

Element 27 – Cobalt
Paul V. Bernhardt AA School of Chemistry and Molecular Biosciences, University of Queensland, Brisbane, Qld 4072, Australia. Email: p.bernhardt@uq.edu.au
Australian Journal of Chemistry 72(4) 241-243 https://doi.org/10.1071/CH19060
Submitted: 6 February 2019 Accepted: 6 February 2019 Published: 15 February 2019
Cobalt was one of the earlier elemental discoveries, having been isolated and named in the 18th century. Therefore, Co was one of the 60 elements comprising Medeleev’s initial periodic table published in 1869.[1] However, archaeology has shown that the unmistakably ‘cobalt blue’ hues in glasses made in ancient Egypt were appreciated long before the origin of the colour was understood. The Swede, Georg Brandt, is credited with the discovery of cobalt around 1735. Its name (derived from Kobold, meaning ‘goblin’ in Germanic folklore) points to the trials and tribulations in isolating the metal in pure form, including the adverse effects on workers whose health suffered in mining its arsenic-containing ores. Although not an uncommon element, it is the rarest first row transition metal in the Earth’s crust.[2]
Co played a key role in our understanding of the field now known as coordination chemistry. The Alsatian-born Alfred Werner (1913 Chemistry Nobel Prize winner) is rightly credited with fundamental breakthroughs in this area. Werner, through a series of both theoretical and experimental papers, introduced the novel proposal of ‘secondary valence’ which we now know as metal-ligand coordinate bonding, thus superseding the conventional theory of the time that oxidation state defined the number of connected functional groups. Werner’s most influential paper was published in 1893 at the age of 26 where he successfully explained the structure and stereochemistry of a series of known CoIII compounds exhibiting different colours and stoichiometries of ammonia and chloride.[3] His deductions led to the revolutionary idea that the compounds CoCl3.3NH3, CoCl3.4NH3, CoCl3.5NH3 and CoCl3.6NH3 all comprised a central Co3+ ion with the same ‘secondary valence’ (six ligands occupying the vertices of an octahedron) which he named coordination compounds. With the addition of each successive ammonia to the formula, one chloride ion (ligand) would be displaced to be outside the coordination sphere. The compounds from that point on were understood to be [Co(NH3)3Cl3], [Co(NH3)4Cl2]Cl, [Co(NH3)5Cl]Cl2 and [Co(NH3)6]Cl3. Werner’s postulates, which met with considerable opposition at the time, were subsequently proven experimentally by both his laboratory and others through conductivity measurements on the compounds, which showed them to vary from charge-neutral non-electrolytes to trications. The ‘free’ chloride counterions were identified and quantified by the immediate formation of a precipitate upon addition of Ag+ ions which distinguished them from the tightly bound chlorido ligands rendered unreactive by the inert CoIII ion.
With the advent of his novel coordination theory, Werner also realised that several stereochemical questions arose. The emergence of six-coordinate complexes led to the possibility of a new form of steroisomerism depending on the spatial relationship of the ligands. Werner’s model was able to explain the distinctly coloured stereoisomers trans-[Co(NH3)4Cl2]Cl (praseocobalt chloride) and cis-[Co(NH3)4Cl2]Cl (violeocobalt chloride) (Fig. 1).
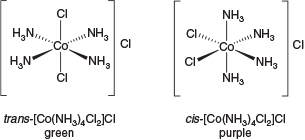
|
The inertness of trivalent cobalt facilitated an additional landmark achievement: the isolation of enantiomers of CoIII compounds bearing achiral ligands, the metal being the sole chirotopic atom (Fig. 2).[4] Up to that point, chirality was believed to be the domain of organic chemistry.
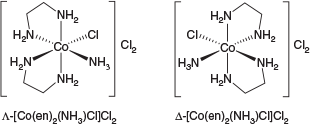
|
Many of these compounds from Werner’s laboratory (mostly crystalline) were subsequently analysed crystallographically[5]; neither X-ray crystallography nor even X-rays themselves were known in 1893 when Werner’s seminal paper[3] was published. In 2015 the author was fortunate to view this remarkable collection of compounds from Werner’s laboratory (Fig. 3) which has been kept at the University of Zürich for more than a century.
