive oxidation through four quasi-stable intermediates (S states). The dark-stable resting state is S1 and the subscripts S0 through to S3 indicate the number of oxidising equivalents stored within the OEC in each intermediate. Electrons are transferred individually to a tyrosine sidechain and then to the P680 chlorophyll reaction centre. On removal of the fourth electron, the metastable S4 state forms, which triggers the concerted water oxidation reaction, releasing dioxygen and protons and resetting the OEC to the S0 state. The manganese ions in the OEC cycle through the +3 and +4 valencies, although the exact valencies of the different S states, and the possible protonation of the oxo ligands, are not clear.[9,10] While early structural information on the OEC came from X-ray absorption spectroscopic data, the first crystal structure was reported in 2001 with a resolution of 3.8 Å.[11] Since then, several more structures have been reported of the S1 and S3 states, achieving a resolution of 1.9 and 2.25 Å, respectively.[12,13] The crystal structures resolved a long standing debate on the structure of the OEC, revealing that the {Mn4CaO5} coordination cluster is a distorted {Mn3CaO4} cubane with an additional manganese centre linked through a fifth μ2-oxo ligand. The peripheral manganese centre and the calcium are coordinated to substrate water molecules as aqua ligands, which ultimately react together to evolve a molecule of dioxygen.

|
Another polynuclear Mn-carboxylate complex has played a significant role in a completely different area – the development of the field of molecular magnetism. While the structure of the mixed-valence manganese(iii)/manganese(iv) dodecanuclear complex [Mn12O12(OAc)16(H2O)4] (Mn12; Fig. 2) was first reported in 1980, it was not until 1993 that evidence was found that this complex exhibits slow magnetic relaxation.[14,15] This ‘single-molecule magnet’ (SMM) behaviour arises from exclusively molecular properties, which is a very different situation to the long-range magnetic ordering that is responsible for slow magnetic relaxation in classical magnets. The implication that arises from slow magnetic relaxation associated with an individual molecule is that a single molecule can act as the tiniest of magnetic units, potentially serving as the ‘1’ or ‘0’ binary digits in a computer hard drive to achieve ultra-high-density molecule-based data storage. Moreover, quantum tunnelling of the magnetisation is also observed for Mn12, as magnetic-field dependent steps in the magnetisation hysteresis profile (Fig. 2).[16] The superposition of ‘spin up’ and ‘spin down’ states associated with such quantum tunnelling suggests a possible role for Mn12 molecules as quantum bits, which serve simultaneously as the processor and memory of a quantum computer.[17] Slow magnetic relaxation in SMMs arises from an energy barrier to reversal of the magnetisation direction. For Mn12 this is due to the large S = 10, giant spin, ground state of the complex and its easy-axis type of magnetic anisotropy. In turn, the molecular magnetic anisotropy arises largely from the approximately parallel arrangement of the Jahn–Teller elongation axes of the eight high spin d4 manganese(iii) ions in the molecule (Fig. 2). Although Mn12 has now been outstripped by SMMs based on lanthanoid(iii) ions that exhibit slow relaxation above liquid nitrogen temperatures, approaching suitability for real applications, it remains the first identified SMM and the molecule from which the entire research field arose.
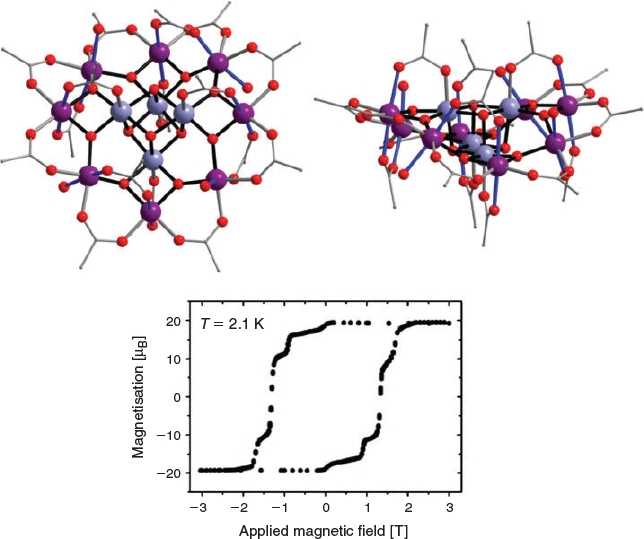
|
While manganese is essential for numerous biological processes, it has been generally rather less employed in catalytic applications than its neighbours iron and cobalt. One exception is the well-known Jacobsen’s catalyst – a N,N′-bis(salicylidene)ethylenediamine (salen) complex of manganese(iii) that catalyses the homogeneous epoxidation of conjugated alkenes. A second widely-employed manganese catalyst system is manganese(iii) acetate for radical-mediated oxidative cyclisation. Indeed, coordination complexes of relatively high-valent manganese have been developed to catalyse C–H oxygenations, nitrogenations, and halogenations.[18] Manganese(i)-catalysed organometallic C–H activations are rarer, but also known.[19,20] Recently, manganese(i) carbonyl compounds with PNP pincer ligands have been shown to have notable activity for homogenous hydrogenation and dehydrogenation catalysis.[21] As for heterogenous catalysis, manganese oxides are very promising for the gaseous catalytic elimination of a range of environmental pollutants. This includes low temperature selective catalytic reduction of NOx with NH3, the catalytic combustion of volatile organic compounds, mercury oxidation and adsorption, and soot oxidation.[22,23]
Manganese is an abundant metal, readily accessible to natural systems and for human activity. Its electronic structure affords a redox versatility that is remarkable and key to its role in many biological systems and industrial applications. In the last couple of years, the scope of manganese-based catalysis has expanded significantly. There is likely much more to come, given the increasing urgency of finding alternatives to the rare and expensive platinum group metals. With global warming and energy production at the forefront of much scientific endeavour, manganese may also offer advantages as a battery material and important advances in this area should be expected in the near future.