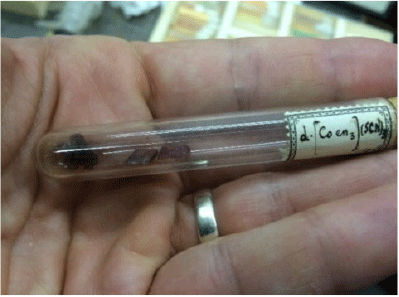
|
A characteristic feature of CoIII chemistry is inherently slow ligand exchange and it remains the classical example of an inert transition metal ion. Throughout the 20th century Co occupied a central place in the development of inorganic reaction mechanisms, including ligand substitution and electron transfer reactions. The classic experiment of Henry Taube (1983 Chemistry Nobel Prize winner) in demonstrating inner sphere electron transfer relied upon the inert CoIII reactant [Co(NH3)5Cl]2+ undergoing rapid reduction by Craq2+ mediated by the bridging chlorido ligand while reduction of the hexaammine homologue [Co(NH3)6]3+ (necessarily via an outer sphere mechanism) was nine orders of magnitude slower (Fig. 4).[6]
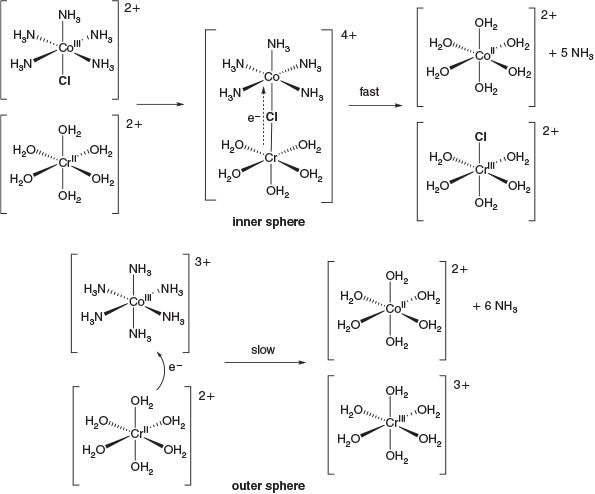
|
There have been many outstanding contributions from Australian laboratories in the area of Co chemistry and space does not allow a comprehensive review here. The mechanistic studies of Alan Sargeson and David Buckingham at the Australian National University, and their co-workers, had a particular focus on the chemistry of Co. Their contributions to our understanding of the mechanisms of ligand substitution reactions include a series of papers during the 1960s in this Journal featuring 1H NMR spectroscopy, a technique still in its infancy at that time, particularly for transition metal compounds but particularly amenable to diamagnetic CoIII complexes. In one paper, they elucidated the rates of proton-deuterium exchange in amine and β-diketonate ligands coordinated to CoIII and also examined the conformations of flexible chelate rings.[7] Another contribution from this team used 1H NMR to deduce the structure of intermediate complexes formed during ligand substitution reactions of CoIII complexes of the type trans-[CoIII(NH3)4(ND3)X]2+ (X = Cl−, Br−, N3−)[8]; selective deuteration of the ammine ligand trans to the leaving group was possible in this case. This paper showed that trigonal bipyramidal trans-[CoIII(NH3)4(ND3)]3+ was not formed though a dissociative process, rather an interchange mechanism was present which led to stereochemical retention of the square pyramidal trans-[CoIII(NH3)4(ND3)]3+ core.
However, the Co chemistry for which the Sargeson group is most recognised is the synthesis of the so-called cage complexes. In an elegant synthesis, the complex [Co(sepulchrate)]3+ (Fig. 5) was prepared in one pot and in high yield from a metal directed reaction between the precursor [Co(en)3]3+, formaldehyde and ammonia.[9] Equally impressive was the fact that the reduced CoII complex [Co(sepulchrate)]2+ was rendered inert to ligand substitution reactions and racemisation, a feat never before achieved in the chemistry of this element.[10]
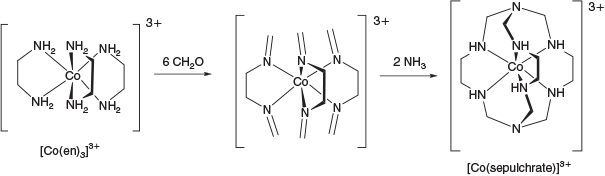
|
The chemistry of Co of course extends beyond ‘classical’ coordination compounds[11] to include organometallics,[12] catalysis,[13–15] and bioinorganic chemistry,[16] not to mention inorganic materials. The diversity of oxidation states, coordination numbers and geometries, reactivity, and its modest cost will ensure that cobalt remains one of the most popularly investigated transition metals in both academic and industrial research settings.
Conflicts of Interest
The author declares no conflicts of interest.
References
[1] Mendeleev on the Periodic Law: Selected Writings, 1869–1905 (Ed. W. B. Jensen) 2005 (Dover Publications: Mineola, NY).[2] CRC Handbook of Chemistry and Physics (90th edn) (Ed. D. R. Lide) 2009 (Taylor and Francis Group: Boca Raton, FL).
[3] A. Werner, Z. Anorg. Chem. 1893, 3, 267.
| Crossref | GoogleScholarGoogle Scholar |
[4] A. Werner, Ber. Dtsch. Chem. Ges. 1911, 44, 1887.
| Crossref | GoogleScholarGoogle Scholar |
[5] K.-H. Ernst, F. R. W. P. Wild, O. Blacque, H. Berke, Angew. Chem. Int. Ed. 2011, 50, 10780.
| Crossref | GoogleScholarGoogle Scholar |
[6] H. Taube, H. Myers, R. L. Rich, J. Am. Chem. Soc. 1953, 75, 4118.
| Crossref | GoogleScholarGoogle Scholar |
[7] D. Buckingham, L. Durham, A. Sargeson, Aust. J. Chem. 1967, 20, 257.
| Crossref | GoogleScholarGoogle Scholar |
[8] D. Buckingham, I. Olsen, A. Sargeson, Aust. J. Chem. 1967, 20, 597.
| Crossref | GoogleScholarGoogle Scholar |
[9] I. I. Creaser, J. M. Harrowfield, A. J. Herlt, A. M. Sargeson, J. Springborg, R. J. Geue, M. R. Snow, J. Am. Chem. Soc. 1977, 99, 3181.
| Crossref | GoogleScholarGoogle Scholar |
[10] I. I. Creaser, R. J. Geue, J. M. Harrowfield, A. J. Herlt, A. M. Sargeson, M. R. Snow, J. Springborg, J. Am. Chem. Soc. 1982, 104, 6016.
| Crossref | GoogleScholarGoogle Scholar |
[11] P. V. Bernhardt, G. A. Lawrance, in Comprehensive Coordination Chemistry II, Volume 6 (Eds J. A. McCleverty, T. J. Meyer) 2004, pp. 1–145 (Elsevier Ltd: Oxford).
[12] Comprehensive Organometallic Chemistry II, Volume 8: Cobalt, Rhodium and Iridium (Ed. J. D. Atwood) 2002 (Elsevier Science: Amsterdam).
[13] G. Cahiez, A. Moyeux, Chem. Rev. 2010, 110, 1435.
| Crossref | GoogleScholarGoogle Scholar | 20148539PubMed |
[14] J. P. A. Heuts, N. M. B. Smeets, Polym. Chem. 2011, 2, 2407.
| Crossref | GoogleScholarGoogle Scholar |
[15] P. Gandeepan, C.-H. Cheng, Acc. Chem. Res. 2015, 48, 1194.
| Crossref | GoogleScholarGoogle Scholar | 25854540PubMed |
[16] S. Okamoto, L. D. Eltis, Metallomics 2011, 3, 963.