Conflicts of Interest
The author declares no conflict of interest.
References
[1] J. Emsley, Nat. Chem. 2013, 5, 978.| Crossref | GoogleScholarGoogle Scholar | 24153378PubMed |
[2] M. E. Weeks, J. Chem. Educ. 1932, 9, 22.
| Crossref | GoogleScholarGoogle Scholar |
[3] J. R. Hein, in Encyclopedia of Marine Geosciences (Eds J. Harff, M. Meschede, S. Peterson, J. Thiede) 2016, pp. 408–412 (Springer: New York, NY).
[4] L. A. Frakes, N. F. Exon, J. W. Granath, Commonwealth of Australia (Geoscience Australia) Bureau of Mineral Resources, Geology and Geophysics 1977. Available at: http://pid.geoscience.gov.au/dataset/ga/80931 (accessed 12 March 2019)
[5] A. J. Bradley, J. Thewlis, Proc. R. Soc. 1927, 115A, 456.
[6] D. Hobbs, J. Hafner, D. Spišák, Phys. Rev. B Condens. Matter Mater. Phys. 2003, 68, 014407.
| Crossref | GoogleScholarGoogle Scholar |
[7] F. A. Armstrong, Philos. Trans. R. Soc. B. 2008, 363, 1263.
| Crossref | GoogleScholarGoogle Scholar |
[8] L. Vogt, D. J. Vinyard, S. Khan, G. W. Brudvig, Curr. Opin. Chem. Biol. 2015, 25, 152.
| Crossref | GoogleScholarGoogle Scholar | 25621456PubMed |
[9] V. Krewald, M. Retegan, N. Cox, J. Messinger, W. Lubitz, S. DeBeer, F. Neese, D. A. Pantazis, Chem. Sci. 2015, 6, 1676.
| Crossref | GoogleScholarGoogle Scholar | 29308133PubMed |
[10] S. Petrie, R. Stranger, R. J. Pace, ChemPhysChem 2018, 19, 3296.
| Crossref | GoogleScholarGoogle Scholar |
[11] A. Zouni, H.-T. Witt, J. Kern, P. Fromme, N. Krauss, W. Saenger, P. Orth, Nature 2001, 409, 739.
| Crossref | GoogleScholarGoogle Scholar | 11217865PubMed |
[12] Y. Umena, K. Kawakami, J. R. Shen, N. Kamiya, Nature 2011, 473, 55.
| Crossref | GoogleScholarGoogle Scholar | 21499260PubMed |
[13] M. Suga, F. Akita, M. Sugahara, M. Kubo, Y. Nakajima, T. Nakane, K. Yamashita, Y. Umena, M. Nakabayashi, T. Yamane, T. Nakano, M. Suzuki, T. Masuda, S. Inoue, T. Kimura, T. Nomura, S. Yonekura, L.-J. Yu, T. Sakamoto, T. Motomura, J.-H. Chen, Y. Kato, T. Noguchi, K. Tono, Y. Joti, T. Kameshima, T. Hatsui, E. Nango, R. Tanaka, H. Naitow, Y. Matsuura, A. Yamashita, M. Yamamoto, O. Nureki, M. Yabashi, T. Ishikawa, S. Iwata, J.-R. Shen, Nature 2017, 543, 131.
| Crossref | GoogleScholarGoogle Scholar | 28219079PubMed |
[14] R. Sessoli, D. Gatteschi, A. Caneschi, M. A. Novak, Nature 1993, 365, 141.
| Crossref | GoogleScholarGoogle Scholar |
[15] R. Sessoli, H. L. Tsai, A. R. Schake, S. Wang, J. B. Vincent, K. Folting, D. Gatteschi, G. Christou, D. N. Hendrickson, J. Am. Chem. Soc. 1993, 115, 1804.
| Crossref | GoogleScholarGoogle Scholar |
[16] L. Thomas, F. Lionti, R. Ballou, D. Gatteschi, R. Sessoli, B. Barbara, Nature 1996, 383, 145.
| Crossref | GoogleScholarGoogle Scholar |
[17] J. R. Friedman, M. P. Sarachik, J. Tejada, R. Ziolo, Phys. Rev. Lett. 1996, 76, 3830.
| Crossref | GoogleScholarGoogle Scholar | 10061120PubMed |
[18] J. R. Carney, B. R. Dillon, S. P. Thomas, Eur. J. Org. Chem. 2016, 3912.
| Crossref | GoogleScholarGoogle Scholar |
[19] W. Liu, L. Ackermann, ACS Catal. 2016, 6, 3743.
| Crossref | GoogleScholarGoogle Scholar |
[20] R. Cano, K. Mackey, G. P. McGlacken, Catal. Sci. Technol. 2018, 8, 1251.
| Crossref | GoogleScholarGoogle Scholar |
[21] F. Kallmeier, R. Kempe, Angew. Chem. Int. Ed. 2018, 57, 46.
| Crossref | GoogleScholarGoogle Scholar |
[22] H. Xu, N. Yan, Z. Qu, W. Liu, J. Mei, W. Huang, S. Zhao, Environ. Sci. Technol. 2017, 51, 8879.
| Crossref | GoogleScholarGoogle Scholar | 28662330PubMed |
[23] S. Zhang, B. Zhang, B. Liu, S. Sun, RSC Adv. 2017, 7, 26226.
| Crossref | GoogleScholarGoogle Scholar |